

Abstract
The AMP-activated serine/threonine protein kinase (AMPK) is a sensor of cellular energy status found in all eukaryotes that is activated under conditions of low intracellular ATP following stresses such as nutrient deprivation or hypoxia. In the past five years, work from a large number of laboratories has revealed that one of the major downstream signaling pathways regulated by AMPK is the mammalian target-of-rapamycin (mTOR pathway). Interestingly, like AMPK, the mTOR serine/threonine kinase plays key roles not only in growth control and cell proliferation but also in metabolism. Recent work has revealed that across eukaryotes mTOR orthologs are found in two biochemically distinct complexes and only one of those complexes (mTORC1 in mammals) is acutely sensitive to rapamycin and regulated by nutrients and AMPK. Many details of the molecular mechanism by which AMPK inhibits mTORC1 signaling have also been decoded in the past 5 years. AMPK directly phosphorylates at least two proteins to induce rapid suppression of mTORC1 activity, the TSC2 tumor suppressor and the critical mTORC1 binding subunit raptor. Here we explore the molecular connections between AMPK and mTOR signaling pathways and examine the physiological processes in which AMPK regulation of mTOR is critical for growth or metabolic control. The functional conservation of AMPK and TOR in all eukaryotes, and the sequence conservation around the AMPK phosphorylation sites in raptor across all eukaryotes examined suggest that this represents a fundamental cell growth module connecting nutrient status to the cell growth machinery. These findings have broad implications for the control of cell growth by nutrients in a number of cellular and organismal contexts.
Keywords: LKB1, AMPK, mTOR, raptor, TSC2, metabolism, checkpoint
AMPK is an energy sensor coupled to growth control
The AMP-activated protein kinase (AMPK) is a highly conserved heterotrimeric kinase complex composed of a catalytic (α) subunit and two regulatory (β and γ) subunits. AMPK is activated under conditions of energy stress, when intracellular ATP levels decline and intracellular AMP increases, as occurs during nutrient deprivation or hypoxia (Kahn et al. 2005). Upon energy stress, AMP directly binds to tandem repeats of crystathionine-β-synthase (CBS) domains in the AMPK γ subunit. Binding of AMP is thought to prevent dephosphorylation of the critical activation loop threonine in the α subunit (Sanders et al. 2007). The phosphorylation of the activation loop threonine is absolutely required for AMPK activation. Biochemical and genetic analyses in worms, flies, and mice have revealed that the serine/threonine kinase LKB1 represents the major kinase phosphorylating the AMPK activation loop under conditions of energy stress across metazoans (Hardie et al. 2007). In Saccharomyces cerevisiae, three related upstream kinases phosphorylate SNF1, the AMPK ortholog, and LKB1 is the human kinase sharing the greatest homology to all three (Hong, SP et al. 2003, Sutherland et al. 2003).
A fundamental requirement of all cells is that they couple the availability of nutrients to signals emanating from growth factors to drive proliferation only when nutrients are in sufficient abundance to guarantee successful cell division. Upon activation under low ATP conditions, AMPK acts a metabolic checkpoint in the cell, halting cell growth and suppressing ATP-consuming biosynthetic processes while stimulating ATP-generating processes to repair the initiating loss of ATP (Shaw et al. 2004a). In addition to its widespread cell-autonomous role as an energy checkpoint, AMPK also plays key roles in glucose and lipid metabolism in specialized metabolic tissues in mammals and higher eukaryotes such as liver, muscle, and adipose (Kahn et al. 2005). Thus AMPK not only governs cellular energetics, but indeed overall organismal bioenergetics by coordinating the response between tissues to nutritional input.
Consistent with a role for AMPK in growth control, its major upstream kinase LKB1 is a human tumor suppressor. LKB1 was originally identified as the human tumor suppressor gene mutated in Peutz-Jeghers syndrome, an autosomal dominant inherited cancer disorder (Hemminki et al. 1998). In addition, somatic LKB1 mutations are prevalent in a large percentage (30–40%) of sporadic non small cell lung cancers (NSCLC) (Sanchez-Cespedes et al. 2002, Ji et al. 2007). Reintroduction of LKB1 into some LKB1-deficient cancer cell lines results in growth arrest and reversion of tumorigenecity (for full review see Hezel & Bardeesy 2008). Moreover, genetically engineered mice bearing a conditionally inactivated allele of LKB1 have been deleted in a number of tissues revealing that loss of LKB1 in prostate, skin, uterus, gut, and pancreas is sufficient to initiate hyperplasia and tumorigenesis in some of these tissues (Hezel & Bardeesy 2008).
