

Abstract
A hallmark feature of Alzheimer's disease pathology is the presence of neurofibrillary tangles (NFTs), which are intracellular aggregates of conformationally abnormal and hyperphosphorylated tau. The presence of NFTs in the forebrain is associated with impairments of cognitive function, supporting a central role for tau in dementia. The significance of the accumulation of NFTs for neuronal and cognitive function is still obscure. It is possible that NFTs disrupt synaptic transmission and plasticity, leading to memory deficits and cognitive malfunction. To elucidate the relationship between the development of tau pathology and synaptic and cognitive functions, we performed behavioral tests and electrophysiological experiments in the htau mouse. Here we report age-dependent cognitive and physiological impairments in htau mice that preceded neurodegeneration. Twelve-month-old htau mice with moderate tau pathology, but not 4-month-old mice with early-stage tau pathology, presented cognitive deficits in an object recognition memory task in which the visual recognition memory of a novel object was disrupted. Moreover, only 12-month-old htau mice exhibit spatial memory deficits, as indicated by the impaired performance in the Morris water maze. In addition, we report that basal synaptic transmission and induction of long-term potentiation with high-frequency stimulation, but not theta burst stimulation, is perturbed in hippocampal CA1 region of old but not young htau mice. Our results suggest that tau pathology may underlie an age-dependent learning impairment through disruption of synaptic function.
Introduction
Tau is a microtubule-associated protein normally found in the axons of neurons and is thought to promote microtubule assembly and stabilization. In postmortem brains of Alzheimer's disease (AD) patients, tau is hyperphosphorylated and conformationally altered, resulting in the formation of intracellular aggregates called neurofibrillary tangles (NFTs) (Iwakiri et al., 2009). NFTs are also present in other dementias, including FTDP-17 (frontotemporal dementia and Parkinsonism linked to chromosome 17), which, unlike AD, can be caused by mutations in the tau gene (Hutton et al., 1998; Spillantini et al., 1998; Poorkaj et al., 2001). Importantly, the extent of tau pathology strongly correlates with the severity of dementia in humans (Arriagada et al., 1992; Bancher et al., 1993; Guillozet et al., 2003). Consistent with the behavioral symptoms of dementia, in AD NFTs are primarily found in brain regions that are critical for memory (Arriagada et al., 1992). Even though tau pathology has been studied in AD and other tauopathies for many years, the direct significance of NFTs accumulation for neuronal and cognitive function is still obscure. We hypothesize that NFTs are responsible for impairing synaptic function, leading to cognitive malfunction.
To investigate whether tau accumulation and aggregation is involved in inducing cognitive impairments by affecting synaptic function, we studied synaptic and cognitive functions of the htau mouse, transgenic mouse line in which the mouse tau gene is replaced by the nonmutated human tau gene (Andorfer et al., 2003). htau mice show an age-dependent development of tau pathology that has similar features to those present in AD cases, such as the regional distribution of tau pathology. Moreover, analogous to the process observed in AD, htau mice exhibit hyperphosphorylated tau, which is progressively redistributed with age from its normal axonal position to somatodendritic regions, and eventually accumulates as insoluble paired helical filaments (PHFs) (Andorfer et al., 2003, 2005). htau mice exhibit a decrease in cortical thickness and a significant reduction in the number of neurons between 8 and 18 months of age. A comparison of ventricle size between htau mice aged 8 months and 18 months revealed a dramatic increase in the older htau mice as well as a dramatic decrease in the thickness of the corpus callosum (Andorfer et al., 2005).
Here we report that 4-month-old htau mice with early-stage tau pathology (pretangles) are unaffected behaviorally and physiologically. Concurrent with the development of moderate tau pathology in the forebrain, 12-month-old htau mice develop learning and memory deficits. These old htau mice show cognitive deficits in object recognition and spatial memory, behavioral tasks that are associated with cortical and hippocampal function (O'Keefe and Dostrovsky, 1971; Morris et al., 1986; Tsien et al., 1996; Winters et al., 2004), precisely the brain regions affected by tau pathology. Behavioral deficits are accompanied by electrophysiological changes that may be the underlying cause of the cognitive deficiencies present in 12-month-old htau mice. Old htau mice also show increased paired pulse facilitation at the Schaffer collateral-to-CA1 pyramidal cell synapse, consistent with a reduction in the probability of release, and lack long-term potentiation (LTP) induced by high-frequency stimulation (HFS) but not by theta burst stimulation (TBS). We found that the inability of Schaffer collateral fibers to follow high-frequency activity can account for the lack of HFS-induced LTP. Our results strongly suggest that tau pathology causes an age-dependent learning impairment through disruption of synaptic function.
Materials and Methods
Generation of htau mice.
htau mice were generated as previously described (Andorfer et al., 2003) by crossing 8c mice that express a tau transgene derived from a human PAC, H1 haplotype driven by the tau promoter (Duff et al., 2000), with tau knock-out (KO) mice that have a targeted disruption of exon 1 of tau (Tucker et al., 2001). The F1 generation of mice that contained the human tau gene was backcrossed to the KO mice to obtain a population of mice that are homozygous for the mouse tau disruption but that also carry and express the human tau transgene. Animals were backcrossed 10 times to C57BL/6J background.
Experimental design.
All animals studied were male mice either young (4 months old) or old (12 months old) and on each individual test, transgenic mice were compared with age-matched C57BL/6J controls. Behavioral tests and electrophysiological experiments were evaluated in the same animal: following behavioral testing, the animals were killed and the brains were divided at the midline; one half of each brain was used for electrophysiology while the remaining half was fixed in 4% paraformaldehyde for immunohistochemistry to confirm that the stage of tau pathology was as expected for the age of the animals studied. The experimenter was blind to genotype for all behavioral tests. Animal handling and use followed a protocol approved by the Animal Care and Use Committee of Albert Einstein College of Medicine, in accordance with National Institutes of Health guidelines.
Immunohistochemistry.
Immunohistochemistry was performed in selected sagittal vibratome sections to assess the extent of tau pathology. In all cases, tau pathology was as expected for the age of the animal. Mouse brains were immunostained using standard streptavidin biotin peroxidase methods as described (Duff et al., 2000) and developed with 3,3′-diaminobenzidine. The following monoclonal antibodies were used to stain free-floating brain sections: CP13 [1:5000; Ps202 (Duff et al., 2000; Lewis et al., 2000)] and PHF1 [1:5000; Ps396/404 (Greenberg et al., 1992; Otvos et al., 1994)].
General behavioral phenotype.
