

Abstract
Alterations in the cerebellum have been described as a neuropathological feature of autism. While numerous studies have focused on the Purkinje cell (PC), the projection neuron of the cerebellar cortex, PC function is critically dependent upon their innervation by the GABAergic basket cells (BCs) and stellate cells (SCs) in the cerebellar molecular layer. The present study was designed to determine if there are differences in the packing density of these inhibitory interneurons or if the ratio of these interneurons to PCs differs in autistic and age-matched control brains. The GABAergic interneurons were identified using immunohistochemistry for parvalbumin (PV) in serial sections from the posterior cerebellar lobe of six autistic and four control brains, and counted using stereological principles. Prior PC counts in the same area on adjacent sections (Whitney et al., 2008) were available and used to calculate the number of BCs and SCs per PC. In this sample of brains, no statistically significant difference was detected between the autistic and control groups in the density of BCs or SCs (p=0.45 and p=0.84, respectively) or in the number of BCs or SCs per PC (p=0.47 and p=0.44, respectively). The preservation of BCs and SCs, in the presence of the reduced PC numbers as found in at least two, and possibly three of these six autistic cases (Whitney et al., 2008), suggests that PCs were generated, migrated to their proper location in the PC layer and subsequently died in the autistic cases that showed a reduction in PCs.
Keywords: cerebellum, basket cells, stellate cells, autism
Introduction
Autism is a neurodevelopmental disorder characterized by deficits in reciprocal social interactions and communication as well as repetitive or stereotypic behaviors or interests (DMS-IV, 1995). Evidence that genetic factors play an important role in autism has been shown in twin studies (Folstein and Rutter, 1977; Steffenburg et al., 1989; Bailey et al., 1995) and in family studies where the risk of having a second child with autism is reported to be 50 to 150 times greater than the general population (Smalley et al., 1988; Folstein and Piven, 1991). It has been suggested that as many as two to fifteen genes (Pickles et al., 1995, Risch et al., 1999), and not necessarily the same genes or same combinations (Folstein and Rosen-Sheidley, 2001), contribute to the expression of autism. Although a single candidate gene has not been identified, several genes that code for GABA receptor subunits have been implicated (Cook et al., 1998; Wolpert et al., 2000; Folstein and Rosen-Sheidley, 2001; Shao et al., 2003; Buxbaum et al., 2000; Ma et al., 2005; Collins et al., 2006). In line with the genetic findings, alterations in components of the GABA system have been identified (Blatt et al., 2001; Fatemi et al., 2002a; Guptill et al., 2007; Yip et al., 2007; Yip et al., 2008). Further evidence of involvement in the GABA system in autism is provided by reports of reduced numbers of GABAergic Purkinje cells (PCs), the sole output neuron of the cerebellar cortex. A recent review by Palmen et al. (2004) indicated that 72% of cases reported in the literature have a decreased number of PCs. In our laboratory, a recent study of six autistic and four control brains revealed that two or three of the autistic cases (33% - 50%) had a reduced density of PCs (Whitney et al., 2008; also see table 1, column 3 [#PC/mm]). In addition to the GABAergic PCs, there are GABAergic interneurons in the cerebellar cortex that innervate PCs, the basket cell (BC) and the stellate cell (SC), but these have not been quantitatively examined in the autistic brain. Although there is some controversy about the classification of cerebellar GABAergic molecular layer interneurons (MLIs), they are most commonly described based on their depth within the molecular layer and their contribution to an axonal plexus (basket) that surrounds the PC soma (Sultan and Bower, 1998) or their innervation of PC cell apical dendrites. Thus, BCs are typically said to reside in the lower one-third of the molecular layer and contribute the axonal plexus surrounding the PC soma (Sultan and Bower, 1998), whereas SCs are typically considered to reside in the upper two-thirds of the molecular layer and synapse with the PC cell dendrite. Both BCs and SCs receive excitatory input from granule cell parallel fibers as well as GABAergic inhibitory input from other BC and SC (Palay and Chan-Palay, 1974) and from the Lugaro cells in the granule cell layer (Laine and Axelrad, 1998). Likewise, efferent projections from BCs and SCs include other BCs, SCs and PCs (Palay and Chan-Palay, 1974).
Table I.
Case Information
| Group | Case # | #PCs /mm |
Age | Sex | Hemisphere | PMI (hours) |
Years in Formalin |
Cause of Death |
|---|---|---|---|---|---|---|---|---|
| Control | Duke-495 | 5.4 | 17 | F | Unknown | Unknown | Unknown | Unknown |
| Control | 4104 | 5.3 | 24 | M | Left | 5 | Unknown | Gun Shot |
| Control | 4334 | 4.0 | 53 | M | Right | 23.75 | 3 | Cancer |
| Control | BCH-13 | 4.0 | 30 | M | Left | Unknown | 6 | Unknown |
| Autism | 44141, 2 | 0.5 | 26 | M | Left | 47.68 | 2.5 | Seizure |
| Autism | 38451, 2 | 2.6 | 32 | M | Left | Unknown | 4 | Pancreatitis |
| Autism | 4099 | 3.3 | 19 | M | Left | 3 | 4.5 | CHF |
| Autism | 2431 | 4.3 | 54 | M | Left | 4 | 8.5 | GI Bleed |
| Autism | 42591, 3 | 4.3 | 13 | F | Left | 18.36 | 4 | Unknown |
| Autism | 35111, 4 | 5.8 | 27 | M | Right | 16 | 7.5 | Trauma |
Case information on autistic and control brains including: age, sex, hemisphere, post-mortem interval (PMI), years in formalin, #PC/mm and cause of death.
Gastrointestinal (GI), congestive heart failure (CHF), Not Applicable (NA)
Seizure disorder;
History of phenytoin [Dilantin ®] use;
History of carbamazepine [Tegretol ®] use;
History of phenobarbital use
The establishment of functional synaptic contacts between PCs and MLIs are of critical importance for the prolonged survival of BCs and SCs (Sotelo and Triller, 1979; Feddersen et al., 1992). Further, PCs are necessary for the proper cellular organization of the developing cerebellar cortex (Sotelo, 1990; Feddersen et al., 1992; Smeyne et al., 1995; Soha et al., 1997). This published work, together with the data obtained in the current study regarding BC and SC density and our prior finding of PC density in these same cases (Whitney et al., 2008; also see table 1, column 3 [#PC/mm]), offers the opportunity to formulate a hypothesis for the timing of the reduced density of PCs in the autistic brain, when present. This hypothesis, which is outline in the discussion section, is based on work in mutant mice and studies of normal cerebellar development.
Given the reported involvement of the GABA system in autism and the functional relationships between PCs and MLIs, we set out to determine the numerical density of these interneurons using stereological methods in sections immunolabeled with parvalbumin. With these data, the relationship of the number of PCs to the number of BCs and SCs could be examined. Based on these data, a hypothesis can be proposed regarding the timing of the reduced number of cerebellar PCs in the autistic brain.
Materials and Methods
Six autistic and four control formalin fixed cerebella were obtained from the Harvard Brain Tissue Resource Center, Kathleen Price Bryan Brain Bank at Duke University Medical Center and University of Maryland Brain Bank. All control brains were free from gross pathology and were obtained from individuals without history of neurological disorders. These same ten cases were previously used in our laboratory to examine the density of PCs (Whitney et al., 2008). Table 1 includes the case number, group, age, sex, hemisphere, post mortem interval (PMI), years in formalin, number of PCs per millimeter (#PCs/mm) and cause of death of each cases and is derived from Whitney et al. (2008).