However, AMPK is not the only substrate of LKB1. LKB1 similarly phosphorylates the activation loop of a family of 12 kinases all related to AMPK, also resulting in their activation (Lizcano et al. 2004; Jaleel et al., 2005). Importantly, of these 14 LKB1-dependent kinases, only AMPKα1 and AMPKα2 are activated under low ATP conditions, probably due to the fact that only they interact with AMPKγ which contains the AMP-binding domains (Al-Hakim et al., 2005). However, two other family members, SNARK/Nuak2 and SIK2 have been reported to be activated under low energy conditions (Susuki et al., 2003; Lefebvre and Rosen, 2005; Du et al., 2008). Further work is needed to examine the regulation of the 12 AMPK-related kinases in additional cell types, and to better examine whether they also can bind the AMP-responsive gamma subunits of AMPK under some conditions. Little is currently known about what stimuli direct LKB1 towards any of these AMPK-related kinases and current evidence suggests that LKB1 is constitutively active and these other kinases may be regulated through phosphorylation at other sites outside of their activation loops. Several of these family members have been shown to be transcriptionally upregulated following specific stimuli, including SIK1 following isoproterenol in a CREB-dependent manner (Berdeaux et al., 2007), and SNARK/Nuak2 following FAS ligand in an NF-kB dependent manner (Legembre et al., 2004). The reported overlap in downstream substrates for several of these kinases suggests that their transcriptional upregulation following specific stimuli would result in the temporally controlled production of active kinases owing to their constitutive activation by LKB1. Whether the dephosphorylation of the activation loop threonine is an acutely regulated step for any of the kinases other than AMPKα1 and AMPKα2 also remains to be determined. Finally, thus far there is no substantial mutational data from human tumors to directly support any of the downstream kinases, including AMPK, as being the critical target for LKB1 in tumorigenesis. One confounding issue with the lack of mutations found in these downstream kinases is that there is a great deal of redundancy and functional overlap amongst them, suggesting that loss of any one of them may be compensated for by other family members, unlike the case for LKB1 where no other specific kinase has been shown to functionally compensate for its regulation of these kinases in vivo.
Despite the lack of direct mutational evidence implicating AMPK in cancer, a number of studies in the past 5 years have revealed key growth suppressive roles for AMPK and the identification of a number of direct substrates for AMPK with well-established roles in growth control is consistent with the concept of AMPK as a metabolic checkpoint. The tumor suppressor p53 (Jones et al. 2005) and the CDK inhibitor protein p27 (Liang et al. 2007) have been reported to be direct substrates of AMPK, though neither of the reported AMPK phosphorylation sites in these proteins exactly conform to the established optimal substrate motif for AMPK, perhaps suggesting they may be regulated indirectly. Activation of p53-dependent transcription has been previously been shown to occur downstream of LKB1 and AMPK activation (Imamura et al. 2001, Karuman et al. 2001, Tiainen et al. 2002).
Notably, Peutz-Jeghers syndrome (PJS) shares a number of clinical features with Cowden’s Disease, which is caused by inactivating mutations in the PTEN tumor suppressor. This phenotypic overlap suggested that LKB1-dependent signaling might negatively regulate some aspect of PI3-kinase signaling, analogous to PTEN function. However, while classic PI3K/Akt signaling is not elevated in LKB1-deficient cells, we and others discovered that mTOR signaling is uniquely hyperactivated in LKB1-deficient murine embryonic fibroblasts (MEFs) and liver (Corredetti et al. 2004, Shaw et al. 2004b, 2005). Consistent with this data from non-cancerous settings, tumors arising in LKB1-heterozygous mice and human lung cancer lines lacking LKB1 similarly exhibit hyperactivation of mTOR (Carretero et al. 2007). Given that mTOR is one of the key downstream targets of PI3-kinase signaling, the observation that mTOR is also elevated in many LKB1-deficient cells and tumors suggested a common biochemical basis for the clinical overlap observed in Peutz-Jeghers syndrome and Cowden’s disease.
mTOR is a central regulator of growth and metabolism which is found in two complexes
mTOR (mammalian target of rapamycin), a serine/threonine kinase highly conserved in all eukaryotes, is a central regulator of cell growth (Wullschleger et al. 2006). Whereas AMPK is active under nutrient-poor conditions and inactive under nutrient-rich conditions, mTOR is activated in the inverse pattern. In higher eukaryotes, mTOR activation requires positive signals from both nutrients (glucose, amino acids) and growth factors. mTOR, like its budding yeast orthologs, is found in two biochemically and functionally discrete signaling complexes (Wullschleger et al. 2006). In mammals, the mTORC1 complex is composed of four known subunits: raptor (regulatory associated protein of mTOR), PRAS40, mLST8, and mTOR. Raptor acts as a scaffold to recruit downstream substrates such as 4EBP1 and ribosomal S6 kinase (p70S6K1), to the mTORC1 complex (Nojima et al. 2004, Schalm et al. 2004). The mTORC2 complex contains rictor (rapamycin insensitive companion of mTOR), mSIN1, PRR5/Protor, mLST8, and mTOR (Guertin & Sabatini 2007). Signaling from mTOR complex 1 (mTORC1) is nutrient-sensitive, acutely inhibited by the bacterial macrolide rapamycin, and controls cell growth, angiogenesis, and metabolism. In contrast, mTORC2 is not sensitive to nutrients, nor acutely inhibited by rapamycin, and its known substrates include the hydrophobic motif phosphorylation (“PDK2”) sites in AGC kinases including Akt, SGK, and PKC family members (Guertin & Sabatini 2007, Garcia-Martinez & Alessi 2008).
