

Abstract
Among the distinguishing characteristics of members of the gamma-2 herpesvirus family is the expression of a mammalian D-type cyclin homolog, termed v-cyclin. Murine gammaherpesvirus 68 (γHV68) is a γ2-herpesvirus that can infect inbred and outbred strains of mice, providing a genetic system for the study of gammaherpesvirus pathogenesis. Disruption of the v-cyclin gene of γHV68 results in a virus that establishes latency in infected mice to wild-type levels, but is severely attenuated for virus reactivation [L.F. van Dyk, H.W. Virgin IV, S.H. Speck (2000) J. Virol. 74:7451-7461]. Transcriptional regulation of the γHV68 v-cyclin has not been defined. We report here the initial characterization of the v-cyclin transcript expressed in permissive murine fibroblasts. Based on 5′ mapping of the v-cyclin transcript, we identified a promoter that is involved in driving v-cyclin expression during virus replication. In addition, we determined that the promoter is responsive to the major viral lytic transactivator, Rta, encoded by orf 50. Using reporter plasmids we have analyzed both basal and Rta-induced v-cyclin promoter activity - initially identifying two regions of the v-cyclin promoter important for both basal and Rta-induced activity. Notably, only one of these regions could be shown to confer Rta responsiveness on a reporter construct containing the hsp70 TATA box. The importance of this region in regulating v-cyclin expression during virus replication was confirmed by introducing these mutations into the context of the viral genome and assessing v-cyclin expression following infection of permissive murine fibroblasts in tissue culture. In addition, we show that mutations that severely cripple Rta-induction of v-cyclin expression did not adversely impact virus reactivation from splenocytes recovered from latently infected mice, indicating that alternatively regulated v-cyclin gene expression is required for virus reactivation.
INTRODUCTION
Infection of laboratory mice with murine gammaherpesvirus 68 (γHV68), a member of the gammaherpesvirus family related to the human gammaherpesviruses Epstein Barr virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV or HHV-8), leads to an initial lytic infection in tissues including the lungs and spleen of the animal. Following resolution of the lytic infection, a lifelong latent infection that is marked by periodic virus reactivation predominates in the host (Speck et al., 1999). As with all members of the herpesvirus family, γHV68 reactivation from latent infection requires efficient transduction of reactivation stimuli and subsequent modulation of the viral and host factors that allow for productive virus infection in reactivating cells (Roizman, 2001). A full understanding of γHV68 reactivation therefore requires identification of not only the viral proteins that are required for reactivation, but also the host and viral proteins that modulate transcription of these viral genes.
Genetic studies of γHV68 have led to the identification of a number of proteins, including M2 and v-cyclin, that play important roles in virus reactivation (Husain et al, 1999; Jacoby et al, 2002; van Dyk et al, 2000; van Dyk et al, 2003; Virgin et al, 1999). M2 function is important not only for virus reactivation, but also for the establishment of latency in the spleen (Herskowitz et al, 2005; Jacoby et al, 2002). In contrast, v-cyclin null viruses establish latency to wild type levels in infected mice, but display a profound defect in virus reactivation from latently infected cells upon explant into tissue culture (Hoge et al, 2000; van Dyk et al, 2000). The in vivo relevance of this phenotype is confirmed by the analysis of γHV68 infection in B cell-deficient mice, where loss of v-cyclin function leads to a progressive loss of latently infected cells and clearance of chronic infection (van Dyk et al, 2003). Because the function of v-cyclin is restricted to reactivation from latency in our system, it is possible to investigate the specific role of v-cyclin cis and trans transcriptional regulatory factors in virus reactivation.
In the following study, we report the characterization of the structure of a v-cyclin transcript found in lytically infected murine NIH 3T12 fibroblasts. In addition, we have identified and characterized the v-cyclin lytic cycle-associated promoter. Using recombinant γHV68 v-cyclin promoter mutants, we have investigated the role of this promoter in regulating v-cyclin expression during virus replication in permissive murine fibroblasts. Finally, we have assessed the impact of mutations that impair lytic cycle-associated v-cyclin expression on virus reactivation from latently infected cells recovered from γHV68 infected mice.
RESULTS
Characterization of the v-cyclin transcript in lytically infected murine fibroblasts
During our initial analysis of v-cyclin function, we determined that v-cyclin protein expression was readily detectable in lytically infected NIH 3T12 fibroblasts by 24 hours post-infection (van Dyk et al, 1999). In addition, transcriptional profiling studies of γHV68 have demonstrated that v-cyclin mRNA is transcribed in the presence of cycloheximide during lytic infection, a characteristic of herpesvirus immediate-early transcripts (Martinez-Guzman et al, 2003). To identify the transcriptional control regions for v-cyclin, we first used RACE and primer extension analyses to identify the termini of the v-cyclin transcript. 5′ RACE was performed on RNA prepared from murine NIH 3T12 fibroblasts (NIH 3T12s) infected with wt virus for 24 hours. Using the primer 72RACE5′ (Figure 1A), RACE products were generated and subcloned. Based on sequencing multiple independent clones, a 5′ end for the v-cyclin transcript was defined between nucleotide positions 103,238 and 103255 (Figure 1A). Primer extension analysis using the 72RACE5′ primer and polyA RNA from productively infected NIH 3T12 fibroblasts, harvested at 12, 24, 36, and 48 hours post-infection, yielded three major bands that mapped to a region extending between nucleotide positions 103,248 and 103,258 in the viral genome (Figure 1B).
Figure 1.

Mapping the v-cyclin transcript in γHV68 infected NIH 3T12 fibroblasts. (A) The locations of the predicted ORFs for v-cyclin and v-Bcl-2(M11) are depicted in their genomic orientation. Filled black arrows are labeled with the genomic coordinates position of the terminal nucleotide of 5′RACE products generated using RNA from infected NIH 3T12 fibroblasts. The position of the v-cyclin-specific PCR primer 72RACE5′ is also indicated. The filled gray arrow is labeled with the genomic coordinate of the terminal nucleotide position of 3′RACE products generated from the same RNA template. Based on proximity to the 3′ terminus, the proposed polyA signal utilized in generation of the v-cyclin lytic transcript is indicated. The position of the PCR primer used in the 3′RACE reactions, 72RACE3′ is also labeled. (B) Primer extension mapping of the 5′end of the v-cyclin transcript in NIH 3T12 fibroblasts infected for 12, 24, 36, 48 hours of uninfected cells. The open circle marks the position of the 3 major extension products detected under these conditions. The deduced position (open circle) and the range of genomic coordinates of the 5′termini of the v-cyclin transcripts are indicated on the genomic depiction in panel A. The position of the primer used in the analysis, 72RACE5′ is also indicated.