To assess the phenotype of the htau mice, we used the SHIRPA protocol as primary screen (Rogers et al., 1997). The SHIRPA primary screen is a battery of tests that provides a behavioral and functional profile. It comprises measures covering various reflexes and basic sensorimotor functions. A total of 30 separate measurements of SHIRPA protocol were recorded for each animal that contribute to an overall assessment of muscle, lower motor neuron, spinocerebellar, sensory and autonomic function (supplemental Table 1, available at www.jneurosci.org as supplemental material). In addition, we measured the body weight of the animals (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The open-field test was used to evaluate locomotor activity and anxiety levels (supplemental Figs. 2, 3A, available at www.jneurosci.org as supplemental material). Anxiety levels were also assessed by using the elevated plus maze test (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material).
Novel object recognition task.
Mice were individually habituated to the open-field box for 6 min. The procedure comprised of a training phase followed by a preference test phase after a delay of 5 min and 30 min. During training trials, two identical objects (A1 and A2) were placed near the two corners at either end of one side of the open field (10 cm from each adjacent wall). The animal was placed into the field facing the center of the opposite wall and allowed a total of 3 min of exploration of A1 and A2. During the preference test trials (3 min duration), the animal was returned to the open field, and presented with two objects in the same positions as during training trials: one object A1, taken from the set of objects used during the training phase (familiar object), and a second novel object (B) of a different shape and color. The time spent exploring each object was scored and the results were expressed by a preference score, calculated as the time spent by each animal exploring the novel object divided by the total time spent exploring both the familiar and novel object. To avoid the presence of olfactory cues, the apparatus and the objects were thoroughly cleaned with ethanol after each individual trial, including between training and test phases. The positions of the objects in the test and the objects used as novel or familiar were counterbalanced between the animals. Exploration was operationally defined as directly attending to the object with the head no more than 2 cm from the object. New objects were used for every procedure. For each mouse, the objects were randomly assigned as either familiar or novel, thus eliminating any effect due to spontaneous preference for an object.
Morris water maze.
Spatial memory was assessed by the Morris water maze test (MWM) (Morris, 1984). MWM included 6 d of learning and memory training period and a probe trial on day 6. Mice were individually trained in a circular pool (110 cm diameter, 60 cm height) filled to a depth of 40 cm with water maintained at 25°C and made opaque using a nontoxic white paint. The maze was located in a lit room with visual cues. An escape platform (10 cm diameter) was placed in the center of one quadrant of the pool. The platform's position was fixed and the animal's starting positions, facing the pool wall, were pseudo-randomized for each trial. Each mouse was separately tested on both visible-platform and hidden-platform versions of the water maze. The hidden-platform version evaluates spatial learning, and it was used to determine the retention of memory to find the platform. During the hidden-platform training trials, the escape platform was placed 1 cm below the surface of the water. On each day, the animal was subjected to three trials with a 20 min interval between trials. Each trial lasted for 60 s unless the animal reached the platform first. The time that elapsed until the mouse reaches the platform (a successful escape) was noted. If an animal failed to find the platform within 60 s, the test was ended and the animal was gently navigated to the platform by hand. Whether a mouse found or failed to find the platform within 60 s, it was maintained on the platform for 30 s. On the last day (day 6), 6 h after the last trial, the platform was removed from its previous location and the animals were given a probe trial in which they had 3 min to search for the platform. The time to reach the missing platform and the number of times the animals crossed the platform location was recorded. All animals underwent nonspatial pretraining during the first 2 training days, which prepared the animals for the spatial learning test. During the visible trials of the water maze, mice were trained to find the platform, made visible to the mice by a small flag (5 cm tall). Data of the escape latency, the distances traveled, swim speed, and number of platform location crossings were collected by the video equipment and processed by a computer equipped with an analysis-management system (Viewer2 Tracking Software, BIOBSERVE).
Electrophysiology.
Transverse hippocampal slices (400 μm thick) were prepared from 4- or 12-month-old male htau and wild-type mice. Animals were deeply anesthetized using isoflurane and killed by decapitation. The brain was quickly removed and the hippocampus from one hemisphere was dissected in an ice-cold cutting solution. Slices were cut on a DTK-2000 microslicer (Dosaka) in ice-cold extracellular solution containing the following (in mm): 215 sucrose, 2.5 KCl, 20 glucose, 26 NaHCO3, 1.6 NaH2PO4, 1 CaCl2, 4 MgCl2, and 4 MgSO4. The cutting medium was gradually switched to the recording solution (ACSF) that contained (in mm) 124 NaCl, 2.5 KCl, 10 glucose, 26 NaHCO3, 1 NaH2PO4, 2.5 CaCl2, and 1.3 MgSO4. The slices were kept at room temperature for at least 1.5 h before being transferred to the recording chamber. Cutting and recording solutions were both saturated with 95% O2 and 5% CO2, pH 7.4. All recording were performed at 25.0°C. A patch-type pipette filled with 1 m NaCl was used to record field EPSPs (fEPSPs) from the middle third of stratum radiatum of the CA1 region. fEPSPs were evoked by stimulation of the Schaffer collateral/commissural pathway at 0.05 Hz with a patch-type pipette (monopolar stimulation) filled with extracellular solution. The stimulating and recording pipettes were positioned at the same depth in the slice and the distance between them was kept constant (∼200 μm). Input/output (I/O) curves were generated using stimulus intensities from 0 to 100 μA in increments of 10 μA and plotted as a function of fiber volley amplitude. Paired-pulse facilitation (PPF) was assessed by using a range of interstimulus intervals (ISIs) (10, 40, 80 120, 200, and 400 ms). Before LTP induction, stable baseline responses were acquired for 15 min at an intensity that evoked a response ∼50% of the maximal evoked response. LTP was induced at baseline stimulus intensity using either HFS or TBS. HFS consisted of four trains of 50 pulses at 100 Hz with 10 s intertrain intervals. TBS consisted of a series of 10 bursts at 5 Hz, with 5 pulses per burst at 100 Hz, delivered four times (5 s apart). The magnitude of LTP was estimated by comparing averaged responses 30–45 min after induction with baseline-averaged responses before induction. To block inhibitory transmission in LTP experiments, 100 μm picrotoxin was added to the external solution. For statistical analysis, responses were collected and every 3 responses (1 min periods) for the LTP experiments or every five responses for the I/O and PPF experiments were averaged. fEPSP slope or amplitude were calculated. To estimate the total charge transfer produced during LTP induction, responses to the burst of synaptic activity during the tetanus (HFS and TBS) were first normalized to the amplitude of the first pulse. Next, the area under the response curve (fEPSP) during the burst was integrated, yielding a normalized measure of total charge transfer. Recordings were performed with a MultiClamp 700A amplifier (Axon Instruments), and output signals were filtered at 3 kHz. Data were digitized (20 kHz) and analyzed online using a macro written in IgorPro (Wavemetrics). All chemicals were obtained from Sigma-Aldrich).