Downstream of the raptor-mTOR (mTORC1) complex are its two well-characterized substrates: 4EBP1 and the p70 ribosomal S6 kinase. Phosphorylation of 4EBP-1 by mTORC1 suppresses its ability to bind and inhibit the translation initiation factor eIF4E. mTORC1 mediates phosphorylation of S6K at a Thr residue in a hydrophobic motif at the C-terminus of the kinase domain. A specific motif (TOS motif) found in both 4EBP1 and S6K was shown to mediate direct binding of these proteins to raptor allowing them to be phosphorylated in the mTORC1 complex. Mechanistic details of how mTORC1 regulates the assembly of translational initiation complexes via a number of ordered phosphorylation events were recently discovered (Holz et al. 2005). mTORC1-dependent translation is known to control a number of specific cell growth regulators, including cyclin D1, the HIF-1α transcriptional factor and c-myc, which in turn promote processes including cell cycle progression, cell growth, glycolysis, and angiogenesis, all contributing to enhanced tumorigenesis (Guertin & Sabatini 2007). Interestingly, HIF-1α is independently negatively regulated by the VHL tumor suppressor, providing another link between the mTOR pathway and cancer. Furthermore, eIF4E is an oncogene upregulated in a number of cancers (Ruggero and Pandolfi 2003), and eIF4E overexpression has been shown to be sufficient to promote rapamycin-resistant growth of tumors (Wendel et al. 2006) and promote increased cyclin D1 (Averous et al. 2008).
Upstream components of the mTORC1 complex were initially discovered through classical cancer genetics. The TSC2 tumor suppressor tuberin and its obligate binding partner hamartin (TSC1), are mutated in a familial tumor syndrome called Tuberous Sclerosis Complex (TSC). TSC patients are predisposed to widespread benign tumors termed hamartomas in kidney, lung, brain, and skin. Genetic studies in Drosophila and mammalian cells identified the Tuberous Sclerosis Complex (TSC) tumor suppressors as critical upstream inhibitors of the mTORC1 complex. TSC2 (also known as tuberin) contains a GTPase activating protein (GAP) domain at its carboxyl terminus that inactivates the small Ras-like GTPase Rheb, which has been shown to associate with and directly activate the mTORC1 complex in vitro (Sancak et al. 2007). Loss of TSC1 or TSC2 therefore leads to hyperactivation of mTORC1. Phosphorylation of TSC1 and TSC2 serves as an integration point for a wide variety of environmental signals that regulate mTORC1 (Shaw & Cantley 2006). One of the key activators of the mTORC1 pathway is PI3-kinase, which plays a key role in promoting cell growth and insulin-mediated effects on metabolism. PI3-kinase via PDK1 activates the serine/threonine kinase Akt, which directly phosphorylates and inactivates TSC2 through a poorly-understood mechanism. In addition to TSC2, Akt also directly phosphorylates and inactivates an inhibitor of the mTORC1 complex named PRAS40 (Sancak et al. 2007, Vander Haar et al. 2007). In addition to being directly phosphorylated by Akt, TSC2 is reportedly phosphorylated and inactivated by Erk as well as its downstream kinase Rsk (Shaw & Cantley 2006).
AMPK inhibits mTORC1 through phosphorylation of TSC2 and raptor
In addition to these growth stimulatory cues that activate mTORC1, the complex is rapidly inactivated by a wide variety of cell stresses, thereby ensuring that cells do not continue to grow under unfavorable conditions. One of the unique aspects of the mTORC1 complex is that unlike many of the aforementioned growth factor activated kinases, it is dependent on nutrient availability for its kinase activity. Withdrawal of glucose, amino acids, or oxygen leads to rapid suppression of mTORC1 activity (Shaw & Cantley 2006). Upon LKB1- and AMP-dependent activation of AMPK by nutrient loss, AMPK directly phosphorylates the TSC2 tumor suppressor on conserved serine sites distinct from those targeted by other kinases, which constitutes one mechanism by which glucose and oxygen control mTORC1 activation (Inoki et al. 2003, Corredetti et al. 2004, Shaw et al., 2004b, Liu et al., 2006). Ser1387 of human TSC2 (Ser1345 in the rat cDNA) is phosphorylated by AMPK in vivo, though there may be multiple additional sites. Phosphorylation of Ser1387 was also shown to serve as a priming site for additional phosphorylations by GSK-3 at Ser1383 and 1379, which is well-established to target serines 4 residues away from a priming phosphorylation site (Inoki et al. 2006). This phosphorylation of TSC2 by GSK-3 is inhibited by the Wnt signaling pathway, creating a signal integration at these sites depending on both the activation state of AMPK and of GSK-3 that dictates the amount of active TSC2 and hence downstream mTORC1 signaling. Like TSC2, we anticipate that many substrates of AMPK may contain phosphorylation sites for additional mitogen-stimulated kinases such as Akt and Rsk allowing for integration of stress and mitogenic signals.