Tissue Processing
In each case, a 2cm × 2cm × 2cm parasagittal block of tissue, cut perpendicular to the folia, was obtained from cerebellar hemisphere lobule crus II, located inferior to the horizontal fissure (Schmahmann et al., 2000). Prior to tissue processing, each case was coded to ensure that the investigators were blind to the case data throughout the experimental process. All tissue blocks were cryoprotected (Rosene et al., 1986) and embedded in albumin-gelatin prior to freezing in order to hold the folia together after cutting and during subsequent processing (Crane and Goldman, 1979). The blocks were then flash frozen in −75°C 2-methylbutane and placed in a −80°C freezer for at least forty-eight hours before being serially sectioned on a sliding microtome from medial to lateral. Eighteen series per brain were cut at thirty microns and two series were cut at sixty microns so that sections within a series were spaced 660 microns apart. Sections were thaw-mounted onto gelatin-subbed slides, air dried, and stored in a −20°C freezer until the day they were stained.
From each brain, ten 30 μm on-the-slide sections, were immunostained using a batch-processing procedure in 4 groups (2 pairs, 2 triplets), matched for age and sex, to ensure that conditions were identical for the autistic and control groups. Immunostaining for PV was conducted using specially designed wells (PolyFab Inc., Avon, MA) that minimized antibody volume. The optimum anti-PV antibody concentration (mouse monoclonal) was predetermined to be 1:750 (Swant Laboratories, Switzerland). Sections were pretreated in a 1% hydrogen peroxide solution to quench endogenous peroxidases and a blocking serum to minimize nonspecific binding. Sections were then incubated in the primary antibody for forty-eight hours at room temperature followed by a one-hour incubation in a biotinylated secondary antibody (Vector Laboratories, Inc.). An avidin-biotinylated peroxidase complex solution (ABC) was then added (Vector Laboratories, Inc., Burlingame, CA) and the antigen was visualized using 3,3′-diaminobenzidine as the chromogen by incubating in hydrogen peroxide (1.5%) for 10 minutes. The reaction was stopped by washing sections in dH20. Sections were air-dried overnight, lightly counter-stained with thionin and coverslipped with Permount.
Basket Cell and Stellate Cell Counts
The equipment and computer software used in this study was identical for all cases. This included a Nikon Eclipse E600 microscope (Nikon Instruments Inc., Melville, NY), Optronics DEI-750 CE camera control unit (Optronics, Goleta, CA), MAC 2002 motor stage control unit (Ludl Electronic Products, Ltd., Hawthorne, NY) and Stereo Investigator® computer software from MircoBrightField Inc. (Williston, VT).
Ten sections from each autistic and control case, equally spaced by 660 microns, were counted for both MLI types, BCs and SCs. The BC and SC counts were performed on sections adjacent to those previously used to obtain total PC counts (Whitney et al., 2008). All sections were cut from tissue blocks taken from lobule crus II, located inferior to the horizontal fissure (Schmahmann et al., 2000). However, because of variability in the tissue samples provided by the brain banks, we were not able to definitively identify the identical folia in all brains that could be counted in its entirety. Consequently, in all brains, the superior-most folium in the most medial section was identified for counting. For that region of interest, stereological sampling principles were maintained across all sections (Gundersen, 1986; West et al., 1991), determining both cell number and the area of molecular layer sampled so that data are presented as cell density measures (#cells/mm3). Additionally, with Stereo Investigator software (MircoBrightField Inc., Williston, VT) and a 2× objective lens, the identical length along the PC layer used for PC counts (Whitney et al., 2008), was measured in the parvalbumin immunostained sections. This measurement defined the region of interest from which we were later able to calculate the number of BCs per PC and the number of SCs per PC. Next, using a 2× objective lens and a transparent ruler superimposed on the computer image, the examiner outlined the lower one-third and upper two-thirds of the molecular layer. These two regions of interest allowed the examiner to obtain two separate density measures, one for BCs (#BCs/mm3) and one for SCs (#SCs/mm3). Within each region of interest, the interneurons were systematically sampled; the approach used was based on the optical disector method (Gundersen, 1986; West et al., 1991) and ensures that all objects, regardless of size, shape and orientation, have an equal chance of being counted.
After the two regions of interest were defined, the magnification was increased to a 60× oil-immersion objective lens. A 600μm × 450μm grid with a 100μm ×75μm counting frame was used to randomly and systematically sample the two ROI. To avoid double counting in the x or y planes, the lower and left borders (x and y) of the counting frame, as well as their extended edges, were considered forbidden and BCs and SCs contacting these lines were not counted (Gundersen, 1986). The BCs or SCs within the counting frame and those contacting the upper and right borders of the counting frame were included in the counts (Gundersen, 1986). To avoid double counting in the z-axis, an exclusionary plane at the top of the section was implemented. This z-axis exclusionary plane was used instead of guard volumes because sections with an original thickness of 30 μm, when thaw-mounted onto slides, shrink in the z-axis to 7.5 to 8.5 μm. At this thickness and with the optics available it was not possible to implement reliable guard volumes. While this does introduce problems due to lost caps (Hedreen, 1998), the same process was used in all cases and therefore, the relative difference between sections is not affected. After completing the counting in one frame, the motorized stage shifted to the next counting frame. This process was repeated until all counting frames within the region of interest were viewed. Then, the second region of interest on the same section was examined.
The numerical density (Nv), was calculated as a ratio of the total number of molecular layer interneurons (BCs or SCs) counted (ΣQ) to the total disector volume, Σv(dis); the latter is determined by the number of times (ΣP ) the disector hits the ROI reference space multiplied by the disector volume v(dis).
The estimated total number of BCs or SCs (N) in the region of interest was then calculated as the product of Nv and the reference volume, V(ref). The V(ref) was calculated as the product of the outlined ROI (area) and section thickness.
Finally, the density of BCs and SCs (NT) within the region of interest, V(ref), was calculated as follows:
where NT represents the estimated number of molecular layer interneurons (BCs or SCs) per cubic millimeter.
Given the controversy over classification of basket and stellate interneurons, the data, in addition to being expressed as #BCs/mm3 and #SCs/mm3, are also expressed as the #MLIs/mm3. Additionally, since we had previously determined the density of PCs in the identical area and along the identical length of the PC layer on adjacent 30 μm calbindin immunolabeled section (Whitney et al., 2008; also see table 1, column 3 [#PC/mm]), data are also expressed as the number of BCs per PC, the number of SCs per PC, and total number of MLIs per PC. To determine the number of these interneurons per PC, we divided the total estimated number of BCs, SCs and MLIs by the total number of PCs along the same length.
Statistical Analysis
The difference in the mean density of interneurons (#BCs/mm3, #SCs/mm3 and #MLIs/mm3) between the autistic and control brains was examined using the t-test statistic. The Pearson correlation was used to examine the relationship between the density of BCs and the density of SCs (Table 2). Pearson correlation was also used to examine the relationship between the density of MLIs (#BCs/mm3, #SCs/mm3) and the density of PCs (#PCs/mm) (Table 2). The relationship between group (autistic, control) and BC and SC density was also explored (Table 2). Further, the t-test analysis was used for comparison of number of BCs per PC (#BCs/PC), number of SCs per PC (#SCs/PC) and number of MLIs per PC (#MLIs/PC) between the autistic and control brains. Finally, to examine the relationship between #MLIs/mm3 and years in formalin as well as #MLIs/mm3 and PMI, Pearson correlation was calculated.