The 3′ end of the v-cyclin message was also mapped by 3′ RACE using RNA prepared from NIH 3T12 fibroblasts in conjuction with the primer 72RACE3′ (Figure 1A). Based on the DNA sequence of the subcloned 3′ RACE products, the polyadenylation signal used in transcription of the v-cyclin message is located within the final two codons of the v-cyclin orf (between nucleotide positions 102,430 and 102,435) and the 3′ end of the message extends to nucleotide position 102,412 (Figure 1A). Thus, during lytic infection, the γHV68 v-cyclin is encoded by an unspliced transcript, typical of lytic herpesvirus transcripts. This is in contrast to the spliced v-cyclin transcripts identified during latent infection of the closely related gammaherpesviruses, herpesvirus saimiri (HVS) and KSHV (Dittmer et al, 1998; Hall et al, 2000; Kedes et al, 1997; Sarid et al, 1999).
Identification of a v-cyclin promoter active in murine fibroblasts
Following the characterization of v-cyclin gene transcription during lytic infection, we sought to identify the v-cyclin lytic cycle-associated v-cyclin promoter. Regions of the viral genome, including the mapped 5′ end of the v-cyclin transcript, were subcloned into a luciferase reporter vector. A construct containing sequences spanning the region between nucleotide 103,183 and 103,982 (72/800) was transiently transfected into NIH 3T12 fibroblasts, and subsequent luciferase assays on protein extracts from these cells indicated promoter activity in this region (Figure 2A). Comparable luciferase activity was detected in cells transfected with constructs containing the region between nucleotide 103,183 and 103,482 (72/300, Figure 2A). Deletion of an additional 100 bp from the 5′ end of the promoter (73/200) resulted in an approximately 5-fold drop in promoter activity (Figure 2A). Only background levels of luciferase activity could be detected in cells transfected with a construct containing sequences extending from nucleotide 103,182 to 103,382 (73/100 Figure 2A). In this manner, the minimal v-cyclin promoter active in NIH 3T12 fibroblasts was mapped to a region between nucleotides 103,182 and 103482 (+57 to −243 relative to the most proximal mapped site of transcription initiation).
Figure 2.

(A) Identification of a v-cyclin promoter active in NIH 3T12 fibroblasts. Reporter constructs (1 μg) containing defined regions of the γHV68 genome upstream of the v-cyclin ATG start codon (see Materials and Methods) were transiently transfected into NIH 3T12 fibroblasts. 48 hours later, transfected cell extracts were prepared and assayed for luciferase activity. The relative light units (RLUs) detected from transfectants containing candidate promoter regions are compared to those detected cells transfected with the empty luciferase control vector, pGL3. Data presented here are representative of three independent experiments. (B) Sequence of the minimal v-cyclin promoter in NIH 3T12 fibroblasts. The minimal promoter extends from +57 to −243 relative to the most proximal mapped site of transcription initiation. 5′ terminal nucleotide positions based on RACE (filled arrows) or primer extension analysis (open arrows) are indicated, as is the putative TATA box (boxed sequence). The position of a selection of transcription factor consensus binding sites (overlines) was determined based on transcription factor binding site analysis using the TESS interface of the TRANSFAC database. AP-1, c/EBPa, CCAAT/enhancer binding protein alpha, c-Fos, GATA-1, MEF2, myocyte enhancer binding factor.
Within the minimal promoter, a candidate TATA box lies between bp 103,280 and 103,285 (Figure 2B). In addition, analysis of the minimal v-cyclin promoter sequence for transcription factor binding sites using the TRANSFAC database (URL: http://www.cbil.upenn.edu/tess) revealed strong consensus binding sites for a number of cellular transcription factors (Figure 2B). Among the consensus sites identified was that for MEF-2 binding. Previous work from our lab, as well as others, has demonstrated a role for human MEF2D binding in the maintenance of a latent EBV infection through MEF-2D mediated recruitment of histone deacetylases to the immediate-early BZLF1 gene promoter (Gruffat et al, 2002; Liu et al, 1997). Assigning a role for this and other transcription factors in v-cyclin lytic cycle-associated promoter function required that we first determine those regions of the promoter that are important for maintaining basal promoter function during lytic infection.
Linker scanning mutagenesis of the lytic cycle-associated v-cyclin promoter and identification of an Rta responsive cis-element
To begin mapping the regulatory regions within the v-cyclin lytic cycle-associated promoter, a consecutive series of nine 10bp linker scanning (LS) mutations were introduced into the region extending upstream from the v-cyclin 5′end, as described in Materials and Methods (Figure 3A). Luciferase constructs carrying these LS mutations (LS1-LS9) in the context of the 72/800 promoter construct were tested for basal promoter activity, compared to the wildtype promoter sequence, in NIH 3T12 fibroblasts (Figure 3B). As can be seen in Figure 3B, the LS4 and LS7 mutations resulted in the greatest loss of v-cyclin promoter activity, with LS4 down by a factor of five as compared to wildtype, and LS7 activity reduced nearly ten-fold.
Figure 3.

Linker scanning mutagenesis of the v-cyclin promoter region. (A)Discrete regions of the v-cyclin minimal promoter, L1-LS9 (overlines), were individually mutated from the wildtype sequence presented in the figure to that of the AscI linker, 5′AGGCGCGCCA3′. 5′ terminal nucleotide positions based on RACE (filled arrows) or primer extension analysis (open arrows) are indicated, as is the putative TATA box (boxed sequence). (B) Characterization of 72/800LS mutant constructs in NIH 3T12 fibroblasts. The control pGL3-basic vector, 72/800, or the Linker Scanning mutants LS1-LS12 (1ug each) were cotransfected into 3T12 fibroblasts along with 1 ug of a control expression vector, pBK. Whole cell lysates from transfected cells were prepared 48 hours after transfection and analyzed for luciferase activity. Results are presented as RLUs. Data presented here are representative of three independent experiments.
Given that the levels of v-cyclin transcript are lower in the presence of cycloheximide, we sought to determine whether the major viral lytic transactivator, Rta, plays a role in regulating v-cyclin lytic cycle-associated promoter function (Martinez-Guzman et al, 2003). NIH 3T12 fibroblasts were co-transfected with v-cyclin promoter-driven reporter constructs and either an empty expression plasmid (PBK), or an expression plasmid into which the γHV68 Rta cDNA had been cloned (PBKRta). Co-transfection of the Rta expression plasmid with the 72/800 promoter construct led to a significant increase in luciferase activity, as compared to cells cotransfected with the 72/800 and the empty control expression vector PBK (Figure 4A). Rta induction of reporter gene expression from the 72/200 construct was comparable to that from the 72/800 construct (Figure 4A). However, a construct that lacked basal promoter activity, 72/100, was not induced by Rta (Figure 4A). Based on this result, we conclude that the v-cyclin promoter active during virus replication in murine fibroblasts is Rta responsive, and that Rta inducibility maps to a region between bp 103,183 and 103,383 in the viral genome.