While TSC2 is clearly a central receiver of a wide variety of positive and negative inputs that regulate mTORC1, cells lacking TSC2 still partially suppress mTORC1 following AMPK activation (albeit less so than wild-type cells) suggesting that additional AMPK substrates may directly or indirectly modulate mTORC1 activity (Hahn-Windgassen et al. 2005; Gwinn et al. 2008). An additional reason to suspect additional AMPK substrates that regulate mTOR is revealed by the fact that glucose inactivation of AMPK orthologs and stimulation of TOR orthologs is conserved across all eukaryotes, including several that lack TSC2 orthologs such as C. elegans and S. cerevisiae. This suggests that either additional mechanisms exist to coordinate the kinase activity of these two master regulators of cell growth and metabolism, or AMPK must target additional conserved components of the pathway. Using an unbiased peptide library screening methodology (Hutti et al. 2004), we recently identified an optimal substrate motif for AMPK which matches well with previous optimal AMPK peptide substrate analyses (Scott et al. 2002). Using the AMPK optimal substrate motif, we performed bioinformatics searches to identify highly conserved substrates of AMPK that may be involved in growth control. We discovered the critical mTOR binding partner raptor as a direct substrate of AMPK, and demonstrated that phosphorylation of raptor by AMPK at two highly conserved serines – Ser722 and Ser792 – induces their direct binding to 14-3-3, which leads to a suppression of mTORC1 kinase activity towards its downstream substrates (Gwinn et al., 2008). Using a phospho-specific antibody generated against Ser792 we demonstrated that endogenous raptor is rapidly phosphorylated at Ser792 in wild-type cells following AMPK agonists and this event is completely absent in AMPK-deficient murine embryonic fibroblasts. Like previous observations in LKB1-deficient MEFs, AMPK-deficient MEFs are also unable to downregulate mTORC1 following energy stress (Gwinn et al., 2008).
Using cells reconstituted with a non-phosphorylatable raptor cDNA or wild-type raptor cDNA, we found that phosphorylation of these two sites in raptor is required for suppression of mTORC1 activity and cell cycle arrest by AMPK agonists including AICAR and phenformin in MEFs. While the p53 tumor suppressor is required for a G1 phase cell cycle arrest following treatment of MEFs with AMPK agonists (Jones et al. 2005), cells lacking both p53 and TSC2 still maintain a G2/M cell cycle arrest following AICAR treatment. We demonstrated that this G2/M arrest is ablated in cells reconstituted with raptor cDNA mutated at the two AMPK phosphorylation sites (Gwinn et al. 2008). These data demonstrate for the first time that downregulation of mTOR is required for efficient cell cycle arrest following AMPK activation. These results also suggest that mTORC1 regulates some step of the G2/M cell cycle progression.
Taken together with previous studies, our findings indicate that energy stress results in LKB-dependent activation of AMPK, which directly phosphorylates both TSC2 and raptor to inhibit mTORC1 activity by a dual-pronged mechanism (see Fig. 1). While the data from murine embryonic fibroblasts suggests that the phosphorylation of TSC2 and raptor are required for AMPK to suppress mTORC1, it remains possible that additional substrates of AMPK also contribute to suppression of mTORC1. Interestingly, mTOR itself has been reported to serve as an AMPK substrate (Cheng et al. 2004), and while the reported site does not conform to the established AMPK substrate consensus, it remains possible there are other bona fide sites for AMPK in mTOR.
Figure 1.
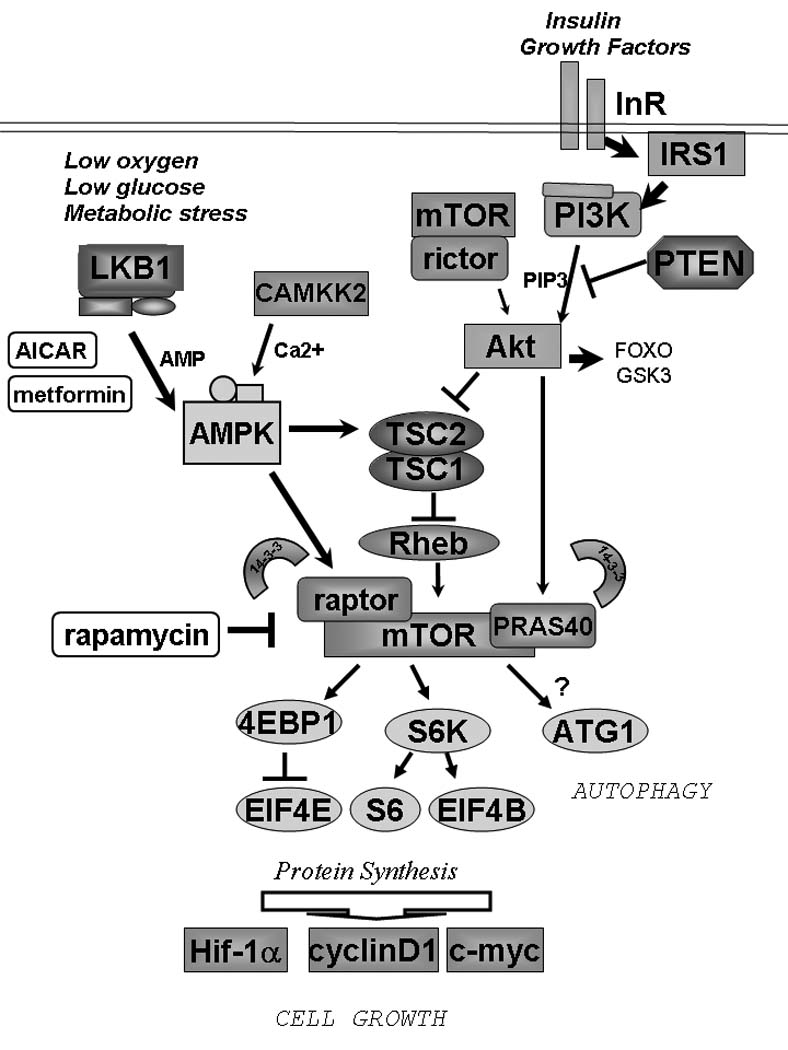
Nutrients and growth factors converge to regulate the mTORC1 complex in mammalian cells. Inherited mutations in LKB1, TSC1. TSC2, and PTEN all result in hamartoma syndromes in humans indicating that hyperactivation of mTORC1 is a common biochemical mechanism underlying these genetic disorders.