Table 2.
Correlation Matrix
| Group | #PCs/mm | #BCs/mm3 | #SCs/mm3 | |
|---|---|---|---|---|
| Group | ---------------- | r=0.40; p=0.25 | r=0.28; p=0.44 | r=0.07; p=0.84 |
| #PCs/mm | r=0.40; p=0.25 | ---------------- | r=0.21; p=0.57 | r=0.35; p=0.34 |
| #BCs/mm3 | r=0.28; p=0.44 | r=0.21; p=0.57 | ---------------- | r=0.96; p<0.0001 |
| #SCs/mm3 | r=0.07; p=0.84 | r=0.35; p=0.34 | r=0.96; p<0.0001 | ---------------- |
This correlation matrix includes the correlation coefficient (r values) and p values for all of the relationships examined. A significant relationship was demonstrated between the density of BCs and SCs with r=0.96 and p< 0.001. No significant relationship was observed between the group (autistic or control) and the density of PCs, density of BCs or density of SCs. Further, no significant relationship was observed between the density of PCs and the density of BCs or density of SCs.
Results
Numbers of Molecular Layer Interneurons
The numerical density of BC interneurons in the lower one-third of the molecular layer is shown in figure 1. In the control brains, the density ranged from 17,219 to 32,129 BCs/mm3 with a mean density ± SD of 23,751 ± 7,658 BCs/mm3. The corresponding densities for the autistic brains were 14,222 to 28,293/mm3 with a mean density ± SD of 20,258 ± 5,980 BCs/mm3. The numerical density of SC interneurons is shown in figure 2. In the control brains, the density ranged from 9,014 to 20,849 SCs/mm3 with a mean density ± SD of 14,794 ± 6,072 SCs/mm3. The corresponding densities for the autistic brains were 8,164 to 22,586 SCs/mm3 with a mean density ± SD of 13,990 ± 6,164 SCs/mm3. Figure 3 represents the density of the combined BC and SC (MLI) interneurons. In the control brains, the density ranged from 13,152 to 26,489 MLIs/mm3 with a mean density ± SD of 19,272 ± 6,853 MLIs/mm3. The corresponding densities for the autistic brains were 11,193 to 25,440 MLIs/mm3 with a mean density ± SD of 17,124± 6,045 MLIs/mm3. With the available number of cases, statistical analysis using the t-test did not detect a significant difference in the density of BC, SC or MLIs between autistic brains as compared with the control brains with p = 0.44, p= 0.84 and p= 0.62, respectively. Additionally, photographs from two autistic and two control cases (fig. 7) visually demonstrate the variability in molecular layer interneuron density within the autistic and control groups.
Figure 1.
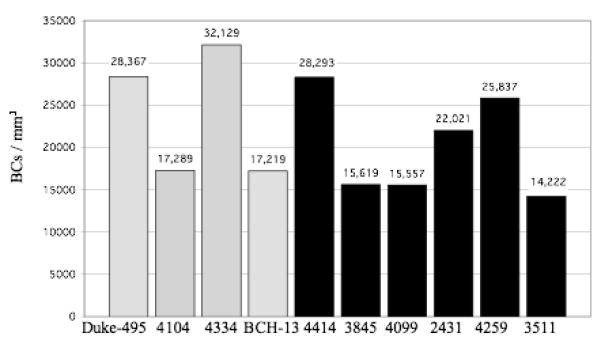
The #BCs/mm3 for each control case (gray bars) and autistic case (black bars) is shown. In the control brains, the density ranged from 17,219 to 32,129/mm3 with a mean density ± SD of 23,751 ± 7,658/mm3. In autistic brains, the density ranged from 14,222 to 28,293/mm3 and 20,258 ± 5,980/mm3. With this sample, no statistically significant difference was identified between the control and autistic groups, with p = 0.44.
Figure 2.
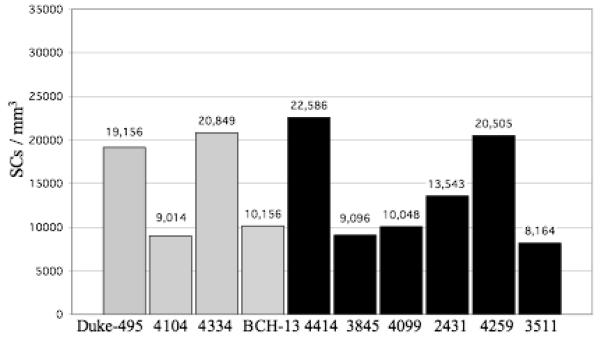
The #SCs/mm3 for each control case (gray bars) and autistic case (black bars) is shown. In the control brains, the density ranged from 9,014 to 20,849/mm3 with a mean density ± SD of 14,794 ± 6,072/mm3. The corresponding densities for the autistic brains were 8,164 to 22,586/mm3 and 13,990 ± 6,164/mm3. With this sample, no statistically significant difference was identified between the control and autistic groups, with p = 0.84.
Figure 3.
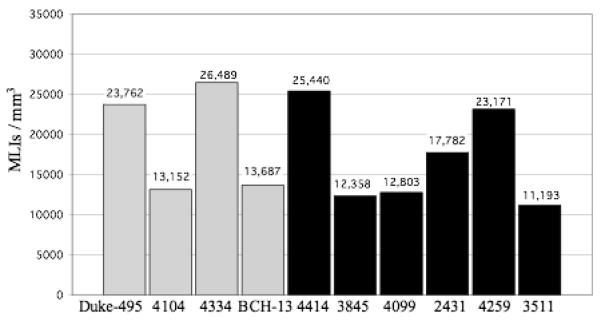
The #MLIs/mm3 for each control case (gray bars) and autistic case (black bars) is shown. In the control brains, the density ranged from 13,152 to 26,489/mm3 with a mean density ± SD of 19,272 ± 6,853/mm3. The corresponding densities for the autistic brains were 11,193 to 25,440/mm3 and 17,124± 6,045/mm3. With this sample, no statistically significant difference was identified between the control and autistic groups, with p= 0.62.
Figure 7.
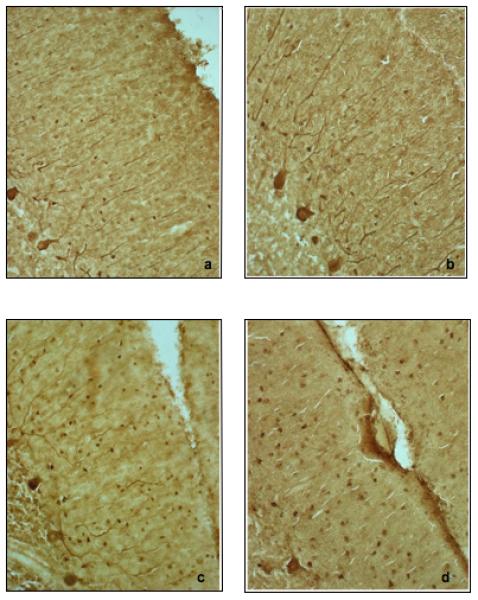
Molecular layer basket and stellate cells are shown in images a through d. Note the variability in molecular layer interneuron density. (a.) control brain 4104; (b.) autistic brain 3511; (c.) control brain Duke-495; (d.) autistic brain 4414. All photographs taken at 200×.