Figure 4.
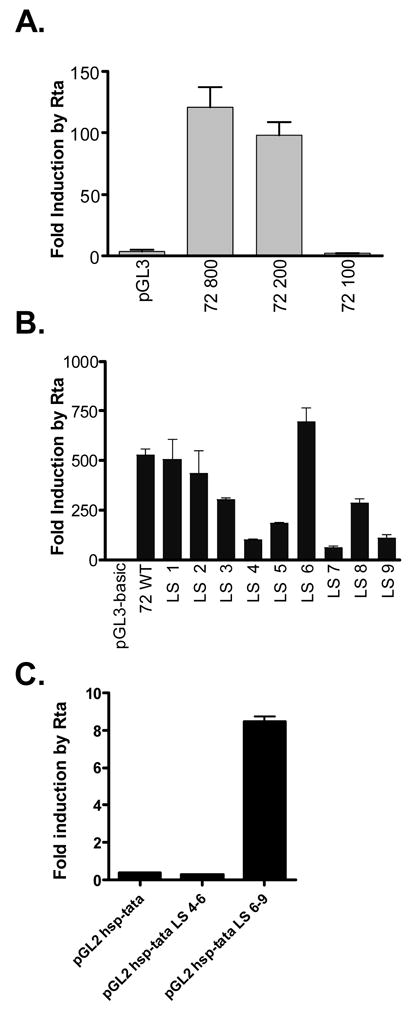
The v-cyclin lytic cycle-associated promoter is Rta-responsive. (A) The minimal Rta-responsive v-cyclin promoter was mapped by cotransfecting 1 μg luciferase reporter constructs containing defined γHV68 genomic regions (see Materials and Methods) with either 1 μg of the empty expression plasmid pBK, or 1 μg of the Rta expression construct pBKRta in NIH 3T12 cells. Luciferase activity for each reporter construct following cotransfection with pBKRta was compared to that following cotransfection with pBK. The quotient of these two values is presented as fold induction by Rta for each construct. (B) Linker scanning mutants LS1-LS9 were assessed for their capacity to be induced by Rta. Luciferase activity for each LS construct following cotransfection with pBK in NIH 3T12 cells (Figure 3B) and fold induction was calculated. (C) Putative Rta response elements within the v-cyclin promoter were assessed independently of the full v-cyclin promoter with use of the heterologous cellular hsp70 basal promoter reporter vector, hsp-tata pGL3. Two μg of the indicated reporter vectors was cotransfected with 2 μg of either pBK or pBKRta in 293T cells. The quotient of these two values is presented as fold induction by Rta for each construct. Data presented here are representative of three independent experiments.
To more precisely map regions of the promoter required for Rta induction, the WT and linker scanning v-cyclin promoter mutants were cotransfected with either the control PBK or PBKRta expression vectors into NIH 3T12 fibroblasts. The relative luciferase activity under each condition was compared, and the fold induction by Rta was plotted for each construct (Figure 4B). Mutant constructs LS1, LS2, LS3, LS6, and LS8 all were induced by Rta to within 2-fold of wildtype v-cyclin promoter levels (Figure 4B). However, inducibility by Rta was reduced by a factor of 10 in promoter mutant LS7 (Figure 4B). Less significant reductions in Rta inducibility were observed with mutants LS4, LS5, and LS9, suggesting that Rta may act to transactivate the v-cyclin promoter through more than one site within the v-cyclin gene promoter (Figure 4B). Alternatively, the reduction of Rta transactivation for some of the linker scanning mutants could reflect disruption of a cis-element(s) critical for basal and induced promoter activity. Indeed, both the LS4 and LS5 mutants are predicted to disrupt the putative TATA box (see Fig. 3A).
Notably, inspection of the DNA sequence of the v-cyclin lytic promoter region did not reveal any consensus Rta binding sites, suggesting that Rta may be modulating promoter activity through an indirect mechanism. Consistent with the hypothesis, the LS mutants that were most impaired for Rta inducibility were the same mutants that displayed impaired basal promoter function (compare Figs. 3B and 4B). If Rta is acting indirectly to modulate v-cyclin promoter function – thereby leading to a general upregulation of basal v-cyclin promoter activity - then it follows that those LS mutations that most affected basal promoter activity would have the greatest impact on Rta inducibility.
To distinguish between a role for the implicated promoter regions in mediating Rta activation of the v-cyclin gene transcription vs. mutation of a critical cis-element(s) required for promoter activity, we cloned these regions upstream of a minimal heterologous promoter-driven reporter construct. This construct contains the TATA box and surrounding basal promoter sequences of the cellular hsp70 gene cloned upstream of the firefly luciferase reporter gene (Lukac et al., 2001). As shown in figure 4C, the wt promoter sequences corresponding to the sequences mutated in the LS4-6 mutants did not confer Rta inducibility on the minimal hsp promoter-driven reporter construct. However, activity of the heterologous reporter construct containing the wt promoter sequences corresponding to the sequences mutated in the LS6-9 mutants was strongly upregulated by Rta expression. This confirms the presence of an Rta responsive cis-element within the v-cyclin promoter mapping within the regions from bp 103,307-103,346 in the viral genome. It is possible that the failure to detect Rta activation of the heterologous reporter construct containing the LS4-6 region was due to failure to include sequences upstream of the LS4 region. However, as shown below, subsequent analysis of recombinant viruses harboring these linker scanning mutations largely rules out this possibility.
Impact of mutating candidate Rta-responsive regions in the v-cyclin promoter on v-cyclin expression during lytic infection of NIH 3T12 fibroblasts
As with any promoter-driven reporter construct, transient transfection analyses can be misleading because it is difficult to assess the influence that contextual factors (e.g., promoter length, neighboring gene effects, and chromatin structure) may have on mutant phenotypes. In an effort to overcome this limitation, and to enable testing of the promoter mutants in a biologically relevant context, the linker scanning mutations were introduced directly into the γHV68 genome. The LS4-LS9 mutations were each engineered into the viral genome by allelic exchange mutagenesis of the γHV68- BAC (described in Materials and Methods) (Adler et al, 2001; Smith et al, 1999). To excise the BAC sequence from the mutant viruses, viral stocks were generated in Vero cells that stably express Cre-recombinase. The floxed γHV68 and γHV68/72LS mutant virus stocks were then used to infect NIH 3T12 fibroblasts at an MOI of 5. Infections were allowed to proceed for 18 hours, at which time whole cell protein lysates were prepared from infected cells. Protein lysates were analyzed by immunoblot for the expression of v-cyclin, using a previously described rabbit polyclonal antisera generated against the v-cyclin (van Dyk et al, 1999), as well as a chicken antibody raised against two peptides from orf 59 encoded early antigen (Fig. 5). Infection with γHV68/72LS4, γHV68/72LS5, and γHV68/72LS6 mutant viruses each yielded wildtype levels of v-cyclin expression as measured at 18 hours post-infection (Figure 5, lanes 1–3). Thus, the changes observed in vitro in either basal promoter function or Rta induction of reporter constructs carrying these mutations did not result in a significant changes in v-cyclin expression during lytic infection. The latter result is also consistent with the failure to observe Rta-inducible activity of the heterologous promoter construct containing the wt sequences corresponding to the LS4-6 mutants. In contrast, v-cyclin expression in cells infected with γHV68/72LS7, γHV68/72LS8 and γHV68/72LS9 mutant viruses was significantly reduced compared to v-cyclin expression following wt virus infection (Fig. 5, lanes 4–6). Notably, expression of the early antigen encoded by orf 59 was unaffected by the alteration in v-cyclin expression – consistent with the absence of any detectable growth defect of v-cyclin mutant viruses under these culture conditions. Thus, the regions disrupted by LS7, LS8, and LS9 are important for the regulation of v-cyclin expression during lytic viral infection of NIH 3T12 fibroblasts, while the regions disrupted by LS4, LS5, and LS6 do not appear to play a critical regulatory role at this stage of virus infection.