Another stress context where AMPK suppresses mTORC1 has recently been uncovered. p53-dependent transcription has been known to result in mTORC1 inhibition in a number of cell types, though the mechanism remained obscure (Levine et al. 2006). Recently, the critical transcriptional targets of p53 that mediate the suppression of mTORC1 were identified as Sestrin1 and Sestrin2 (Budanov & Karin 2008). Overexpression of sestrin1 or sestrin 2 led to increases in AMPK activation and suppression of mTORC1 signaling, whereas mice lacking sestrin2 failed to downregulate mTORC1 following the carcinogen DEN. The molecular mechanism by which sestrins activate AMPK remains to be elucidated and whether sestrins themselves behave as tumor suppressors and in what tissues they are rate-limiting for suppression of mTORC1 following genotoxic stress remains to be fully explored.
Importantly, we expect that the relative contribution of TSC2 as compared to raptor in suppressing mTORC1 downstream of AMPK will vary from tissue to tissue depending on TSC2 and raptor expression patterns and crosstalk on these two proteins from other signaling pathways in individual tissues. Future studies will be needed to define the relative contribution of AMPK-mediated phosphorylation of raptor and TSC2 in various tissue settings to define the physiological contexts in which each plays a significant role in controlling endogenous mTORC1 activity. We explore below some of the tissue settings where AMPK and mTOR have been shown to play specialized roles.
AMPK and mTOR have opposing roles in specialized metabolic tissues in mammals
In addition to broad roles in controlling cell growth in all mammalian cell types, mTOR and AMPK play key roles in a number of “professional” metabolic tissues in mammals. Here we briefly examine a few tissue settings where AMPK and mTOR have been shown to demonstrate opposing control of metabolic function.
In the hypothalamus, food intake is controlled by neurons in the arcuate nucleus. Hypothalmic AMPK is activated in response to low glucose, endocannibinoids, AgRP, or the gastric hormone ghrelin, all of which are increased during fasting. Conversely, AMPK activity is decreased upon refeeding or adminstration of insulin or leptin. Consistent with its suppressive effect on AMPK, leptin induces mTORC1 activity in the neurons of the arcuate nucleus (Woods et al. 2008). Mice bearing disruptions in AMPK or core mTORC1 components exhibit a variety of defects in food intake and organismal energy metabolism (Cota et al. 2006, Claret et al. 2007).
Skeletal and cardiac muscle are additional tissues where AMPK and mTOR play key roles in glucose metabolism, hypertrophy, and the response to exercise. In skeletal muscle, AMPK activation has been shown to promote mitochondrial biogenesis at least in part through transcription effects downstream of PGC-1α and PPARδ (Jager et al. 2007, Narkar et al. 2008). Consistent with an observed loss of mitochondrial mass, mice lacking AMPK function in muscle, either from expression of a dominant-negative AMPK or deletion of LKB1, exhibit dramatic reduction in voluntary exercise (Mu et al. 2001, Thomson et al. 2007). Resistance exercise in humans has been shown to decrease mTORC1-dependent phosphorylation of 4EBP1 coincident with maximal activation of AMPK (Dreyer et al. 2006). Whether AMPK-deficient mice show elevations in mTOR within subtypes of muscle following exercise and whether mTORC1 plays any role in the metabolic reprogramming of muscle fiber type remains to be examined. The picture in skeletal muscle is likely to be complex, as both AMPK and mTOR have been reported to stimulate PGC-1α-dependent mitochondrial biogenesis in this tissue, albeit via distinct mechanisms (Jager et al. 2007; Cunningham et al., 2007). Indeed, previous observation suggest isoform specific AMPK activation in individual muscle types (McGee et al, 2008), suggesting a thorough analysis of all fiber types and muscle groups in the individual AMPKα1 and AMPKα2 knockout mice and ultimately a skeletal muscle tissue-specific deletion of both, will prove necessary to define where and when AMPK is most rate-limiting for mTOR suppression and PGC-1α regulation following specific stimuli. Notably, mTORC1 signaling following insulin or leucine or electrical stimulation is suppressed by AICAR pretreatment in EDL, gastrocnemius, and extensor digitorum longus muscles, respectively (Deshmukh et al. 2008; Pruznak et al., 2008; Thomson et al., 2008). Importantly, utilizing muscle from knockout mice lacking AMPKα2 or AMPKγ3, it was recently demonstrated that each of these AMPK isoforms are required for AICAR to suppress mTORC1 activity (Deshmukh et al. 2008). Indeed AMPK activation by metformin or AICAR or by overexpression of activated LKB1 inhibits protein synthesis and hypertrophy in neonatal rat cardiac myocytes coincident with suppression of mTORC1 signaling (Chan et al. 2004, Noga et al. 2007). Consistent with these findings, induction of hypertrophy by angiotensin II is accompanied by inhibition of AMPK and activation of mTORC1 (Stuck et al. 2008). In critical genetic tests of the involvement of AMPK in cardiac hypertrophy, two independent studies found that following isoproterenol (Zarrinpashneh et al. 2008), or transverse aortic constriction (Zhang et al. 2008), increased hypertrophy was observed in AMPKα2-deficient mice, which correlated with dramatic increases in mTORC1 signaling in the AMPKα2-deficient hearts (Zhang et al. 2008). Although much remains to be elucidated in these models and the molecular interplay between AMPK and mTOR signaling in skeletal and cardiac muscle, it is clear that AMPK modulation of mTOR may play a central role in cardiac hypertrophy.