Correlations of Analyses of Molecular Layer Interneurons
Pearson correlation, used to examine the relationship between the #BCs/mm3 and the #SCs/mm3, returned a value of r = 0.96 (p < 0.0001) (Table 2). Thus, as BC density increased, SC density also increased. Using the Pearson correlation, a statistically significant relationship was not identified when examining the relationship between the densities BCs and PCs or the relationship between the densities of SCs and PCs, with r = 0.21 (p = 0.57) and r = 0.35 (p = 0.34), respectively (Table 2). Analysis of the relationship between group (autistic, control) and the #BCs/mm3, using the Pearson correlation, was not statistically significant with r = 0.28 (p = 0.44) (Table 2). Similarly, analysis of the relationship between group (autistic, control) and the #SCs/mm3 was not statistically significant with r = 0.07 (p = 0.84) (Table 2).
Number of Molecular Layer Interneurons Relative to Purkinje Cells
The number of BCs per PC is shown in figure 4. In the controls, the number of BCs per PC ranged from 16.0 to 34.9 with a mean ± SD of 22.7 ± 8.5 BCs/PC. In the autistic group, these numbers ranged from 10.6 to 124.5 with a mean ± SD of 39.2 ± 42.7 BCs/PC. Compared to the other cases, autistic case 4414 appears as an outlier with 124.5 BCs per PC (fig. 4). This high number can be explained by the fact that autistic case 4414 had the lowest density of PCs (0.5 PC/mm, see table 1), but a normal density of BCs that fell within the control range (28,293BCs/mm3, see fig. 1). The reduced PC numbers, coupled with normal BC numbers, yielded a high number of BCs per PC and shows that BCs were preserved in this case with decreased PCs. The number of SCs per PC is shown in figure 5. In the control cases, the number of SCs per PC ranged from 16.7 to 41.9 with a mean ± SD of 27.5 ± 11.1 SCs/PC. In the autistic cases, these numbers ranged from 12.3 to 195.3 with a mean ± SD of 56.3 ± 68.9 SCs/PC. Again, autistic case 4414 appears as an outlier with 195.3 SCs per PC (fig. 5) with this case having the lowest density of PCs (0.5 PC/mm, see table 1) coupled with a high density of SCs (22,586 BCs/mm3, see fig. 2). Thus, the reduced PC numbers, coupled with high SC numbers, yielded a high number of SCs per PC and shows that SCs were preserved in this case with decreased PCs. The number of combined BCs and SCs (MLIs) per PC is shown in figure 6. In the controls, the number of MLIs per PC ranged from 32.7 to 76.8 with a mean ± SD of 50.1 ± 19.5 MLIs/PC. In the autistic group, these numbers ranged from 22.9 to 319.8 with a mean ± SD of 95.5 ± 111.5 MLIs/PC. With the available number of cases, statistical analysis using the t-test did not detect a significant difference in the number of BCs, SCs or MLIs per PC between autistic brains as compared with the control brains with p = 0.47, p = 0.44 and p = 0.45, respectively.
Figure 4.
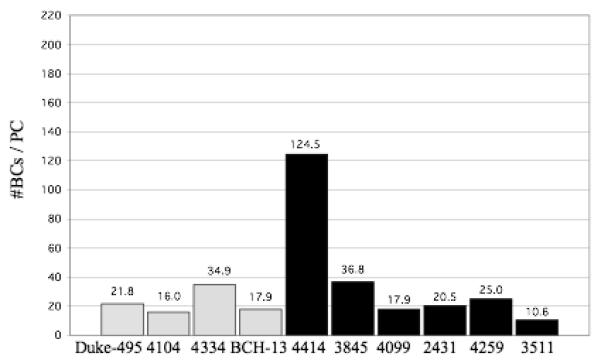
Number of BCs per PC for each control case (gray bars) and autistic case (black bars) is shown. In the controls, the number of BCs per PC ranged from 16.0 to 34.9 with a mean ± SD of 22.7 ± 8.5 BCs/PC. In the autistic group, these numbers ranged from 10.6 to 124.5 with a mean ± SD of 39.2 ± 42.7 BCs/PC. With this sample, no statistically significant difference was identified between the control and autistic groups, with p = 0.47.
Figure 5.
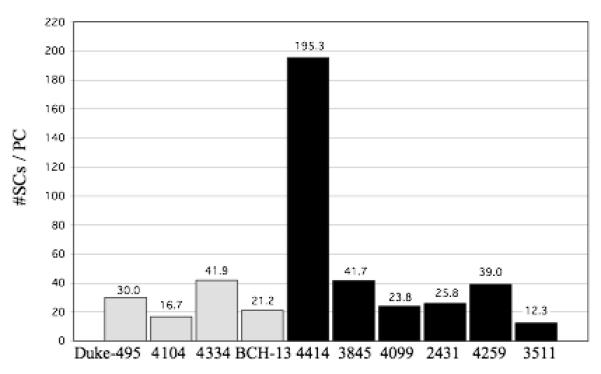
Number of SCs per PC for each control case (gray bars) and autistic case (black bars) is shown. In the controls, the number of SCs per PC ranged from 16.7 to 41.9 with a mean ± SD of 27.5 ± 11.1 SCs/PC. In the autistic group, these numbers ranged from 12.3 to 195.3 with a mean ± SD of 56.3 ± 68.9 SCs/PC. With this sample, no statistically significant difference was identified between the control and autistic groups, with p = 0.44.
Figure 6.
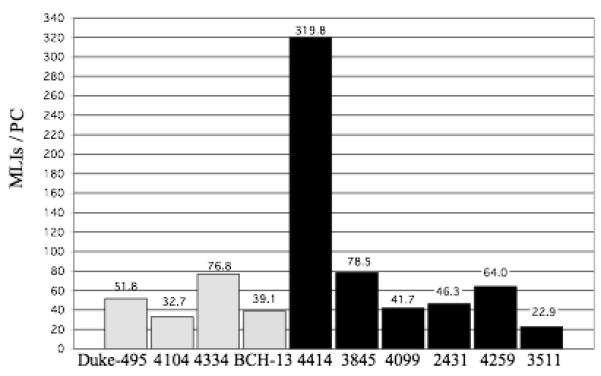
Number of MLIs per PC for each control case (gray bars) and autistic case (black bars) is shown. In the controls, the number of MLIs per PC ranged from 32.7 to 76.8 with a mean ± SD of 50.1 ± 19.5 MLIs/PC. In the autistic group, these numbers ranged from 22.9 to 319.8 with a mean ± SD of 95.5 ± 111.5 MLIs/PC. With this sample, no statistically significant difference was identified between the control and autistic groups, with p = 0.45.