Statistical analyses.
Data were analyzed with appropriate statistics including Student's t test, one- or two-way ANOVAs, or, in the case of the water maze test, repeated-measures ANOVA (RMANOVA), followed by Tukey post hoc test (when necessary). p values <0.05 were considered to be statistically significant. Data were reported as mean ± SEM.
Results
In this study, behavioral, electrophysiological and histological analyses were conducted on the same htau and wild-type mice. We first tested young (4-month-old) and old (12-month-old) male htau mice in behavioral tests followed by electrophysiological experiments using acute hippocampal slices from one of the hemispheres. The remaining hemisphere of each animal was used for immunocytochemical staining for phosphorylated tau. Hence, we were able to evaluate the distribution and qualitatively assess the degree of the pathology of all the animals.
htau mice develop tau pathology in an age-dependent manner
We confirmed by immunohistochemistry that all htau animals from the two age groups exhibited an age-dependent tau pathology with very similar features to those present in human AD cases, as previously reported (Andorfer et al., 2003, 2005). Phospho-tau antibodies were used to detect different aspects of tau pathology. The monoclonal antibody CP13, specific to phosphorylated serine 202 (Ps202) on tau, is commonly used to detect tau pathology in both early (pretangle) and more advanced stages of NFTs accumulation. PHF1 is a marker for later stage tangles that is specific for phosphorylation at serines 396 and 404 (Ps396/404). Both markers indicated that the development of tau pathology was restricted to the forebrain, with the hippocampal formation being one of the first regions to be affected. An age-dependent change in the localization of CP13-positive tau was observed in the htau mice (Fig. 1). The pathology developed early in the CA1 and CA3 regions of the hippocampus. Young htau mice (4 months old) show modest somatodendritic accumulation of CP13-reactive tau (Fig. 1 A,B) and very few PHF1-positive neurons in CA3 (Fig. 1 C) and CA1 (Fig. 1 D), a typical staining pattern of early (pretangle) tau pathology. As htau mice aged, at 12 months, the redistribution of tau and CP13 reactivity was notably enhanced in both CA3 (Fig. 1 E) and CA1 (Fig. 1 F). Somatodendritic accumulation of tau was evident in that 12-month-old htau mice have many PHF1-positive cells in the CA3 (Fig. 1 G) and CA1 (Fig. 1 H) regions of the hippocampus. At 12 months, htau mice exhibit hippocampal CP13 and PHF1 reactivity that resembles reactivity of human AD at moderate stages of tau pathology.
Figure 1.

Tau pathology in the hippocampus of htau mice. There was an age-dependent change in the localization of CP13 (Ps202)-positive tau in the htau mice. Young (4-month-old) htau mice show modest somatodendritic accumulation of CP13 (Ps202)-reactive tau in CA3 (A) and CA1 (B). Four-month-old mice show very few PHF1 (Ps396/404)-positive neurons in CA3 (C) and CA1 (D). In 12-month-old htau mice, the redistribution of tau and CP13 reactivity was notably enhanced in both CA3 (E) and CA1 (F). Somatodendritic accumulation of tau in old (12-month-old) htau mice have many PHF1-positive cells in CA3 (G) and CA1 (H). Scale bars: A–H, 40 μm.
htau mice develop learning and memory deficitsin an age-dependent manner
htau mice were evaluated in the SHIRPA primary screening. htau mice showed no differences in general health, basic reflexes, sensory responses, or gross motor function compared with age-matched controls (supplemental Table 1, available at www.jneurosci.org as supplemental material). Transgenic mice did not have differences in weight compared with controls (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In addition, locomotor activity (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) and anxiety levels (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material) assessed by open field and elevated plus maze were unaffected. Therefore, at 4 months and 12 months of age, htau mice exhibit normal general behavioral phenotype and no sensorimotor deficits, making them an excellent model for studying cognitive behavior.
To establish whether htau mice exhibit learning and memory deficits, we conducted learning tasks relevant to the forebrain structures, such as hippocampus and neocortex. To examine whether htau mice have deficits in visual recognition memory, we measured familiarity discrimination by an object recognition memory test that measured spontaneous preference for a novel object compared with a familiar object (Ennaceur and Delacour, 1988). Object recognition memory was unaffected in young htau mice: the animals spent significantly more time exploring the novel object compared with the familiar object that yields a preference score higher than 50% (Fig. 2). In contrast, object recognition memory was disrupted in old htau mice compared with age-matched controls: old htau mice had no preference for the novel object compared with the familiar object at both 5 min and 30 min delays, showing preference score of ∼50%, thus performance at chance levels (Fig. 2).
Figure 2.

Object recognition memory was disrupted in aged htau mice (black bars) compared with age-matched wild type (white bars). Comparison of preference score, calculated as the time spent exploring the novel object divided by the total time spent exploring both the familiar and novel object, multiplied by 100. Dotted line represents performance at chance (50%). Aged htau mice had no preference for the novel object compared with the familiar object at both 5 min and 30 min delays, showing score preference of ∼50%, thus performance at chance (12-month-old mice: wild type, n = 13; htau, n = 8). Object recognition memory was unaffected in young htau mice. Young htau and young wild-type mice exhibit similar preference toward the novel object, spending significantly more time exploring the novel object than the familiar object, which yields a preference score higher than 50% (4-month-old mice: wild type, n = 5; htau, n = 4). All values are expressed as means ± SEM. * indicates significant difference from age-matched wild type, p < 0.05.