In liver, AMPK plays key roles in glucose and lipid metabolism. Hormones that activate AMPK in liver including glucagon (Kimball et al. 2004) and adiponectin (Wang et al. 2007) have been reported to suppress mTORC1 signaling. AMPK-dependent effects on hepatic gluconeogenesis are mediated through direct phosphorylation of transcription factors and coactivators that control transcription of gluconeogenic enzymes (Yang et al., 2001; Hong, YH et al., 2003; Koo et al. 2005; Inoue and Yamauchi, 2006). Hence regulation of gluconeogenesis by AMPK is likely independent of effects on mTOR. In contrast, lipogenesis is controlled in part by mTORC1-dependent signals. One key regulator of lipogenesis is the SREBP1 transcription factor. SREBP-1 is a sterol-sensing transcription factor that drives lipogenesis not only in liver, but also in a large variety of mammalian cells. Recently, mTORC1 signaling was shown to be required for nuclear accumulation of SREBP1 and the induction of SREBP1 target genes (Porstmann et al. 2008). Similar to rapamycin, treatment with AMPK agonists including AICAR and 2DG resulted in suppression of nuclear SREBP1 accumulation (Porstmann et al. 2008). Similarly, nuclear SREBP1 is suppressed in the liver of mice treated with metformin (Zhou et al. 2001). Metformin treatment or overexpression of an activated allele of AMPK was found to be sufficient to reduce triglyceride content in insulin resistant HepG2 cells (Zang et al. 2004). Mice lacking hepatic AMPK function due to liver-specific LKB1 deletion show elevated SREBP1 and SREBP1 target genes resulting in lipid accumulation and hepatic steatosis (Shaw et al. 2005). Metformin treatment of mice leads to robust phosphorylation of raptor Ser792 in murine liver, which is ablated in the LKB1-liver specific knockout mice further illustrating that this molecular event may be relevant in the context of AMPK-mediated control of lipid metabolism (Gwinn et al. 2008). Beyond hepatic lipogenesis, SREBP1 has been shown to be critical for cell growth in both Drosophila and mammalian cells (Porstmann et al. 2008) suggesting that it may be a critical target of AMPK and mTOR signaling not only in the context of metabolic disease, but also in tumorigenesis. Consistent with this idea, expression the SREBP-1 transcriptional target Fatty Acid Synthase (FASN) has been linked to breast cancer proliferation and FASN inhibitors are beginning to be explored clinically as anti-cancer agents (Menendez and Lupu, 2007). In future studies, it will be important to define how much of the lipid-reducing effects of AMPK are due to direct phosphorylation of lipogenic enzymes such as Acetyl-CoA Carboxylase (ACC), and how much are due to effects on SREBP-1 dependent transcription through effects of AMPK on mTORC1.
One final metabolic function that AMPK downregulation of mTORC1 may help explain is the effect of AMPK activators as an insulin sensitizer. The ability of metformin and other AMPK-activating drugs to act as insulin sensitizers is well-documented (Towler & Hardie 2007), and one explanation for that effect is that by AMPK lowering hepatic gluconeogenesis independently from insulin, it relieves the amount of insulin required to be made by the pancreas to reduce circulating blood glucose. Another, cell-autonomously molecular explanation for increased insulin resistance observed in the metabolic syndrome has also been decoded in the past 5years. Upon activation by insulin, the insulin receptor dimerizes and transphorylates the IRS1 and IRS2 scaffolding proteins which bind which then serve as docking sites for the regulatory subunit of PI3-kinase, and are responsible for insulin-dependent PI3-kinase activation (Manning 2004). A large number of laboratories in the past 5 years have discovered that one effect of hyperglycemia, hyperlipidemia, and hyperinsulinemia in the circulation is chronic hyperactivation of the mTORC1 complex, and that mTORC1 and its downstream effector S6K1 both directly phosphorylate the IRS family of scaffolds leading to their targeted degradation (Harrington et al. 2004, Shah et al., 2004). This negative feedback loop suppressing of IRS-PI3-kinase activation can explain effects of overnutrition in the organism as well as in cultured cells (Um et al. 2004). Since mTORC1 activity is dictating the extent of the feedback and suppression of PI3-kinase activity, AMPK activation actually serves to attenuate the feedback and promotes cell autonomous restoration of IRS protein levels and IRS signaling to PI3-kinase. The net effect is most clearly observed on IRS protein levels and Akt activation. When mTORC1 activity is high, IRS proteins are low and Akt is inhibited. When AMPK is activated, it suppresses mTORC1, restoring IRS protein levels and Akt activation.