Effect of Time in Fixative and Post-mortem Delay
Pearson correlation, used to examine the relationship between density of MLIs (BCs and SCs) and years in formalin (2 control, 6 autistic), returned a value of r = 0.58 (p = 0.13). This data, although not statistically significant, suggests that 33% of the variability in the density of MLIs may be accounted for by the number of years in formalin (r2 = 0.33). It is important to note, however, that this is a positive correlation; as years in formalin increases, the #MLIs/mm3 also increases. Thus, longer time periods in formalin did not appear to have a detrimental effect on the number of molecular layer interneurons identified with PV immunohistochemistry. Pearson correlation, also used to examine the relationship between density of MLIs and PMI (2 control, 5 autistic), returned a value of r = 0.71 (r2 = 0.50) (p = 0.07). Again, this is a positive correlation; as PMI increases, the #MLIs/mm3 also increases. Thus, the longer PMI did not appear to have a detrimental effect on the MLIs.
Discussion
Summary of Results
This study presents the first stereological cell counting data of cerebellar MLIs in the autistic brain. In both the autistic and control cases there was a significant correlation between the density of BCs and SCs (p < 0.001) without a significant difference in the density of BC and SC interneurons, with p = 0.44 and p= 0.84, respectively. There was no significant relationship between the density of these interneurons (BCs, SCs) and group (autistic, control) (Table 2; Figs 1-3).
Discussion of MLI Counts
Although the sample size in this study was modest, the density and variability of MLIs in both the autistic and control cases in the current study are in close agreement with Andersen et al. (1992). In their study of the normal human brain, the density ranged from approximately 8,000 to 32,000 MLIs/mm3. In the current study, the density of the MLIs in the autistic cases ranged from 11,193 to 25,440 MLIs/mm3 and in the control cases from 13,152 to 26,489 MLIs/mm3.
There was no significant relationship between the density of the interneurons (#BCs/mm3, #SCs/ mm3) and the density of PC (#PCs/mm) (p=0.57, p=0.34) (Table 2). A significant difference in the #BCs per PC and the #SCs per PC between the control and autistic cases was also not identified (p = 0.47 and p = 0.44, respectively) (Fig. 4-5). The number of MLIs per PC in the control brains (Fig. 6) is in close agreement with Andersen et al. (1992). They reported a mean of 50.0 MLIs/PC in the human cerebellum. In the current study, the mean number of MLIs/PC in the control brains was 50.1. In the autistic brains, the mean number of MLIs/PC was higher, at 95.5, due to a decreased number of PCs in at least two, and possibly three, of the six autistic cases (table 1, figures 3- 6). This data demonstrates that MLIs were preserved in cases with reduced PCs numbers.
The finding that autistic cases with a reduced density of PCs had preservation MLIs was the basis from which we developed our hypothesis for the timing of PC loss in autistic cases with reduced PC numbers. Specifically, we look to studies of mutant mice, normal rat cerebellar development and normal human cerebellar development to formulate this hypothesis. Collectively, this work reveals the importance of the early presence of PCs for BC and SC survival.
Contribution of mutant mice studies to our hypothesis of the timing of PC reduction
Up to this point in time, many researchers have been careful to indicate that there is reduction in PCs in the autistic brain, as there was no direct evidence that PCs were generated and then died at a later time. However, published work in mutant mice with PC loss at varying developmental time periods lends evidence that, in autistic brains with reduced PC numbers, PCs were generated during the prenatal period and subsequently died. This research has shown that PCs are the critical for the proper cellular organization and development of the cerebellar cortex (Sotelo, 1990; Feddersen et al., 1992; Smeyne et al., 1995; Soha et al., 1997). Specifically, in mice with disruption in PC development, it has been shown that the cerebellum does not possess the normal three-layer cortex and that there are gross abnormalities in cerebellar foliation (Feddersen et al., 1992; Smeyne et al., 1995). In the L7ADT transgenic mouse, for example, PC death occurs early in development and results in a small cerebellum that lacks both normal foliation and cytoarchitecture (Smeyne et al., 1995). Three transgenic mouse strains (SV4, SV5, SV6) examined by Fedderson et al. (1992) provide further evidence that PCs are necessary for normal cerebellar development. Fedderson et al. (1992) found that the SV5 transgenic mouse, which experience disrupted PC development at a time when these cells are still immature, had an overall reduced cerebellar size, abnormal foliation and abnormal cerebellar cytoarchitecture. In contrast, death of mature PCs in the SV4 and SV6 mice was associated normal cerebellar foliation and normal cerebellar cortical architecture. In the autistic brain, although there have been inconsistent reports of atrophy of selective folia (Courchesne et al., 1988; Murakami et al., 1989; Garber et al., 1989; Garber & Ritvo, 1992; Hashimoto et al., 1992; Holttum et al., 1992; Kleiman et al., 1992; Courchesne et al., 1994a; Courchesne et al., 1994b; Piven et al., 1997; Hardan et al., 2001; Sparks et al., 2002), there is no evidence of cerebellar cortical malformations (Williams et al., 1980; Bauman and Kemper, 1985; Kemper and Bauman, 1993; Rivto et al., 1986; Bailey et al., 1998; Lee et al., 2002; Fatemi et al., 2002b; Whitney et al., 2008).
Additionally, the autistic cases in the current study with a reduced density of PCs and concomitant normal density of MLIs, provide further evidence for the timing of the PCs loss in the autistic brain. Based on work in mutant mice, it is proposed that the survival of cerebellar interneurons is dependent on the establishment of functional synaptic contacts prior to PC death (Sotelo and Triller, 1979; Feddersen et al., 1992). Thus, this suggests that the survival of BCs and SCs in brains with reduced PC numbers, as seen in cases 4414, 3845 and possibly 4099, requires that BCs and SCs established functional synaptic contacts prior to PC death. Once these synaptic contacts are established, it is proposed that BCs and SCs are able to persist without the PC target (Sotelo and Triller, 1979). In contrast to the L7ADT and S5V mice that lose PC prior to synaptogenesis, PCs in the nervous mutant mouse develop normally until P23, at a time when major synaptic contacts have been established (Sotelo and Triller, 1979). These mutants then experience marked PC loss (Sotelo and Triller, 1979). Qualitative analysis, more than a year later, reveals that the BC and SC populations are “relatively unaffected” (Sotelo and Triller, 1979). The pogo (Jeong et al., 2000) and toppler (Duchala et al., 2004) mutant mice also lose PCs following the period of synaptogenesis. Both of these mutants have preservation of their BC and SC populations (Jeong et al., 2000; Duchala et al., 2004). Thus, based on these data, we propose that in autistic brains where a reduction in PC is observed, that the PCs are lost following the period when major synaptic contacts have been established. The timing of these synaptic contacts will now be discussed.
Cerebellar Synaptogenesis
In the human cerebellum, BCs are first observed in the molecular layer between 16 and 19 weeks gestation, with SCs observed five weeks later (Bayer et al., 1993; Rakic and Sidman, 1970). Based on developmental patterns in the rat, Bayer et al. (1993) have speculated on the timing of synaptogenesis in the human cerebellum. They speculate that, in the human brain, BC-parallel fiber synaptogenesis begins in the lower molecular layer at approximately 28 weeks of gestation and the BC-PC synapse begins soon thereafter, by 30 weeks of gestation. Synaptogenesis then proceeds in an inside-out pattern, and by approximately 32 weeks gestation, both SC-parallel fiber and SC-PC synapses begin to form in the upper molecular layer. Rakic and Sidman (1970), from direct observation of the developing human cerebellum, place the development of the basket cell plexus on the Purkinje cell in the human brain at some time after 32 weeks.