To test spatial learning and memory, we used the MWM. During the cued platform version of MWM, the mice learn to find a visible platform using a visual strategy. The time to escape to the visible platform (escape latency) of both young and old htau mice did not differ from the escape latency of age-matched controls (Fig. 3 A,C). htau mice performed similarly to controls in the cued platform version of the MWM, indicating that they have no visual or swimming deficiency. To test whether htau mice have spatial memory deficits, the mice were evaluated in the hidden-platform version of the MWM. The performance of young htau mice did not differ from young controls (Fig. 3 B). Old control animals (12 months old) showed decreased escape latency on the first day of training and further improved their performance by the second day, when they seemingly learned the task. In contrast, old htau mice showed impaired spatial memory by performing significantly worse than age-matched controls from days 1 to 3 and did not learn the task until the fourth day of training (Fig. 3 D). Swimming speed during the hidden platform test was similar in controls and htau mice, indicating that the prolonged escape latency of the htau mice does not reflect impaired motor control (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), consistent with the results from the SHIRPA exam. Rather, we can conclude that old htau mice have a delay in learning the location of the platform. Probe trials, in which the platform was removed and mice were given 3 min to explore the pool, showed impaired spatial memory in old htau mice compared with age-matched controls (Fig. 3 E,F). Old htau mice crossed the target platform location (platform location before its removal) significantly less often than controls (Fig. 3 E). Moreover, old htau mice took significantly longer than controls to find the target platform location (Fig. 3 F). Results from the probe trial indicate that htau mice did not recall where the platform was located, suggesting a deficit in memory retention. Together, our results suggest that 12-month-old htau mice have learning and memory deficits as shown by impaired object recognition memory and deficits in spatial memory.
Figure 3.
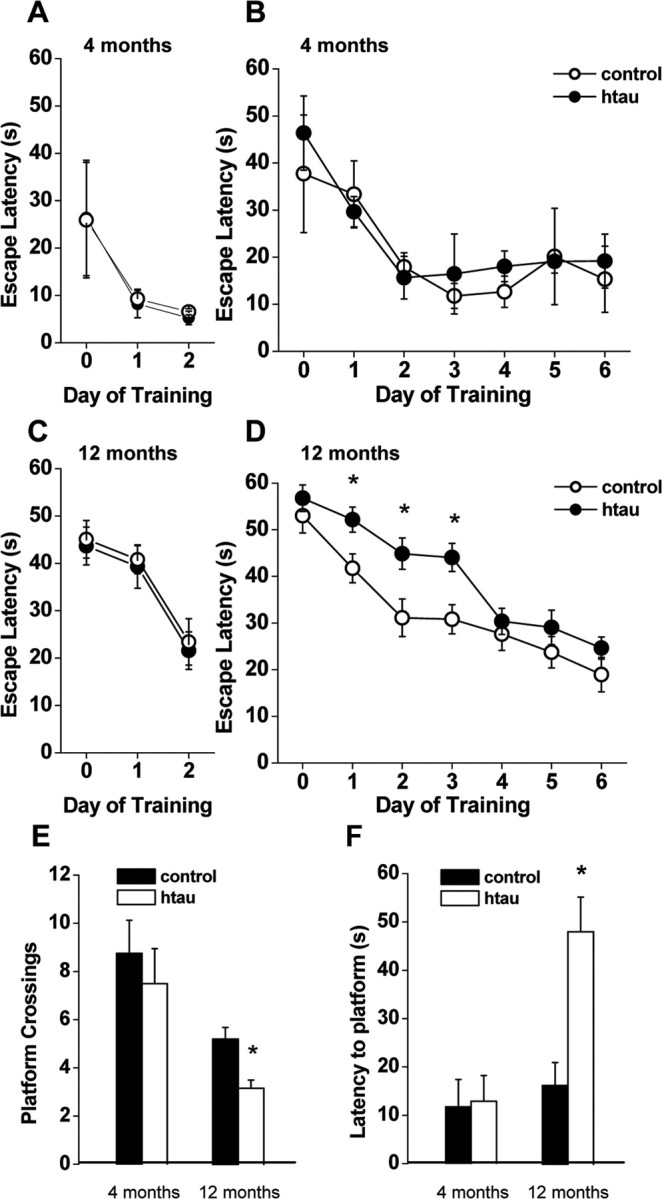
Impaired spatial memory in aged htau mice. A, C, Cued platform learning curves. Day 0 indicates performance on the first trial, and subsequent points represent average of all daily trials. The performance of young and aged htau mice (filled circles) did not differ from age-matched wild type (open circles). A, Four-month-old animals (wild type, n = 5; htau, n = 4). C, Twelve-month-old animals (wild type, n = 16; htau, n = 17). B, D, Hidden platform learning curves. The performance of young htau mice did not differ from young wild type (B). D, Aged htau mice: hidden platform learning curves differed by genotype (RMANOVA: p = 0.0011). In post hoc comparison, aged wild-type mice gradually improved their performance, starting to improve during the first day of training (p = 0.025) and further improving by the second day (p = 0.04) when they learned the task, whereas aged htau mice did not showed evidence of learning until the fourth day of training (p = 0.002). Aged htau mice showed impaired spatial memory by performing significantly worse than age-matched wild type on training days 1 (p = 0.015), 2 (p = 0.0124), and 3 (p = 0.0027). E, F, Probe trials 6 h after completion of 6 d of hidden platform training. E, Number of target platform crossings: aged htau mice crossed the target platform location significantly less often than age-matched wild type. F, Escape latency to target platform location: aged htau mice took significantly longer than wild type to find the target platform location. All values are expressed as means ± SEM. * indicates significant difference from age-matched wild type, p < 0.05.
htau mice develop synaptic dysfunctionin an age-dependent manner
LTP is a widely accepted cellular model for learning and memory (Bliss and Lomo, 1973; Bliss and Collingridge, 1993). LTP is a use-dependent enhancement in synaptic efficacy that can be induced by activating synapses briefly at high frequency. The behavioral deficits we observed suggested impairment in synaptic transmission and plasticity in the hippocampus. To test this hypothesis, we performed electrophysiological field recordings in acute slices obtained from the animals used for behavioral tests.
We first examined basic synaptic properties in young (4-month-old) and old (12-month-old) htau mice. We tested paired-pulse ratio (PPR), a form of short-term plasticity which is traditionally used as a measure of the probability of transmitter release (Pr). PPR is inversely correlated with Pr, such that synapses with low Pr show PPF, whereas synapses with high Pr show paired-pulse depression (PPD) (Manabe et al., 1993; Thomson, 2000). PPR was unaffected in young htau mice (Fig. 4 A), whereas old htau mice showed an increase in PPR compared with controls (Fig. 4 B), suggesting a decrease in probability of transmitter release. Therefore, the finding that old htau mice have increased PPR is indicative of a presynaptic deficit. To further investigate basic synaptic properties, we determined I/O curves by stimulation of Schaffer collaterals with a range of stimulus intensities. Young htau mice did not show changes in I/O curve compared with controls. Intriguingly, despite the reduction in the probability of transmitter release, 12-month-old htau mice showed an increase in the I/O function in which responses were larger for any given fiber volley amplitude (except the first) compared with controls (Fig. 5 A,B). This increase could be a direct result of tau accumulation on CA1 cells or a compensatory response to reduced excitatory input by increasing excitability (see Discussion).