AMPK and TOR function in model organisms to control growth, metabolism, autophagy, and aging
The central role of AMPK in growth and metabolic regulation is widely conserved across eukaryotes. First studied in budding yeast, the AMPK ortholog Snf1 coordinates the response to altered carbon sources as well as diverse environmental stresses including salt stress, nutrient deprivation, and hypoxia (Hong and Carlson 2007). In a manner parallel to its effects on mammalian physiology, under low glucose conditions Snf1 is activated and promotes a reprogramming of glucose metabolism via regulation of a number of transcription factors. Snf1 is also activated upon nitrogen starvation, paralleling inactivation of TOR (Orlova et al. 2006). Further emphasizing the convergence of these pathways across evolution, Snf1- and TOR- dependent signals have been shown to converge in the transcriptional regulation of stress-responsive transcription factors Gln3 and Msn2 (Bertram et al. 2002, Mayordomo et al. 2002). In addition, deletions in AMPK α or γ subunit orthologs result in shortened lifespan in response to nutrient deprivation in yeast (Ashrafi et al. 2000). Opposing the effects seen for AMPK, depletion of TOR or its effector S6K ortholog Sch9 results in lifespan extension in budding yeast (Kaeberlein et al. 2005). The lifespan extension effects of TOR in yeast have been further linked to suppression of protein translation (Steffen et al. 2008, Kaeberlein & Kennedy 2008).
Similarly to yeast, AMPK orthologs in plants (KIN10/KIN11) also play a key role in the response to nutrient deprivation, salt, and herbicide stress (Baena-Gonzalez et al. 2007). Depletion of AMPK results in shortened lifespan in response environmental stresses including constant darkness in plants (Baena-Gonzalez et al. 2007, Thelander et al. 2004). The TOR pathway is also conserved in photosynthetic eukaryotes including the model plant Aribidopsis thaliana and the green algae Chlamydomonas reinhardtii, and studies using genetic TOR mutants have revealed that TOR similarly controls cell growth (Diaz-Troya et al. 2008). Many components of the TORC1 pathway are conserved in plants, and the AMPK phosphorylation sites are found in the Arabidopsis ortholog of raptor.
The strongest support for opposing roles of AMPK and TOR in growth control, physiology, and aging amongst lower eukaryotes come from studies in the nematode C. elegans. AMPK (aak-2) and LKB1 (par-4) orthologs are required for the extended cell cycle arrest of germ cells in dauer worms (Narbonnay & Roy, 2006) as well as the arrest of L1 stage V lineage cells under starvation conditions (Baugh & Sternberg, 2006). In both lineages, AMPK or LKB1 loss causes inappropriate proliferation under nutrient poor conditions. Furthermore, dauer worms lacking AMPK or LKB1 expend their lipid stores resulting in premature death (Narbonnay & Roy, 2008). In addition, AMPK activation is required in C. elegans for lifespan extension by daf-2, heat shock, and glycolytic inhibitors (Apfeld et al. 2004, Curtis et al. 2006, Schulz et al. 2007). Conversely, suppression of TOR promotes lifespan extension in C. elegans (Vellai et al. 2003, Jia et al. 2004, Kapahi P et al. 2004). Lifespan extension has also been observed with depletion of the TORC1 downstream effectors S6K and eIF4E (Hansen et al. 2007, Pan et al. 2007, Syntichaki et al. 2007), as well as with TOR-dependent autophagy components and the Pha-4 Foxa transcription factor (Hansen et al. 2008, Sheaffer et al. 2008), which was previously found as a key lifespan extension gene required for caloric restriction to extend lifespan in C. elegans (Panowski et al. 2007)
It is likely that conserved and opposing roles for AMPK and TOR will be uncovered in other model organisms as well. TOR has a central role in cell growth in all eukaryotes examined to date and recently AMPK activation has been shown to suppress cell proliferation in both Drosophila and Dictyostelium (Mandal et al. 2005, Bokko et al. 2007).
In addition to effects on protein translation that impact cell growth, metabolism, and aging, TOR also has an independent role in the suppression of autophagy under nutrient-rich conditions. Autophagy is the process by which cells consume their own proteins and organelles to maintain levels of essential building blocks and promote survival under nutrient-poor conditions (Levine & Kroemer 2008, Mizushima et al. 2008). The conserved roles of AMPK to promote autophagy from yeast to fly to mammals (Meijer, A.J., & Codogno 2007; Lippai et al., 2008; Hoyer-Hansen & Jaattela 2007) and of TOR to suppress autophagy across those species (Noda & Ohsumi 1998, Dubouloz et al. 2005, Arsham & Neufeld 2006), suggest that these two central metabolic integrators play key roles in the molecular regulation of this process. A conserved role for TOR in autophagy control is well established from budding yeast studies demonstrating that TOR controls the first committed step of autophagy through regulation of the ATG1 serine/threonine kinase (Kamada et al. 2004). mTOR is also known to control mammalian autophagy, and the ATG1 orthologs ULK1 and ULK2 also have reported roles in autophagy in different mammalian cell types (Stephan & Herman 2006) though much further work is needed to dissect the molecular details in higher eukaryotes. The ability of AMPK to promote cell survival under conditions of environmental stress may lie in part in its ability to stimulate autophagy. This cell survival mechanism is likely to play a significant role in tumor cells facing a variety of energetic and environmental stresses as well as in the control of stress resistance and lifespan of the aforementioned species. Notably, autophagy has been tied to lifespan extension as well (Jia & Levine, 2007, Hansen et al. 2008).