Hypothesis: The timing of PC loss in the autistic brain
Based on observations in mutant mice and the timing of the synaptogenesis in the developing human cerebellum, taken together with the preserved density of interneurons in the autistic brain with decreased number of PCs, suggest the earliest timing of the cerebellar pathology is the latter quarter of the gestational period or early postnatal period. This gestational timing for the loss of PCs is slightly later than that proposed by Kemper of Bauman (1993). They speculate that the loss of PCs may have occurred at or before about 30 weeks of gestation. This timing was based on two observations. One was that the decreased number of PCs was not accompanied by loss of neurons in the inferior olive, a loss that regularly occurs in the human brain with loss of PCs after 39 weeks of gestation (Holms and Stewart, 1908; Norman, 1940). At this late gestational stage climbing fibers from the contralateral inferior olive densely innervates the PC dendritic tree. The other was the initiation of the complex pattern of climbing fiber innervation of the PC at about 30 weeks of gestation. According to Marin-Padilla (1985), the olivocerebellar climbing fiber initially forms a pericellular plexus around the Purkinje cell body at 29-31 weeks of gestation and, at 34-36 weeks are concentrated above Purkinje cell body, and then start to climb up the Purkinje cell dendrites at about 36 weeks of gestation. The timing of the dependency of the inferior olive could conceivably be any time during this trajectory.
The current study, taken together with prior published work on the preservation of inferior olivary neurons in the autistic brain (Kemper and Bauman, 1993), overwhelmingly favors a late prenatal loss of PCs, during the latter quarter of the gestation period. Despite the strong evidence for a prenatal loss of PCs, it remains possible that PC loss, when present in the autistic brain, may extend into the postnatal period. This potential scenario is based on the transient innervation pattern of immature PCs. In the developing rodent cerebellum, a single PC is innervated by multiple climbing fibers (Mariani and Changeux, 1981; Crepel, 1982; Kakizawa et al., 2000). As the rodent cerebellum matures, however, surplus climbing fibers regress and, by the second to third postnatal week, a time that corresponds to the final few weeks of human gestation (Bayer et al., 1993), each PC is innervated by a single climbing fiber (Mariani and Changeux, 1981; Kano et al., 1997; Hashimoto and Kano, 2005). Experimental manipulation of metabotropic glutamate receptors, type 1 (mGluR1) (Kano et al., 1997) and NMDA receptors (Kakizawa et al., 2000), however, have been shown to halt the regression of the surplus climbing fibers. Consequently, in these experimental animals, mature PCs persist with innervation from multiple olivocerebellar climbing fibers. This alteration interferes with the one-to-one relationship between climbing fibers and PCs and perhaps, alters the dependency of inferior olivary neurons on PC survival. Although there is currently no evidence to suggest that a similar process occurs in autistic brains with reduced PC numbers, we cannot discount the possibility that some unknown alternative process is responsible for the survival of the inferior olivary neurons in these brains. In this regard, the age-related changes in the inferior olive and deep cerebellar nuclei, as described by Kemper and Bauman (1993), may signify an alternate circuitry and thus, an alternate process by which inferior olivary neurons are preserved in autistic cases with reduced PC numbers.
Conclusion
With the data from the current study we conclude that despite genetic and receptor binding evidence of involvement of the GABAergic system in autism, there is no evidence of an abnormality in the number or distribution of the GABAergic BC and SC interneurons in the cerebellar molecular layer. Additionally, data from the current study, taken together with prior published work on the preservation of inferior olivary neurons in the autistic brain, evidence from studies on mutant mice and normal rat and human cerebellar development, provides the strongest evidence to date that in autistic brains with PC reduction, that PCs were generated, migrated to their proper location in the PC layer and then died. Although, the evidence overwhelmingly supports a loss of PCs in the latter part of the gestational period, we cannot rule out the possibility that PC loss persists into the postnatal period.
Acknowledgement
We thank Michael Bowley for assistance with the stereology software instruction and maintenance of the system. We are also grateful to several members of the laboratory for their assistance with tissue processing: Rita Marcon, Sandy Thevarkunnel, Melissa Martchek, Matthew Stoker and Matthew Fields. This work was supported by NIH-NICHD HD39459, National Alliance for Autism Research (NAAR), the Nancy Laurie Marks Foundation and a grant from the John and Lisa Hussman Foundation.
Grant Information: This work was supported by NIH-NICHD HD39459, National Alliance for Autism Research (NAAR), the Nancy Laurie Marks Foundation and a grant from the John and Lisa Hussman Foundation.
References
- Bayer SA, Altman J, Russo RJ, Zhang X. Timetables of neurogenesis in the human brain based on experimental determined patterns in the rat. Neurotoxicology. 1993;14:83–144. [PubMed] [Google Scholar]
- Bailey A, Luthert P, Dean A, Harding B, Janota I, Montgomery M, Rutter M, Lantos P. A clinicopathological study of autism. Brain. 1998;121:889–905. doi: 10.1093/brain/121.5.889. [DOI] [PubMed] [Google Scholar]
- Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- Bauman ML, Kemper TL. Histoanatomic observations of the brain in early infantile autism. Neurology. 1985;35:866–874. doi: 10.1212/wnl.35.6.866. [DOI] [PubMed] [Google Scholar]
- Blatt GJ, Fitzgerald CM, Guptill JT, Booker AB, Kemper TL, Bauman ML. Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J Autism Dev Disord. 2001;31:537–543. doi: 10.1023/a:1013238809666. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Silverman JM, Smith CJ, Greenberg DA, Kilifarski M, Reichert J, Cook EH, Jr, Fang Y, Song CY, Vitale R. Association between a GABRB3 polymorphism and autism. Mol Psychiatry. 2002;7:311–316. doi: 10.1038/sj.mp.4001011. [DOI] [PubMed] [Google Scholar]