Figure 5.
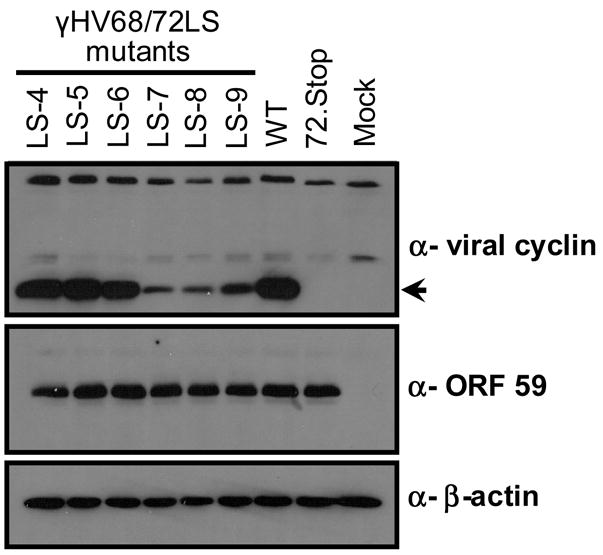
v-cyclin lytic promoter function is necessary for expression of v-cyclin in lytically infected NIH 3T12 fibroblasts. Cells were infected with γHV68/LS4 (LS-4, lane 1), γHV68/LS5 (LS-5, lane 2), γHV68/LS6 (LS-6, lane 3), γHV68/LS7 (LS-7, lane 4), γHV68/LS8 (LS-8, lane 5), γHV68/LS9 (LS-9, lane 6), γHV68 (WT, lane 7), γHV68 72.Stop (v-cyclin stop mutant, lane 8), or mock infected (lane 9) at an MOI of 5. Eighteen hours post infection, cell lysates were prepared, quantitated, and 30 μg of total protein was loaded onto a 15% polyacrylamide gel. Following incubation with anti-v-cyclin antibody, immunoblots were stripped and re-probed first with the anti-ORF 59 antibody to control for effects of the mutations on other viral proteins and then with the anti-β-actin antibody to control for protein loading.
Role of the v-cyclin lytic cycle-associated promoter in virus reactivation from latency
Previous reports have demonstrated that v-cyclin expression is dispensable during lytic replication both in vitro and in vivo (van Dyk, Virgin and Speck 2000). More recently we have shown that following low dose intranasal inoculation that there is a role for the v-cyclin in acute virus replication in vivo (Upton and Speck 2006). In addition, we have shown that the v-cyclin does play a critical role in the reactivation of virus from latent infection in vivo (van Dyk, Virgin and Speck, 2000; Upton and Speck, 2006). Thusfar, we have established that disruption of the v-cyclin lytic cycle-associated promoter can result in diminished v-cyclin expression during virus replication in vitro. To extend these results, we investigated the role of the v-cyclin lytic cycle-associated promoter in directing v-cyclin expression during reactivation from latency. If the lytic cycle-associated promoter plays a critical role in driving transcription of the v-cyclin gene during reactivation from latency, then we would expect that disruption of this promoter would result in a decrease in γHV68 reactivation similar to that seen with the v-cyclin null virus (72Stop) (van Dyk, Virgin and Speck, 2000; Upton and Speck, 2006). In addition, because there are fewer viral genomes in latently infected cells than in lytically infected cells, one might expect that defects in v-cyclin expression would have a more profound effect on reactivation than on lytic replication.
To determine whether those promoter mutations that decrease v-cyclin expression during lytic infection might also disrupt virus reactivation, C57BL/6 mice were infected intranasally with 1000 pfu of WT, 72Stop, γHV68/72LS7, γHV68/72LS8, or γHV68/72LS9 viruses. 16 days post-infection, spleens were harvested and analyzed for the frequency of splenocytes that reactivated virus ex vivo (Figure 6). Splenocytes infected with the 72Stop virus (Figure 6, filled triangles) reactivated virus at an approximately 10-fold lower frequency than WT infected splenocytes (Figure 6, filled squares). However, splenocytes infected with γHV68/72LS7, γHV68/72LS8, or γHV68/72LS9 viruses all reactivated to wildtype levels (Figure 6, open symbols). Notably, the v-cyclin linker scanning mutant viruses also behaved as wildtype following intraperitoneal infection and measurement of reactivation from infected peritoneal exudate cells 42 days post-infection (data not shown). These results indicate that mutations in those regions of the v-cyclin lytic cycle-associated promoter that are most important for regulating v-cyclin expression during lytic infection, do not lead to a loss of reactivation efficiency in vivo. It is possible that the control of virus reactivation from latency maps to other regions of the v-cyclin lytic cycle-associated promoter. However, none of the other v-cyclin LS mutants we have tested (γHV68/72LS4, γHV68/72LS5, and γHV68/72LS6) exhibit diminished reactivation from latency compared to WT virus (data not shown). This could reflect the fact that none of the linker scanning mutants completely abrogates v-cyclin expression, at least as determined in lytically infected murine fibroblasts (see Figure 5). Alternatively, the failure of the LS promoter mutations to recapitulate the 72Stop reactivation phenotype may point to the existence of a separate v-cyclin promoter which is responsible for the regulation of v-cyclin expression during virus reactivation.
Figure 6.