Figure 4.
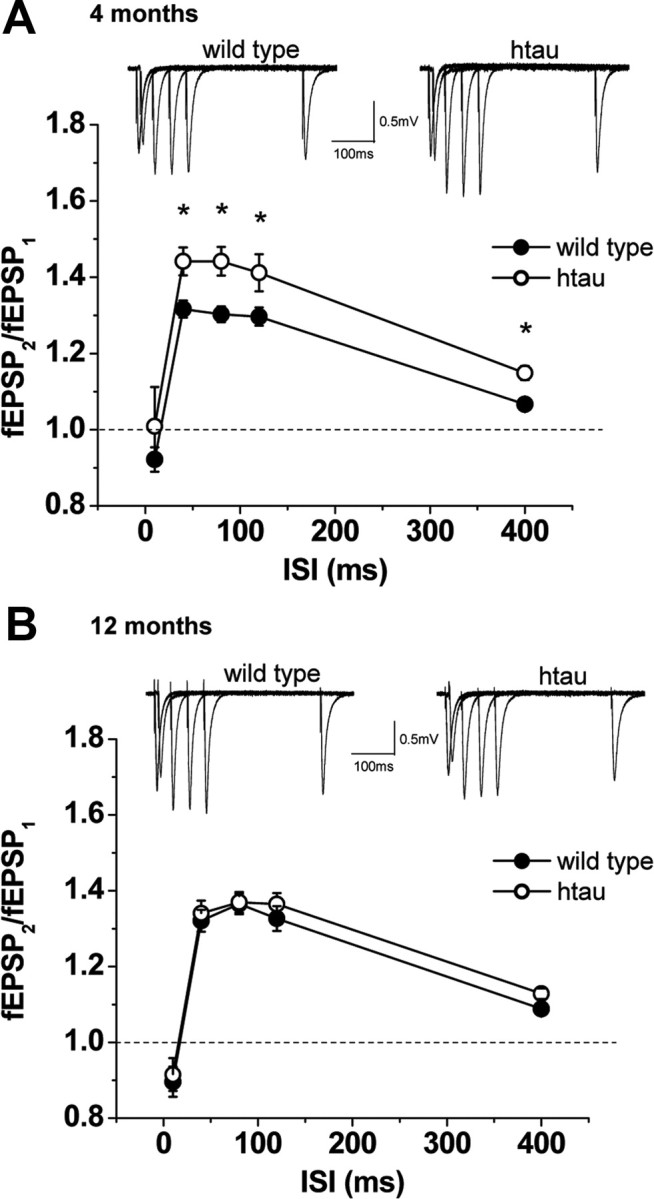
Excitatory synaptic responses to paired-pulse stimulation in htau mice. PPF and representative fEPSPs at different ISIs. PPF (fEPSP2/fEPSP1) was recorded as a function of different ISIs at Schaffer collateral/CA1 pyramidal cell synapses for wild-type (open circles) and htau (filled circles) mice at 4 months [wild type, n = 10 (4); htau, n = 10 (4)] (A) and 12 months [wild type, n = 10 (7); htau, n = 8 (5)] (B). A, At 4 months of age, htau mice exhibit normal PPF compared with age-matched wild type, whereas at 12 months of age, htau mice exhibited significantly enhanced PPF. All values are expressed as means ± SEM. * indicates significant difference from age-matched wild type, p < 0.05. n = number of slices (number of animals).
Figure 5.
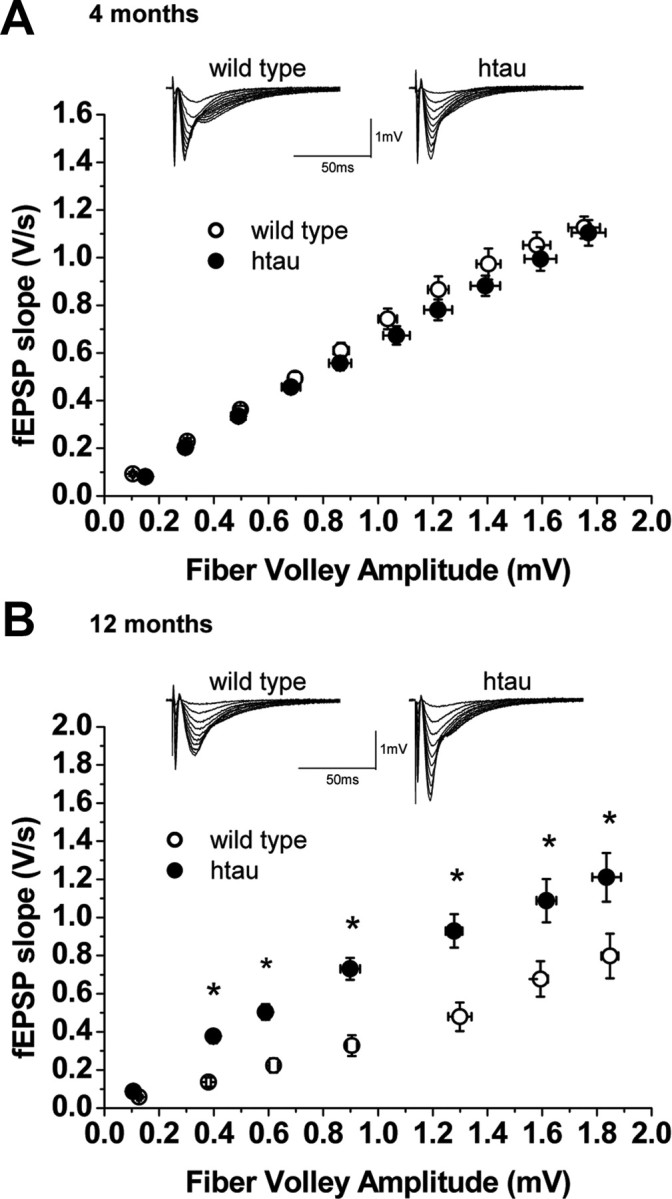
Synaptic efficacy at Schaffer collateral/CA1 pyramidal cell synapses in htau mice. Input–output curves and representative fEPSPs at increasing stimulus strengths are shown for wild-type (open circles) and htau (filled circles) mice at 4 months [wild type, n = 11 (4); htau, n = 10 (4)] (A) and 12 months [wild type, n = 10 (4); htau, n = 8 (4)] (B). fEPSP slope is plotted as a function of fiber volley amplitude showing no differences in basal synaptic transmission between wild-type and htau slices at 4 months. All values are expressed as means ± SEM. * indicates significant difference from age-matched wild type, p < 0.05. n = number of slices (number of animals).