- Collins AL, Ma D, Whitehead PL, Martin ER, Wright HH, Abramson RK, Hussman JP, Haines JL, Cuccaro ML, Gilbert JR, Pericak-Vance MA. Investigation of autism and GABA receptor subunit genes in multiple ethnic groups. Neurogenetics. 2006;7:167–174. doi: 10.1007/s10048-006-0045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook EH, Courchesne RY, Cox NJ, Lord C, Gonen D, Guter SJ, Lincoln A, Nix K, Haas R, Leventhal BL, Courchesne E. Linkage-disequilibrium mapping of autistic disorder with 15q11-13 marker. Am J Hum Genet. 1998;62:1077–1083. doi: 10.1086/301832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, Yeung-Courchesne R, Press GA, Hesselink JR, Jernigan TL. Hypoplasia of cerebellar vermal lobules VI and VII in autism. N Engl J Med. 1988;318:1349–1354. doi: 10.1056/NEJM198805263182102. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Townsend J, Saitoh O. The brain in infantile autism: posterior fossa structures are abnormal. Neurology. 1994a;44:214–223. doi: 10.1212/wnl.44.2.214. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Saitoh O, Yeung-Courchesne R, Press GA, Lincoln AJ, Haas RH, Schreibman L. Abnormality of cerebellar vermian lobules VI and VII in patients with infantile autism: identification of hypoplastic and hyperplastic subgroups with MR imaging. Am J Roentgenol. 1994b;162:123–130. doi: 10.2214/ajr.162.1.8273650. [DOI] [PubMed] [Google Scholar]
- Crane AM, Goldman PS. An improved method for embedding brain tissue in albumin-gelatin. Stain Technol. 1979;54:71–75. doi: 10.3109/10520297909112637. [DOI] [PubMed] [Google Scholar]
- Crepel F. Regression of functional synapses in the immature mammalian cerebellum. Tends Neurosci. 1982;5:266–269. [Google Scholar]
- Diagnostic and Statistical Manual of Mental Disorders. 4th ed. American Psychiatric Association; Washington, DC: 1994. (DSM IV) [Google Scholar]
- Duchala CS, Shick HE, Garcia J, Deweese DM, Sun S, Stewart VJ, Macklin WB. The toppler mouse: a novel mutant exhibiting loss of Purkinje cells. J Comp Neurol. 2004;476:113–129. doi: 10.1002/cne.20206. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Halt AR, Stary JM, Kanodia R, Schulz SC, Realmuto GR. Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol Psychiatry. 2002a;52:805–810. doi: 10.1016/s0006-3223(02)01430-0. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Halt AR, Realmuto G, Earle J, Kist DA, Thuras P, Merz A. Purkinje cell size is reduced in cerebellum of patients with autism. Cell Mol Neurobiol. 2002b;22:171–175. doi: 10.1023/A:1019861721160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feddersen RM, Ehlenfeldt R, Yunis WS, Clark HB, Orr HT. Disrupted cerebellar cortical development and progressive degeneration of Purkinje cells in SV40 T antigen transgenic mice. Neuron. 1992;9:955–966. doi: 10.1016/0896-6273(92)90247-b. [DOI] [PubMed] [Google Scholar]
- Folstein SE, Piven J. Etiology of autism: genetic influences. Pediatrics. 1991;87:767–773. [PubMed] [Google Scholar]
- Folstein SE, Rosen-Sheidley B. Genetics of autism: complex aetiology for a heterogeneous disorder. Nat Rev Genet. 2001;2:943–955. doi: 10.1038/35103559. [DOI] [PubMed] [Google Scholar]
- Folstein SE, Rutter M. Infantile autism: A genetic study of 21 twin pairs. J Child Psychol Psychiatry. 1977;18:297–321. doi: 10.1111/j.1469-7610.1977.tb00443.x. [DOI] [PubMed] [Google Scholar]
- Garber HJ, Ritvo ER, Chiu LC, Griswold VJ, Kashanian A, Freeman BJ, Oldendorf WH. A magnetic resonance imaging study of autism: normal fourth ventricle size and absence of pathology. Am J Psychiatry. 1989;146:532–534. doi: 10.1176/ajp.146.4.532. [DOI] [PubMed] [Google Scholar]
- Garber HJ, Ritvo ER. Magnetic resonance imaging of the posterior fossa in autistic adults. Am J Psychiatry. 1992;149:245–247. doi: 10.1176/ajp.149.2.245. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ. Stereology of arbitrary particles. J Microsc. 1986;143:3–45. [PubMed] [Google Scholar]
- Guptill JT, Booker AB, Gibbs TT, Kemper TL, Bauman ML, Blatt GJ. [3H]-flunitrazepam-labeled benzodiazepine binding sites in the hippocampal formation in autism: a multiple concentration autoradiographic study. J Autism Dev Disord. 2007;37:911–920. doi: 10.1007/s10803-006-0226-7. [DOI] [PubMed] [Google Scholar]
- Hardan AY, Minshew NJ, Harenski K, Keshavan MS. Posterior fossa magnetic resonance imaging in autism. J Am Acad Child Adolesc Psychiatry. 2001;40:666–672. doi: 10.1097/00004583-200106000-00011. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Murakawa K, Miyazaki M, Tayama M, Kurdoa Y. Magnetic resonance imaging of the brain structures in the posterior fossa in retarded autistic children. Acta Paediatr. 1992;81:1030–1034. doi: 10.1111/j.1651-2227.1992.tb12169.x. [DOI] [PubMed] [Google Scholar]
- Hedreen JC. Lost caps in histological counting methods. Anat Rec. 1998;250:366–372. doi: 10.1002/(SICI)1097-0185(199803)250:3<366::AID-AR11>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Holttum JR, Minshew NJ, Sanders RS, Phillips NE. Magnetic resonance imaging of the posterior fossa in autism. Biol Psychiatry. 1992;32:1091–1101. doi: 10.1016/0006-3223(92)90189-7. [DOI] [PubMed] [Google Scholar]
- Holms G, Stewart TG. On the connection of the inferior olives with the cerebellum in man. Brain. 1908;31:125–137. [Google Scholar]
- Jeong YG, Hyun BH, Hawker R. Abnormalities in cerebellar Purkinje cells in the novel ataxic mutant mouse, pogo. Devel Brain Res. 2000;125:61–67. doi: 10.1016/s0165-3806(00)00114-0. [DOI] [PubMed] [Google Scholar]
- Kakizawa S, Yamasaki M, Watanabe M, Kano M. Critical period for activity-dependent synapse elimination in developing cerebellum. J Neurosci. 2000;20:4954–4961. doi: 10.1523/JNEUROSCI.20-13-04954.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Kurihara H, Watanabe M, Inoue Y, Aiba A, Tonegawa S. Persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking mGluR1. Neuron. 1997;18:71–79. doi: 10.1016/s0896-6273(01)80047-7. [DOI] [PubMed] [Google Scholar]
- Kemper TL, Bauman ML. The contribution of neuropathologic studies to the understanding of autism. Neurol Clin. 1993;11:175–187. [PubMed] [Google Scholar]
- Laine J, Axelrad H. Lugaro cells target basket and stellate cells in the cerebellar cortex. Neuroreport. 1998;9:2399–2340. doi: 10.1097/00001756-199807130-00045. [DOI] [PubMed] [Google Scholar]
- Lee M, Martin-Ruiz C, Graham A, Court J, Jaros E, Perry R, Iversen P, Bauman M, Perry E. Nicotinic receptor abnormalities in the cerebellar cortex in autism. Brain. 2002;125:1483–1495. doi: 10.1093/brain/awf160. [DOI] [PubMed] [Google Scholar]
- Ma DQ, Whitehead PL, Menold MM, Martin ER, Ashley-Koch AE, Mei H, Ritchie MD, Delong GR, Abramson RK, Wright HH, Cuccaro ML, Hussman JP, Gilbert JR, Pericak-Vance MA. Identification of significant association and gene-gene interaction of GABA receptor subunit genes in autism. Am J Hum Genet. 2005;77:377–388. doi: 10.1086/433195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Padilla M. Neurogenesis of the climbing fibers in the human cerebellum: a Golgi study. J Comp Neurol. 1985;235:82–96. doi: 10.1002/cne.902350107. [DOI] [PubMed] [Google Scholar]