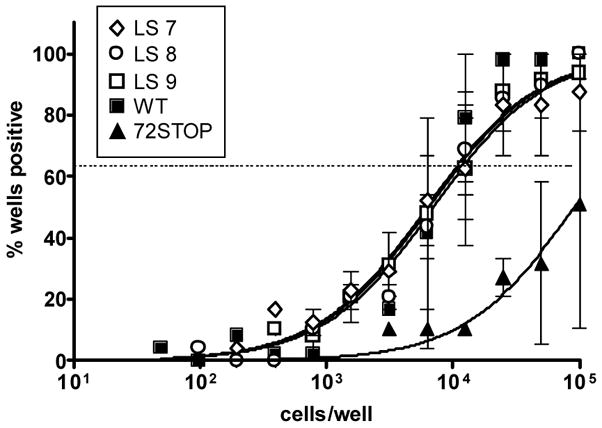
The v-cyclin lytic promoter regions mutated in γHV68/72LS7, γHV68/72LS8, and γHV68/72LS9 viruses are dispensable for virus reactivation. Mice were infected intranasally with 1000 pfu of virus in 20 μl of DMEM. Splenocytes were harvested 16 days after infection and plates on MEF monolayers to assay for virus reactivation (see Materials and Methods) Results from infections with the following viruses are shown: WT (filled squares), 72Stop (filled triangles), γHV68/72LS7 (LS7, open diamonds), γHV68/72LS8 (LS8, open circles), and γHV68/72LS9 (LS9, open squares). The data were compiled from at least two independent experiments, each containing four or five mice per group, and standard error of the mean is indicated.
DISCUSSION
Using RT-PCR or microarray techniques, the v-cyclin transcript has been detected during both lytic and latent virus infection in γHV68 and the related gammaherpesviruses, KSHV and HVS (Dittmer, 2003; Ebrahimi et al, 2003; Jenner et al, 2001; Martinez-Guzman et al, 2003). Published work on v-cyclin transcript structure and gene regulation has focused on those transcripts that are produced in latently infected cells (Dittmer et al, 1998; Hall et al, 2000; Kedes et al, 1997; Sarid et al, 1999). During KSHV latency, the v-cyclin gene is transcribed as an alternatively spliced message that is 3′ coterminal with messages encoding the KSHV LANA and v-FLIP, all driven by a common promoter (Dittmer et al, 1998; Kedes et al, 1997; Sarid et al, 1999). The transcriptional analysis of the v-cyclin gene reported here has focused on identifying transcripts from lytically infected cells. We have characterized a v-cyclin transcript present during lytic infection of murine NIH 3T12 fibroblasts, as well as the promoter that directs transcription of this message. In addition, we have presented data that demonstrate the v-cyclin lytic cycle-associated promoter is Rta responsive. We have yet to determine experimentally whether Rta induction of the v-cyclin lytic cycle-associated promoter is mediated by a direct or indirect mechanism.
Both Rta and v-cyclin fall into the immediate-early class of γHV68 transcripts in lytically infected cells (Martinez-Guzman et al, 2003). As well, lytic infection of BHK cells with a virus that overexpresses Rta leads to a 4-fold increase in v-cyclin transcription by 4 hours post-infection as compared to cells infected with WT virus (Martin-Guzman et al, 2003). It is important to note that Rta expression is necessary for lytic infection and also sufficient to trigger virus reactivation in latently infected cells (Pavlova et al, 2003;Wu et al, 2001; Wu et al, 2000). Thus, among the genes required for γHV68 reactivation are those genes expressed at immediate-early times in lytic infection. It follows that the regulation of genes by Rta during lytic infection indicates a regulatory role of Rta in virus reactivation. Taken together, these results suggest v-cyclin is positioned downstream of Rta in the regulatory cascade that results in γHV68 reactivation. Using a γHV68 Rta-deficient virus, we are currently characterizing the changes in transcription of specific immediate-early gene transcripts in the absence of Rta. By this method we hope to identify additional genes, like v-cyclin, that are dispensable for lytic replication, but required for γHV68 reactivation.
Through systematic mutagenesis of the v-cyclin lytic promoter, we have identified promoter regions that are critical for transactivation by Rta, as well as basal expression of v-cyclin during lytic infection. The LS7, LS8, and LS9 linker scanning mutations define regulatory regions that are required for both transactivation by Rta and v-cyclin expression during lytic infection. Conversely, the LS5 and LS6 mutations defined promoter regions that are required for transactivation by Rta in transient transfection reporter assays, but have no detectable role in directing v-cyclin expression during lytic infection. Thus, by placing the linker scanning mutations in the context of the viral genome, we were able to identify those mutations which, by virtue of their effects on lytic cycle-asociated v-cyclin expression, had the greatest chance of affecting v-cyclin expression during γHV68 reactivation. Because v-cyclin protein function is dispensable for the establishment of latent infection in vivo, but plays an important role for efficient virus reactivation – both in reactivation from latently infected PECs (van Dyk, Virgin and Speck, 2000) and splenocytes (Upton and Speck, 2006) – we examined the in vivo role of the lytic cycle-associated v-cyclin promoter in regulating v-cyclin expression during reactivation. Results from these experiments suggest that the v-cyclin lytic cycle-associated promoter is dispensable for regulation of v-cyclin during virus reactivation. However, because the mutations introduced did not completely abrogate lytic cycle-associated v-cyclin expression, we cannot rule out a role for the v-cyclin lytic promoter during virus reactivation. Nevertheless, we hypothesize that v-cyclin expression during γHV68 reactivation is regulated in a manner independent of v-cyclin lytic cycle-associated expression. Indeed, there is evidence that there are alternative mechanisms for regulating v-cyclin expression. We have recently identified an alternatively spliced v-cyclin transcripts present in a γHV68 latently infected cell line (Allen et al, 2006). These transcripts share 5′ exons with a transcript encoding the γHV68 LANA protein (orf 73). We are currently pursuing experiments to determine the role of this latency-associated v-cyclin transcript in virus reactivation. It will also be important to establish the relative contribution of the different v-cyclin gene promoters in the early and late stages of virus reactivation in various infected cell types in vivo.
MATERIALS AND METHODS
Viruses and tissue culture
NIH 3T12 fibroblasts and murine embryonic fibroblasts were maintained as previously described (Virgin et al, 1997). Vero Cre cells were maintained in Dulbecco’s modified Eagles media (DMEM) supplemented with 10% fetal bovine serum, 100U/ml concentration of antibiotics, 2mM L -glutamine and 300 μg of hygromycin B (Calbiochem) per ml. BAC-derived wildtype and mutant virus stocks were grown and titered on Vero–Cre cells as previously described (Moorman et al, 2004). The generation of the 72Stop virus has been described previously (van Dyk et al, 2000). For analysis of v-cyclin expression during lytic infection, 2x105 NIH 3T12 fibroblasts were plated in 6 well plates and the following day were infected at an moi of 5 with wildtype or mutant virus stock in 200 μl of DMEM supplemented with 10% fetal bovine serum, 100U/ml concentration of antibiotics, and 2mM L-glutamine (CMEM). Plates were incubated at 37°C for 1h and rocked every 15 minutes. Following incubation, 2 mls of prewarmed CMEM was added and plates were incubated for at 37°C for 18 hours prior to harvesting of infected cells for Western Blot analysis (see below).