We next assessed whether LTP is altered in acute hippocampal htau slices by delivering either HFS or TBS. HFS-LTP was normal in young htau mice (Fig. 6 A). In contrast, HFS-LTP was abolished in old htau mice but normal in age-matched controls (Fig. 6 B). Surprisingly, LTP induced by TBS was not compromised in old htau mice and did not differ from age-matched controls (Fig. 6 C). Twelve-month-old controls show no difference in the amount of HFS-LTP and TBS-LTP (Fig. 6 B,C).
Figure 6.
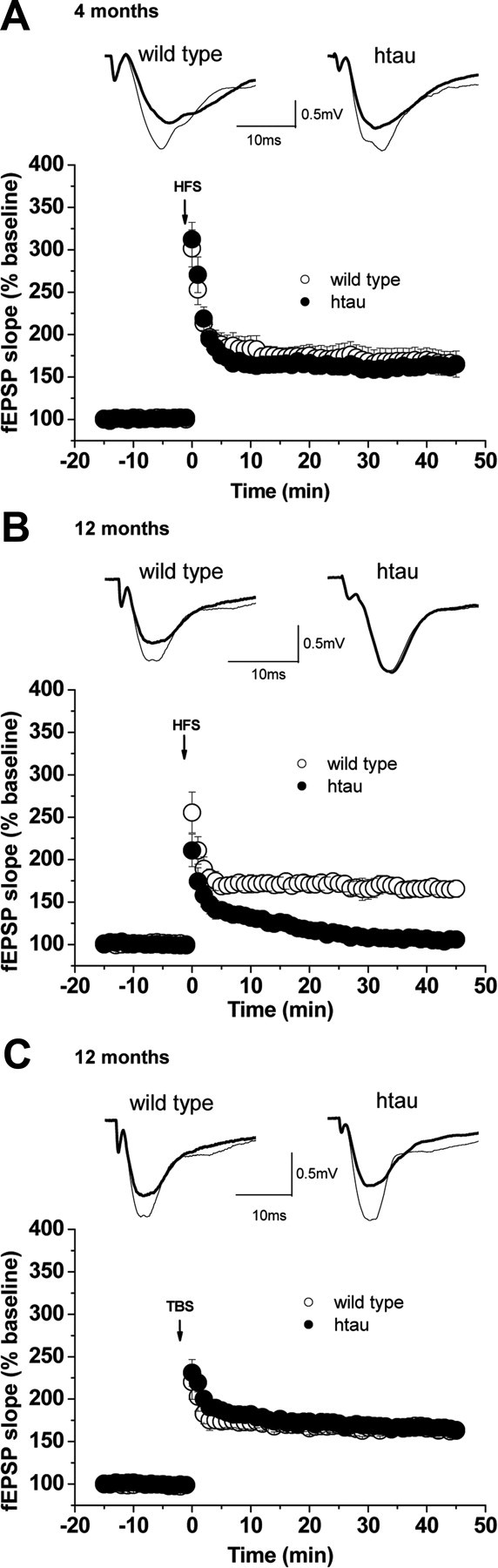
LTP in htau mice. LTP was induced by HFS trains and TBS. fEPSP slopes were recorded and were expressed as the percentage of the pretetanus baseline. Representative fEPSP traces before (bold line) and 30–45 min after (thin line) the induction of LTP are shown for wild-type (open circles) and htau (filled circles) mice at 4 months [wild type, n = 9 (4); htau, n = 8 (4)] (A) and 12 months [wild type, n = 7 (5); htau, n = 8 (5)] (B). A, LTP was normal in htau mice at 4 months of age. B, HFS-induced LTP was markedly impaired in aged htau mice. The amount of potentiation between 30 and 45 min after HFS was 167 ± 9.5% in 12-month-old wild type, while there was no significant potentiation in aged htau mice (107 ± 2.2%, p = 0.00001). C, TBS-induced LTP was normal in 12-month-old htau mice [wild type, n = 12 (7); htau, n = 12 (8)]. The amount of potentiation between 30 and 45 min after TBS was 164 ± 6.8% in wild-type and 166 ± 5.6% in htau mice. All values are expressed as means ± SEM. * indicates significant difference from age-matched wild type, p < 0.05. n = number of slices (number of animals).
The fact that TBS-LTP is normal in htau 12-month-old mice indicates that the expression of LTP is conserved in these mice. We therefore hypothesized that the absence of HFS-LTP may reflect an induction deficit. Given the presynaptic deficits we observed in the old htau mice, the deficit in HFS-LTP in old htau mice may be a result of the inability of Schaffer collateral fibers to sustain high-frequency activity. To test this possibility, we compared fEPSP responses from old htau mice and controls during the tetanus (HFS and TBS). We estimated the total charge transfer produced during LTP induction by integrating the area under the response curve during the burst, the responses of which were first normalized to the amplitude of the first pulse. Interestingly, we found that while bursts of synaptic activity induced by TBS did not differ between wild-type and htau mice (Fig. 7 A), htau mice showed a significant reduction of HFS-bursts compared with control animals (Fig. 7 B). Because HFS consists of four trains, we separately calculated the total charge transfer for responses to individual trains and found that the reduction of HFS-burst responses in htau mice emerged during the first train and persisted for the second and third trains (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Such a reduction likely underlies the inability of HFS to induce LTP in old htau mice.
Figure 7.