- Mariani J, Changeux J-P. Ontogenesis of olivocerebellar relationships I. Studies by intracellular recordings of the multiple innervations of Purkinje cells by climbing fibers in the developing rat cerebellum. J Neurosci. 1981;1:696–702. doi: 10.1523/JNEUROSCI.01-07-00696.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami JW, Courchesne E, Press GA, Yeung-Courchesne R, Hesselink JR. Reduced cerebellar hemisphere size and its relationship to vermal hypoplasia in autism. Arch Neurol. 1989;46:689–694. doi: 10.1001/archneur.1989.00520420111032. [DOI] [PubMed] [Google Scholar]
- Norman RM. Cerebellar atrophy associated with etat marbre of the basal ganglia. J Neurol Psychiatry. 1940;3:311–318. doi: 10.1136/jnnp.3.4.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palay SL, Chan-Palay V. Cerebellar cortex; cytoloty and organization. Springer-Verlag; New York: 1974. [Google Scholar]
- Palmen S, van Engeland H, Hof PR, Schmitz C. Neuropathological findings in autism. Brain. 2004;127:2572–2583. doi: 10.1093/brain/awh287. [DOI] [PubMed] [Google Scholar]
- Pickles A, Bolton P, MacDonald H, Bailey A, Le Couteur A, Sim CH, Rutter M. Latent-class analysis of recurrence risks for complex phenotypes with selection and measurement error: a twin and family history study of autism. Am J Hum Genet. 1995;57:717–726. [PMC free article] [PubMed] [Google Scholar]
- Piven J, Saliba K, Bailey J, Arndt S. An MRI study of autism: the cerebellum revisited. Neurology. 1997;49:546–551. doi: 10.1212/wnl.49.2.546. [DOI] [PubMed] [Google Scholar]
- Rakic P, Sidman DL. Histogenesis of the cortical layers in human cerebellum particularly the lamina dissecans. J Comp Neurol. 1970;139:473–500. doi: 10.1002/cne.901390407. [DOI] [PubMed] [Google Scholar]
- Risch N, Spiker D, Lotspeich L, Nouri N, Hinds D, Hallmayer J, Kalaydjieva L, McCague P, Dimiceli S, Pitts T, Nguyen L, Yang J, Harper C, Thorpe D, Vermeer S, Young H, Hebert J, Lin A, Ferguson J, Chiotti C, Wiese-Slater S, Rogers T, Salmon B, Nicholas P, Petersen PB, Pingree C, McMahon W, Wong DL, Cavalli-Sforza LL, Kraemer HC, Myers RM. A genomic screen of autism: evidence for a multilocus etiology. Am J Hum Genet. 1999;65:493–507. doi: 10.1086/302497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritvo ER, Freeman BJ, Scheibel AB, Duong T, Robinson H, Guthrie D, Ritvo A. Lower Purkinje cell counts in the cerebella of four autistic subjects: initial findings of the UCLA-NSAC autopsy research report. Amer J Psychiatry. 1986;146:862–866. doi: 10.1176/ajp.143.7.862. [DOI] [PubMed] [Google Scholar]
- Rosene DL, Roy NJ, Davis BJ. A cryoprotection method that facilitates cutting frozen sections of whole monkey brains for histological and histochemical processing without freezing artifact. J Histochem Cytochem. 1986;34:1301–1315. doi: 10.1177/34.10.3745909. [DOI] [PubMed] [Google Scholar]
- Schmahmann JD, Doyon J, Toga AW, Petrides M, Evans AC. MRI Atlas of the Human Cerebellum. Academic Press; San Diego, CA: 2000. pp. 3–20. [DOI] [PubMed] [Google Scholar]
- Shao Y, Cuccaro ML, Hauser ER, Raiford KL, Menold MM, Wolpert CM, Ravan SA, Elston L, Decena K, Donnelly SL, Abramson RK, Wright HH, DeLong GR, Gilbert JR, Pericak-Vance MA. Fine mapping of autistic disorder to chromosome 15q11-q13 by use of phenotypic subtypes. Am J Hum Genet. 2003;72:539–548. doi: 10.1086/367846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalley SL, Asarnow RF, Spence MA. Autism and genetics. A decade of research. Arch Gen Psychiatry. 1988;45:285–295. doi: 10.1001/archpsyc.1988.01800340081013. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Chu T, Lewin A, Bian F, Crisman SS, Kunsch C, Lira SA, Oberdick J. Local control of granule cell generation by cerebellar Purkinje cells. Mol Cell Neurosci. 1995;6:230–251. doi: 10.1006/mcne.1995.1019. [DOI] [PubMed] [Google Scholar]
- Soha JM, Kim S, Crandall JE, Vogel MW. Rapid growth of parallel fibers in the cerebella of normal and staggerer mutant mice. J Comp Neurol. 1997;389:642–654. doi: 10.1002/(sici)1096-9861(19971229)389:4<642::aid-cne7>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Sotelo C, Triller A. Fate of presynaptic afferents to Purkinje cells in the adult nervous mutant mouse: a model to study presynaptic stabilization. Brain Res. 1979;175:11–36. doi: 10.1016/0006-8993(79)90511-0. [DOI] [PubMed] [Google Scholar]
- Sotelo C. Cerebellar synaptogenesis: what we can learn from mutant mice. J Exp Biol. 1990;153:225–249. doi: 10.1242/jeb.153.1.225. [DOI] [PubMed] [Google Scholar]
- Sparks BF, Friedman SD, Shaw DW, Aylward EH, Echelard D, Artru AA, Maravilla KR, Giedd JN, Munson J, Dawson G, Dager SR. Brain structural abnormalities in young children with autism spectrum disorder. Neurology. 2002;59:184–192. doi: 10.1212/wnl.59.2.184. [DOI] [PubMed] [Google Scholar]
- Steffenburg S, Gillberg C, Hellgren L, Andersson L, Gillberg IC, Jakobsson G, Bohman M. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J Child Psychol Psychiatry. 1989;30:405–416. doi: 10.1111/j.1469-7610.1989.tb00254.x. [DOI] [PubMed] [Google Scholar]
- Sultan F, Bower JM. Quantitative Golgi study of the rat cerebellar molecular layer interneurons using principal component analysis. J Comp Neurol. 1998;393:353–373. [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Whitney ER, Kemper TL, Bauman ML, Rosene DL, Blatt GJ. Cerebellar Purkinje Cells are Reduced in a Subpopulation of Autistic Brains: A Stereological Experiment Using Calbindin-D28k. Cerebellum. 2008 doi: 10.1007/s12311-008-0043-y. DIO 10.1007/s12311-008-0043-y: in press. [DOI] [PubMed] [Google Scholar]
- Williams RS, Hauser SL, Purpura DP, DeLong GR, Swisher CN. Autism and Mental Retardation: neuropathologic studies performed in four retarded persons with autistic behavior. Arch Neurol. 1980;37:749–753. doi: 10.1001/archneur.1980.00500610029003. [DOI] [PubMed] [Google Scholar]
- Wolpert CM, Menold MM, Bass MP, Qumsiyeh MB, Donnelly SL, Ravan SA, Vance JM, Gilbert JR, Abramson RK, Wright HH, Cuccaro ML, Pericak-Vance MA. Three probands with autistic disorder and isodicentric chromosome 15. Am J Med Genet. 2000;96:365–372. doi: 10.1002/1096-8628(20000612)96:3<365::aid-ajmg25>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Yip J, Soghomonian JJ, Blatt GJ. Decreased GAD67 mRNA levels in cerebellar Purkinje cells in autism: pathophysiological implications. Acta Neuropathol. 2007;112:559–568. doi: 10.1007/s00401-006-0176-3. [DOI] [PubMed] [Google Scholar]
- Yip J, Soghomonian JJ, Blatt GJ. Increased GAD67 mRNA expression in cerebellar interneurons in autism: implications for Purkinje cell dysfunction. J Neurosci Res. 2008;86:525–530. doi: 10.1002/jnr.21520. [DOI] [PubMed] [Google Scholar]