RACE
RNA was harvested from infected NIH 3T12 fibroblasts by lysis of infected cells with guanidine isothiocyanate/phenol solution (GITC-Phenol, 2M Guanidine Isothiocyanate, 0.05M β-mercaptoethanol, 0.25% Sarcosyl 0.1M NaOAc) followed by extraction with chloroform. RNA was then precipitated with isopropanol and resuspended in RNase-free water. RACE was carried out using the GeneRacer system (Invitrogen). The sequence of the gene specific oligonucleotide primer used for 5′RACE, 72RACE5′, was 5′AGGTCTTTGCACACACAAAACATCCACGTG3′. The sequence of the gene specific oligonucleotide primer used for 3′RACE, 72RACE3′, was 5′ACCCAGTTGGCATACCTTTG3′. PCR products were electrophoresed on 1% agarose gels, and specific bands were excised and prepared for ligation using GeneClean (Q-Biogene). Products were then T-A subcloned into either pGEM-T (Promega) or pCR4-TOPO vector (Invitrogen) and multiple subclones were analyzed by DNA sequencing.
Primer Extension
Primer extension reactions were carried out essentially as described in Current Protocols in Molecular Biology (Ausubel et al, 1992). Briefly, total cellular RNA was harvested from infected or uninfected cells using the GITC-Phenol extraction described above. Polyadenylated RNA was then prepared from total RNA using GenElute mRNA oligodT columns (Sigma). PolyA RNA was hybridized with radiolabeled oligonucleotide probe 72RACE5′ by incubating the mixture at 65°C for 90 minutes. Following incubation, 30 μl of the following reaction mixture was added to the hybridization reaction, (0.9 μl 1M Tris-Cl, pH 8.3, 0.9 μl 0.5M MgCl2, 0.25 μl 1M dithiothreitol, 6.75 μl 1mg/ml antinomycin D, 1.33 μl 5mM mixture of dATP, dTTP, dGTP, and dCTP, 20 μl RNase free dH20, and 0.2 μl 25U/μl AMV reverse transcriptase). Samples were incubated at 42°C for 1 hour, and reactions were stopped by adding 105 μl of RNase reaction mix (100 μg/ml salmon sperm DNA, 20 μg/ml RnaseA in TEN 100 buffer). Samples were loaded onto 9% acrylamide/7M urea gel along with a lamda gt11 sequencing reaction that served as a size marker. Following electrophoresis, the gel was dried, exposed on a phosphoimager screen, and screens were read on a Typhoon 9410 variable mode imager (GE Amersham).
Plasmids and constructs
Luciferase reporter constructs
For wild type v-cyclin promoter constructs, PCR using WT γHV68 BAC DNA template (Adler, Messerle, and Koszinowski, 2001; Adler, Messerle, and Koszinowski, 2003) was performed with an oligonucleotide primer containing nucleotides 103,182 to 103,201 and one of the following oligonucleotide primers: 72/100, nucleotide 103,282-103,262; 72/200, nucleotide 103,382-103,362; 72/300, 103,482-103,462; 72/800, 103,982-103,962. Products were digested with NheI and XhoI (restriction sites were incorporated into the oligonucleotide primer sequences), ligated into pGL3, and the resultant subclones were confirmed by restriction enzyme digestion. For reporter constructs containing putative Rta responsive elements, sense and antisense oligonucleotide pairs comprising LS4-6 (103,287-103,316) or LS6-9 (103,307-103,346) were annealed and cloned into the NheI and BglII site of the heterologous TATA luciferase reporter vector, hsp-tata pGL3 as previously described (Lukac et al., 2001). The sequences of the oligonucleotide primer pairs are as follows with the BglII or NheI overhangs underlined: LS4-LS6_R_GL3, 5′-CTAGCTTTAGTACCTGAAACTAATTACATCATCTG-3′; LS4-LS6_D_GL3, 5′-GATCTCAGATGATGTAATTAGTTTCAGGTACTAAA-3′; LS6-LS9_R_GL3, 5′-CTAGCAAATAGCTGCGCACGCACAACACACCACTTT TTAGTACCT-3′; LS6-LS9_D_GL3, 5′-GATCTAGGTACTAAAAAGTGGTGTG TTGTGCGTGCGCAGCTATTT-3′. Subclones were screened by PCR with the vector oligonucleotide primers pGL3_C (5′-GTGATGTCCACCTCGATATGTGCATC-3′) and pGL3_D (5′-CTAGCAAAATAGGCTGTCCCCAGTG-3′), which generate a 301 bp product without an insert and increase in size concomitant with the insert size. Clones with inserts were sequenced with the oligonucleotide primer pGL3_C for verification. Linker scanning mutations were generated by Quick Change mutagenesis (Stratagene) using 72/800 as a template. In each case, the 10 bp mutated region was replaced by the 10 bp linker sequence, 5′-AGGCGCCCA-3′, which contains an AscI restriction site. Candidate mutants were screened by restriction enzyme digestion and confirmed by DNA sequencing. The promoter sequences and coordinates replaced in each linker scanning mutant are as follows: LS1, 5′-GGTTCGGGTG-3′ (103,257-103,266); LS2, 5′-GAATACAAAA-3′ (103,267-103276); LS3, 5′-GACTTTATTC-3′ (103,277-103,286); LS4, 5′-CAGATGATGT-3′ (103,287-103,296); LS5, 5′-AATTAGTTTC-3′ (103,297-103,306); LS6, 5′-AGGTACTAAA-3′ (103,307-103,316); LS7, 5′-AAGTGGTGTG (103,317-103,326); LS8, 5′-TTGTGCGTGC-3′ (103,327-103,336); LS9, 5′-GCAGC TATTT-3′ (103,337-103,346).
Allelic exchange targeting vectors
All BAC targeting constructs were generated on the backbone of the suicide vector pGS284 (Smith and Enquist, 1999) and inserts were subcloned into the SphI restriction site of the pGS284 polylinker. The linker scanning mutations were as described above in the context of wild type sequences extending from nucleotide 102,702 to 103,881. Allelic exchange mutagenesis was carried out as previously described (Moorman, Willer, and Speck, 2003) and the resultant gHV68 72 LS BAC mutants were screened by AscI digestion of PCR products amplified with primers 72CPR1 (5′-GCGGCCGCTAGCGAAACAGTCTTC AAGAAGGCT-3′) and 72CPR2 (5′-GCGGCCGCTAGCTCTGCAGTCTTCTTCATT TAA-3′). The presence of the intended mutation was confirmed by DNA sequencing of the targeted region using viral DNA as template. Briefly, primers 102750-102773_D (5′-AGCTACCCACGAGAGGGTATCCAG-3′) and 103750-103728_C (5′-ATCCTCCGTCCAACTGCCGGGAC-3′) were used to PCR amplify a 1000 bp fragment from each mutant that was subsequently cloned into pCR Blunt (Invitrogen Corporation) and submitted for sequencing. The Rta expression plasmid pBKRta was constructed using the eukaryotic expression vector pBK-CMV (Stratagene) as previously described (Liu et al., 2000).