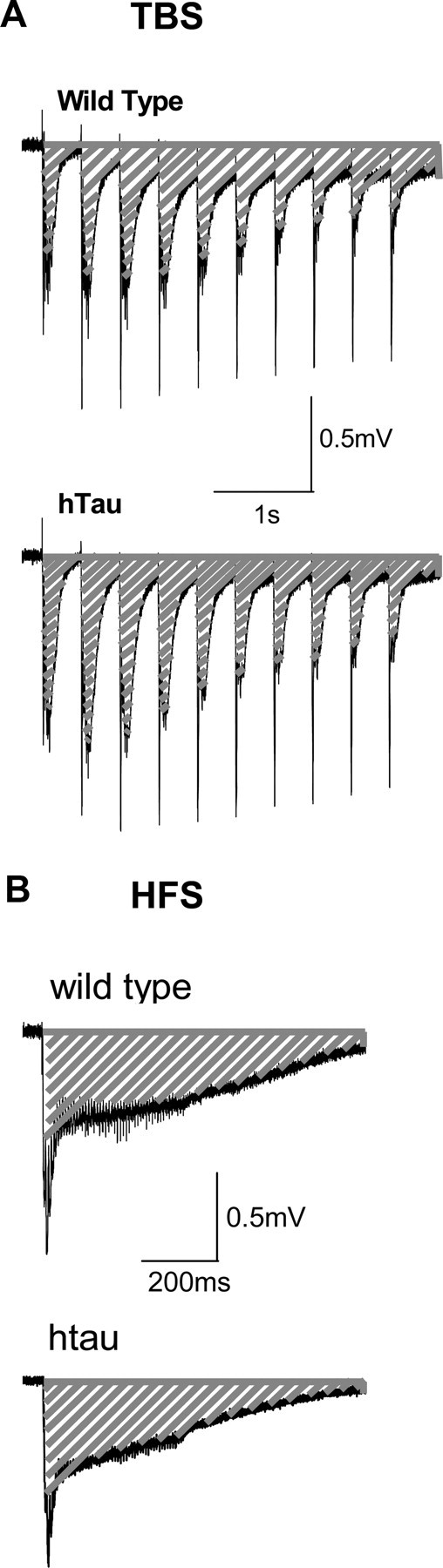
Total charge transfer during induction of LTP. Representative fEPSP traces corresponding to the first burst of TBS and HFS are shown for wild-type and htau mice at 12 months. The total charge transfer was estimated by the fEPSP integral (dashed area) during the delivery of TBS (A) and HFS (B). A, TBS produced similar total charge transfer in wild-type (100 ± 12.22) and htau (111.6 ± 1.43) mice. B, HFS produces greater charge transfer in slices from wild-type mice (100 ± 11.63) than in those from htau mice (68.3 ± 1.95, p < 0.05) [wild type, n = 3 (3); htau, n = 3 (2)]. n = number of slices (number of animals).
Discussion
The results of the present study suggest that progressively developing tau pathology may cause an age-dependent learning impairment, possibly through disruption of synaptic transmission. As previously described, and further confirmed in the present study, htau mice develop age-dependent and progressive tau pathology (Andorfer et al., 2003). Behavioral tests assessing learning and memory showed that young htau mice, with early stages of tau pathology (pretangles), did not present any cognitive deficits. As tau accumulation progresses to a moderate stage of tau pathology, characterized by the presence of NFTs, cognitive function also declines. Specifically, old htau mice have spatial memory deficits in the MWM, which is known to be dependent on the integrity of Schaffer collateral to CA1 pyramidal cell synapse (O'Keefe and Dostrovsky, 1971; Morris et al., 1986; Tsien et al., 1996). In addition, old htau mice have a deficit in memory retention as assessed by the probe trial. Furthermore, object discrimination was impaired as shown by a deficit in object recognition test. Together, the behavioral data suggest hippocampal and neocortical-dependent memory deficits.
We show that basic synaptic function was dysregulated at the Schaffer collateral to CA1 pyramidal cell synapse of htau mouse in an age-dependent manner. Only the old htau mice exhibited a change in PPR that is indicative of a decrease in probability of glutamate release. Given that old htau mice present cognitive impairment as shown by deficits in hippocampal related behavioral tasks, and changes in basic synaptic function, we tested whether synaptic plasticity was also altered in an age-dependent manner in htau mice. HFS failed to induce LTP in old htau mice, while LTP induced by TBS was unchanged. Hence, unaffected TBS-LTP implies that the necessary machinery for LTP expression by CA1 pyramidal neurons may be intact. Together, our results strongly suggest a presynaptic deficiency at the induction level but not at the expression level. We propose that diminished charge transfer during HFS is a result of the inability of Schaffer collateral fibers to sustain high-frequency activity, which in turn is responsible for the inability of HFS to induce LTP.
Cognitive and synaptic functions are normal in young htau mice, when there is no evidence of significant NFT formation. Upon the development of moderate tau pathology, old htau mice develop learning and memory deficits as well as synaptic dysfunction. We provide evidence that NFT formation (or alternatively a process upstream of NFT formation) may underlie the synaptic dysfunction and, perhaps, the cognitive decline. Future studies will need to clarify precisely how tau accumulation can affect the function of neurons in the brain of htau mice. Our findings support the notion that tau pathology may result in abnormal neuronal function presynaptically in the axons from CA3. Tau is normally an axonal soluble protein that promotes microtubule assembly and stabilization. In tau pathology, tau is redistributed from the normal axonal location (presynaptic) to the cell body and dendrites (postsynaptic). The observed increase in input–output curve could be a direct effect of tau accumulation (or NFT formation) postsynaptically or an attempt to compensate for the reduction in transmitter release by increasing excitability. Thus, the deleterious effects of tau pathology in cognitive and physiological functions may be due to a gain of function by tau. However, tau pathology could also reflect a loss-of-function defect (i.e., the inability of tau to bind and promote the assembly of microtubules). The absence of tau in the axons of affected neurons could cause an axonal cytoskeleton disruption which may lead to impaired axonal transport (Kanai et al., 1989; Knops et al., 1991; Esmaeli-Azad et al., 1994; Liu et al., 1999; Takei et al., 2000; Dawson et al., 2001). Therefore, the loss of function of tau in Schaffer collateral fibers could be responsible for presynaptic dysfunction. We found evidence to support the notion of a presynaptic impairment such as increased PPR, suggesting a decrease in presynaptic transmitter release. Moreover, we report a decrease of total charge transfer during HFS which advocates for the possibility that Schaffer collateral fibers are incapable of sustaining high-frequency activity. Thus, rather than being simply a consequence of a gain of function by abnormal tau, pathological tau may also induce cognitive deficits by disruption of synaptic function through a loss-of-function defect.