Transient Transfections
NIH 3T12 fibroblasts or 293T cells were plated at 3 x 105 or 2 x 105 cells per well, respectively in 6 well plates and were transfected using Superfect reagent (Qiagen) the following day. Transfected cells were harvested after incubation at 37°C for 48 hours. For luciferase assays, transfected cells were resuspended in 200 μl 1X Passive Lysis Buffer (Promega) and incubated 15 minutes at room temperature. Lysates were added to 100 μl luciferase substrate (Promega) and analyzed for luciferase activity with a Turner 20/20 Luminometer (Turner Systems).
Immunoblotting and antibodies
NIH 3T12 cells were infected at an MOI of 5 for 1 hour, washed once with phosphate-buffered saline (PBS), and replaced with complete medium for 18 hours. Cells were harvested by scraping and centrifugation; cell pellets were frozen at −80°C. Whole-cell lysates were prepared from cell pellets resuspended in ELB with protease inhibitors (Upton and Speck, 2006). Protein concentration was determined by A280 measurement on a NanoDrop spectrophotometer (NanoDrop Technologies) and a total of 30ug of total protein was loaded per lane on a 15% sodium dodecyl sulfate (SDS)-polyacrylamide gel and semi-dry transferred to nitrocellulose membrane for immunoblotting. Membranes were blocked at room temperature in blocking buffer (PBS with 0.05% Tween-20 [PBS-T] and 5% nonfat milk) for 1 hour before incubation with the indicated primary antibody for 1 hour. Blots were washed with three changes of PBS-T before incubation with horseradish peroxidase (HRP)-conjugated donkey anti-rabbit, anti-mouse (Jackson Immunochemicals) or anti-chicken (Gallus Immunotech) secondary antibodies at 1:2000, 1:2000, and 1:10,000 dilution, respectively, in blocking buffer for 1 h at room temperature. Bound antibody complexes were detected by enhanced chemiluminescence reagent (Amersham Biosciences) and exposed to film. Primary antibodies used were rabbit polyclonal anti-v-cyclin antiserum (van Dyk et al., 1999), chicken poly-clonal anti-ORF59 (Upton, van Dyk, and Speck, 2005), and mouse anti-β-actin (Sigma).
Mice, infections, and organ harvests
Mice maintained at Yerkes National Primate Research Center in accordance with all federal and university policies. C57BL/6 mice were purchased from Jackson Laboratories. Mice were age and sex matched and were infected at between 8 and 12 weeks of age. Mice were infected by intranasal injection with 1x103 PFU of virus in 20 μl of complete DMEM. Upon sacrifice, spleens were harvested and prepared for plating on MEF monolayers as previously described (29).
Limiting dilution ex vivo reactivation assay
Detection of γHV68 reactivation was performed as previously described (Weck et al., 1996; Weck et al., 1999). Briefly, splenocytes were plated in two-fold serial dilutions, starting at 105 cells per well, onto MEF monolayers in 96-well tissue culture plates. Twenty-four wells were plated per dilution. 21 days post plating, wells were scored for the presence of cytopathic effect (CPE). To detect preformed infectious virus, parallel samples were resuspended in 1/3x DMEM in the presence of 0.5-mm silica beads and treated as previously described (Weck et al., 1996). Disrupted cells were plated in a similar series of twofold dilutions and scored for CPE as described above.
Statistical analyses
All data were analyzed using GraphPad Prism (GraphPad Software, San Diego, Calif.). Titer data were statistically analyzed using the nonparametric Mann-Whitney test. The frequencies of reactivation were statistically analyzed using the paired t test. To accurately obtain the frequency for each limiting dilution, data were subjected to nonlinear regression (using a sigmoidal dose curve with nonvariable slope to fit the data). Frequencies of reactivation were obtained by calculating the cell density at which 63% of the wells scored positive for reactivating virus based on a Poisson distribution.
Acknowledgments
The γHV68-BAC was a generous gift from H. Adler and U. Koszinowski. This work was supported by a Leukemia and Lymphoma Society grant to RDA (5575-02) and a National Institutes of Health grant to SHS (CA87650). We thank past and current members of the Speck laboratory and Dr. Skip Virgin’s laboratory for their helpful advice on experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen RD, Dickerson S, Speck SH. Identification of spliced gammaherpesvirus 68 LANA and v-cyclin transcripts and analysis of their expression in vivo during latent infection. J Virol. 2006;80:2055–2062. doi: 10.1128/JVI.80.4.2055-2062.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler H, Messerle M, Koszinowski UH. Cloning of herpesviral genomes as bacterial artificial chromosomes. Rev Med Virol. 2003;13:111–21. doi: 10.1002/rmv.380. [DOI] [PubMed] [Google Scholar]
- Adler H, Messerle M, Koszinowski UH. Virus reconstituted from infectious bacterial artificial chromosome (BAC)-cloned murine gammaherpesvirus 68 acquires wild-type properties in vivo only after excision of BAC vector sequences. J Virol. 2001;75:5692–6. doi: 10.1128/JVI.75.12.5692-5696.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, Ganem D. A cluster of latently expressed genes in Kaposi’s sarcoma-associated herpesvirus. J Virol. 1998;72:8309–15. doi: 10.1128/jvi.72.10.8309-8315.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer DP. Transcription profile of Kaposi’s sarcoma-associated herpesvirus in primary Kaposi’s sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 2003;63:2010–5. [PubMed] [Google Scholar]
- Ebrahimi B, Dutia BM, Roberts KL, Garcia-Ramirez JJ, Dickinson P, Stewart JP, Ghazal P, Roy DJ, Nash AA. Transcriptome profile of murine gammaherpesvirus-68 lytic infection. J Gen Virol. 2003;84:99–109. doi: 10.1099/vir.0.18639-0. [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DM, Seidman JG, Smith JA, Struhl K. Current Protocols in Molecular Biology. John Wiley and Sons, Inc; 1992. [Google Scholar]