The present study showed for the first time that accumulation and aggregation of nonmutant human tau isoforms may cause cognitive and synaptic dysfunctions. Although tau pathology has been extensively studied for years, until now, the impact of tau pathology on neuronal and cognitive function was unknown. A probable explanation for this gap in the literature is the deficiency of a good experimental model. To date, the studies that have attempted to address the functional significance of tau pathology in neurons were performed in transgenic mouse models that express mutant human tau genes, making them good models for inherited tauopathies. A problem with these mouse models is that they develop brainstem and spinal cord tau inclusions, which leads to progressive motor disturbances (Goedert et al., 1999; Lewis et al., 2000; Arendash et al., 2004), thereby imposing limitations on the use of these animals for behavioral and physiological studies. Ramsden et al. (2005) created the rTg(tauP301L)4510 mouse that expressed transgenic mutant human tau (P301L) driven by Ca2+/calmodulin kinase II promoter, thus expressing the transgene exclusively in the forebrain. Even though these mice do not have spinal cord pathology, they do show motor impairments, such as hunched posture with limb dysfunction and tail rigor. These mice also develop behavioral deficits in the Morris water maze as early as 2.5 months and, although this deficit gets worse with age, it coincides with neural loss at 5–10 months. Moreover, these mice show decreased swim speed at 9.5 months, and therefore the behavioral deficits are not clear and could be a consequence of dystonic posture with tail rigor (Ramsden et al., 2005). In contrast, htau mice develop forebrain tau pathology exclusively without brainstem and spinal cord tau involvement. In this study, evaluation of htau mice using a broad-based screening approach revealed that these htau mice exhibit normal general behavioral phenotype with no gross motor deficits, and similar swimming abilities compared with age-matched controls. The absence of sensorimotor deficits, in conjunction with the regional distribution of tau pathology that matches well with that observed in AD patients, makes the htau mouse a good model for studying the functional consequences of tau pathology.
Here we showed that htau mice have cognitive deficits and physiological impairment at 12 months. It was previously reported that htau mice do not show significant neuronal death until after 14 months of age (Andorfer et al., 2005). Therefore, problems in learning and memory precede neurodegeneration in htau mice, providing evidence that tau accumulation and aggregation into NFTs may be involved in inducing cognitive impairments by affecting synaptic function rather than merely causing neuronal death. Defining the relevant mechanisms involved in tau pathology induced neuronal dysfunction could lead to therapies aimed at living neurons, a strategy with greater potential for success than intervening once neurons are dead.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant NS048447. We thank Dr. Chiayu Chiu, Dr. Boris Heifets, Dr. Maria Gulinello, and Dr. Vivien Chevaleyre for their assistance in this work.
References
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendash GW, Lewis J, Leighty RE, McGowan E, Cracchiolo JR, Hutton M, Garcia MF. Multi-metric behavioral comparison of APPsw and P301L models for Alzheimer's disease: linkage of poorer cognitive performance to tau pathology in forebrain. Brain Res. 2004;1012:29–41. doi: 10.1016/j.brainres.2004.02.081. [DOI] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Bancher C, Braak H, Fischer P, Jellinger KA. Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer's and Parkinson's disease patients. Neurosci Lett. 1993;162:179–182. doi: 10.1016/0304-3940(93)90590-h. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci. 2001;114:1179–1187. doi: 10.1242/jcs.114.6.1179. [DOI] [PubMed] [Google Scholar]
- Duff K, Knight H, Refolo LM, Sanders S, Yu X, Picciano M, Malester B, Hutton M, Adamson J, Goedert M, Burki K, Davies P. Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol Dis. 2000;7:87–98. doi: 10.1006/nbdi.1999.0279. [DOI] [PubMed] [Google Scholar]
- Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- Esmaeli-Azad B, McCarty JH, Feinstein SC. Sense and antisense transfection analysis of tau function: tau influences net microtubule assembly, neurite outgrowth and neuritic stability. J Cell Sci. 1994;107:869–879. doi: 10.1242/jcs.107.4.869. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Crowther RA. Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of tau filaments. FEBS Lett. 1999;450:306–311. doi: 10.1016/s0014-5793(99)00508-6. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P, Schein JD, Binder LI. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem. 1992;267:564–569. [PubMed] [Google Scholar]
- Guillozet AL, Weintraub S, Mash DC, Mesulam MM. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol. 2003;60:729–736. doi: 10.1001/archneur.60.5.729. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Iwakiri M, Mizukami K, Ikonomovic MD, Ishikawa M, Abrahamson EE, Dekosky ST, Asada T. An immunohistochemical study of GABA receptor gamma subunits in Alzheimer's disease hippocampus: relationship to neurofibrillary tangle progression. Neuropathology. 2009;29:263–269. doi: 10.1111/j.1440-1789.2008.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y, Takemura R, Oshima T, Mori H, Ihara Y, Yanagisawa M, Masaki T, Hirokawa N. Expression of multiple tau isoforms and microtubule bundle formation in fibroblasts transfected with a single tau cDNA. J Cell Biol. 1989;109:1173–1184. doi: 10.1083/jcb.109.3.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knops J, Kosik KS, Lee G, Pardee JD, Cohen-Gould L, McConlogue L. Overexpression of tau in a nonneuronal cell induces long cellular processes. J Cell Biol. 1991;114:725–733. doi: 10.1083/jcb.114.4.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Liu CW, Lee G, Jay DG. Tau is required for neurite outgrowth and growth cone motility of chick sensory neurons. Cell Motil Cytoskeleton. 1999;43:232–242. doi: 10.1002/(SICI)1097-0169(1999)43:3<232::AID-CM6>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-d-aspartate receptor antagonist, AP5. Nature. 1986;319:774–776. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- O'Keefe J, Dostrovsky J. The hippocampus as a spatial map. Preliminary evidence from unit activity in the freely-moving rat. Brain Res. 1971;34:171–175. doi: 10.1016/0006-8993(71)90358-1. [DOI] [PubMed] [Google Scholar]
- Otvos L, Jr, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39:669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Kas A, D'Souza I, Zhou Y, Pham Q, Stone M, Olson MV, Schellenberg GD. A genomic sequence analysis of the mouse and human microtubule-associated protein tau. Mamm Genome. 2001;12:700–712. doi: 10.1007/s00335-001-2044-8. [DOI] [PubMed] [Google Scholar]
- Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, Guimaraes A, Yue M, Lewis J, Carlson G, Hutton M, Ashe KH. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L) J Neurosci. 2005;25:10637–10647. doi: 10.1523/JNEUROSCI.3279-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome. 1997;8:711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei Y, Teng J, Harada A, Hirokawa N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J Cell Biol. 2000;150:989–1000. doi: 10.1083/jcb.150.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson AM. Facilitation, augmentation and potentiation at central synapses. Trends Neurosci. 2000;23:305–312. doi: 10.1016/s0166-2236(00)01580-0. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Tucker KL, Meyer M, Barde YA. Neurotrophins are required for nerve growth during development. Nat Neurosci. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- Winters BD, Forwood SE, Cowell RA, Saksida LM, Bussey TJ. Double dissociation between the effects of peri-postrhinal cortex and hippocampal lesions on tests of object recognition and spatial memory: heterogeneity of function within the temporal lobe. J Neurosci. 2004;24:5901–5908. doi: 10.1523/JNEUROSCI.1346-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]