- Gruffat H, Manet E, Sergeant A. MEF2-mediated recruitment of class II HDAC at the EBV immediate early gene BZLF1 links latency and chromatin remodeling. EMBO Rep. 2002;3:141–6. doi: 10.1093/embo-reports/kvf031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall KT, Giles MS, Goodwin DJ, Calderwood MA, Carr LM, Stevenson AJ, Markham AF, Whitehouse A. Analysis of gene expression in a human cell line stably transduced with herpesvirus saimiri. J Virol. 2000;74:7331–7. doi: 10.1128/jvi.74.16.7331-7337.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskowitz J, Jacoby MA, Speck SH. The murine gammaherpesvirus 68 M2 gene is required for efficient reactivation from latently infected B cells. J Virol. 2005;79:2261–73. doi: 10.1128/JVI.79.4.2261-2273.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoge AT, Hendrickson SB, Burns WH. Murine gammaherpesvirus 68 cyclin D homologue is required for efficient reactivation from latency. J Virol. 2000;74:7016–23. doi: 10.1128/jvi.74.15.7016-7023.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain SM, Usherwood EJ, Dyson H, Coleclough C, Coppola MA, Woodland DL, Blackman MA, Stewart JP, Sample JT. Murine gammaherpesvirus M2 gene is latency-associated and its protein a target for CD8(+) T lymphocytes. Proc Natl Acad Sci U S A. 1999;96:7508–13. doi: 10.1073/pnas.96.13.7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby MA, Virgin HW, Speck SH. Disruption of the M2 gene of murine gammaherpesvirus 68 alters splenic latency following intranasal, but not intraperitoneal, inoculation. J Virol. 2002;76:1790–801. doi: 10.1128/JVI.76.4.1790-1801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner RG, Alba MM, Boshoff C, Kellam P. Kaposi’s sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J Virol. 2001;75:891–902. doi: 10.1128/JVI.75.2.891-902.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedes DH, Lagunoff M, Renne R, Ganem D. Identification of the gene encoding the major latency-associated nuclear antigen of the Kaposi’s sarcoma-associated herpesvirus. J Clin Invest. 1997;100:2606–10. doi: 10.1172/JCI119804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Liu P, Borras A, Chatila T, Speck SH. Cyclosporin A-sensitive induction of the Epstein-Barr virus lytic switch is mediated via a novel pathway involving a MEF2 family member. Embo J. 1997;16:143–53. doi: 10.1093/emboj/16.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Pavlova LV, Virgin HW, Speck SH. Characterization of gammaherpesvirus 68 gene 50 transcription. J Virol. 2000;74:2029–37. doi: 10.1128/jvi.74.4.2029-2037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukac DM, Garibyan L, Kirshner JR, Palmeri D, Ganem D. DNA binding by Kaposi’s sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J Virol. 2001;75(15):6786–99. doi: 10.1128/JVI.75.15.6786-6799.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Guzman D, Rickabaugh T, Wu TT, Brown H, Cole S, Song MJ, Tong L, Sun R. Transcription program of murine gammaherpesvirus 68. J Virol. 2003;77:10488–503. doi: 10.1128/JVI.77.19.10488-10503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman NJ, Lin CY, Speck SH. Identification of candidate gammaherpesvirus 68 genes required for virus replication by signature-tagged transposon mutagenesis. J Virol. 2004;78:10282–90. doi: 10.1128/JVI.78.19.10282-10290.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman NJ, Willer DO, Speck SH. The gammaherpesvirus 68 latency-associated nuclear antigen homolog is critical for the establishment of splenic latency. J Virol. 2003;77:10295–303. doi: 10.1128/JVI.77.19.10295-10303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova IV, Virgin HW, Speck SH. Disruption of gammaherpesvirus 68 gene 50 demonstrates that Rta is essential for virus replication. J Virol. 2003;77:5731–9. doi: 10.1128/JVI.77.10.5731-5739.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman B. The Family Herpesviridae: A Brief Introduction. In: David PMH, Knipe W, editors. Fields Virology. 4. Vol. 2. Lippincott Williams and Wilkins; 2001. pp. 2381–2397. [Google Scholar]
- Sarid R, Wiezorek JS, Moore PS, Chang Y. Characterization and cell cycle regulation of the major Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) latent genes and their promoter. J Virol. 1999;73:1438–46. doi: 10.1128/jvi.73.2.1438-1446.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GA, Enquist LW. Construction and transposon mutagenesis in Escherichia coli of a full-length infectious clone of pseudorabies virus, an alphaherpesvirus. J Virol. 1999;73:6405–14. doi: 10.1128/jvi.73.8.6405-6414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck SH, Virgin HW. Host and viral genetics of chronic infection: a mouse model of gamma-herpesvirus pathogenesis. Curr Opin Microbiol. 1999;2:403–9. doi: 10.1016/s1369-5274(99)80071-x. [DOI] [PubMed] [Google Scholar]
- Upton JW, van Dyk LF, Speck SH. Characterization of murine gammaherpesvirus 68 v-cyclin interactions with cellular cdks. Virology. 2005;341(2):271–83. doi: 10.1016/j.virol.2005.07.014. [DOI] [PubMed] [Google Scholar]
- Upton JW, Speck SH. Evidence for CDK-dependent and CDK-independent functions of the murine gammaherpesvirus 68 v-cyclin. J Virol. 2006;80(24):11946–59. doi: 10.1128/JVI.01722-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dyk LF, Hess JL, Katz JD, Jacoby M, Speck SH, Virgin HW. The murine gammaherpesvirus 68 v-cyclin gene is an oncogene that promotes cell cycle progression in primary lymphocytes. J Virol. 1999;73:5110–22. doi: 10.1128/jvi.73.6.5110-5122.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dyk LF, Virgin HW, Speck SH. Maintenance of gammaherpesvirus latency requires viral cyclin in the absence of B lymphocytes. J Virol. 2003;77:5118–26. doi: 10.1128/JVI.77.9.5118-5126.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dyk LF, Virgin HW, Speck SH. The murine gammaherpesvirus 68 v-cyclin is a critical regulator of reactivation from latency. J Virol. 2000;74:7451–61. doi: 10.1128/jvi.74.16.7451-7461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin HW, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol. 1997;71:5894–904. doi: 10.1128/jvi.71.8.5894-5904.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin HW, Presti RM, Li XY, Liu C, Speck SH. Three distinct regions of the murine gammaherpesvirus 68 genome are transcriptionally active in latently infected mice. J Virol. 1999;73:2321–32. doi: 10.1128/jvi.73.3.2321-2332.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weck KE, Barkon ML, Yoo LL, Speck SH, Virgin HW. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J Virol. 1996;70:6775–80. doi: 10.1128/jvi.70.10.6775-6780.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weck KE, Kim SS, Virgin HW, Speck SH. B cells regulate murine gammaherpesvirus 68 latency. J Virol. 1999;73:4651–61. doi: 10.1128/jvi.73.6.4651-4661.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TT, Tong L, Rickabaugh T, Speck S, Sun R. Function of Rta is essential for lytic replication of murine gammaherpesvirus 68. J Virol. 2001;75:9262–73. doi: 10.1128/JVI.75.19.9262-9273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TT, Usherwood EJ, Stewart JP, Nash AA, Sun R. Rta of murine gammaherpesvirus 68 reactivates the complete lytic cycle from latency. J Virol. 2000;74:3659–67. doi: 10.1128/jvi.74.8.3659-3667.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]