Given this plethora of conserved functions for AMPK and TOR, it will be interesting to determine if the predicted AMPK phosphorylation sites in raptor similarly dictate nutrient dependent responses governing cell growth, aging, and stress response in lower organisms.
Therapeutic implications
The suppression of mTORC1 signaling by AMPK has two immediate therapeutic implications for the treatment of human cancer using existing FDA-approved agents:
Tumors in Peutz-Jeghers patients or sporadic non small cell lung cancers lacking LKB1 may be sensitive to mTORC1 inhibitors including rapamycin analogs or mTOR kinase inhibitors
Any tumor exhibiting hyperactivation of mTORC1 may be sensitive to growth suppression by AMPK agonists
Rapamycin as a therapeutic for hamartomas and other LKB1-deficient tumors
Aberrant activation of the mTORC1 pathway has been observed in spontaneously arising tumors in mice genetically engineered for loss of the tumor suppressors Pten, Nf1, Tsc2, or Lkb1 (Shaw and Cantley 2006). Mutations in these genes are responsible for the inherited cancer syndromes Cowden’s disease, Neurofibromatosis Type I, Tuberous Sclerosis Complex, and Peutz-Jeghers syndrome, collectively referred to as phakomatoses and all sharing overlapping clinical features including the development of hamartomas and pigmentation defects. As illustrated in Figure 1, biochemical and cell biological studies from the past decade have revealed that these tumor suppressors all are direct components of the mTOR signaling pathway that serve to inhibit mTORC1 activity.
The underlying hypothesis is that mutational inactivation of these tumor suppressors in individual cells lead to cell-autonomous hyperactivation of mTORC1, promoting cell growth and ultimately resulting in tumors that are subsequently reliant on mTORC1 signaling for tumor maintenance. Consistent with this possibility, rapamycin analogs have been examined for their therapeutic efficacy in the suppression of tumors that arise in a number of the aforementioned mouse models. The Pten+/−, Nf1+/−, Tsc+/−, and activated Akt transgenic mouse models have also proven to be responsive to rapamycin analogs. These drugs have been proven to effectively inhibit mTORC1 in vivo and reduce tumor burden through mTORC1 dependent mechanisms, including suppression of cyclin D, Mcl-1, or HIF-1α and its targets (Podsypanina et al. 2001, Majumder et al. 2004, Wendel et al. 2006, Johannessen et al. 2008).
In recent clinical trials, rapamycin analogs were shown to have palliative success on patients with PTEN-deficient glioblastomas and metastatic renal cell carcinoma (Hudes et al. 2007, Cloughesy et al. 2008) Furthermore, in a pair of phase II clinical trials involving tuberous sclerosis (TSC) and lymphangioleiomyomatosis (LAM) patients, partial responses to the rapamycin analogs were observed, including regression of angiomyoliomas (Bissler et al. 2008, Davies et al. 2008). Combined with data from mouse models, these clinical data suggest that hamartoma syndromes with hyperactivation of mTORC1 may be particularly responsive to rapamycin analogs as a single agent. To date there are no therapies to treat Peutz-Jeghers syndrome (PJS) and the only course of treatment is resection of arising gastrointestinal hamartomatous polyps. We and others have recently completed a preclinical trial using rapamycin to treat the Lkb1+/− mouse model of PJS which displayed great efficacy (Wei et al. 2008, Shackelford et al. submitted). While these results are encouraging for the use of rapamycin analogs as therapeutics for PJS, like the recent Phase II clinical trial findings with TSC patients, removal of the drug may result in rapid return of the initial tumor due to the largely cytostatic nature of the response (Bissler et al. 2008). Perhaps new, targeted inhibitors directed at the kinase domain of mTOR will produce greater therapeutic response with targeted cytotoxicity, or perhaps kinase inhibitors that dually inactivate mTOR and PI3K would be even more effective, as PI3K provides a survival signal in most epithelial cell types. As observed in most cancers studied to date, combinations of targeted therapeutics, or of targeted and traditional chemo-therapeutics may find the ultimate utility in the treatment of this disease. Importantly, it is worth noting here that rapamycin treatment may not only be therapeutically useful for the hamartomas that arise in Peutz-Jeghers patients, but also in preventing and reducing any secondary malignancies that arise in these patients at additional sites (breast, pancreas, ovary).
In terms of human disease however, the number of patients with Peutz-Jeghers syndrome is dwarfed by the number of people with sporadic lung tumors containing LKB1 mutations. Recent studies have revealed that LKB1 mutations are among the most common in smoking-associated lung tumors, which are still the most common form of cancer in the United States and worldwide. LKB1 mutations are found in non-small cell lung carcinomas, which represent 85% of lung cancer, and mutation rate estimates vary between 20% and 40% depending on the study and the methodology employed for detecting LKB1 alterations (Ji et al. 2007, Hezel & Bardeesy 2008). Analysis of human NSCLC cell lines and primary tumors have revealed loss of AMPK and hyperactivation of mTORC1 (Carretero et al. 2005), indicating that the AMPK/ mTOR pathway is likely a relevant target of LKB1 for the suppression of tumorigenesis in the setting of the lung. Interestingly, the K-ras oncogene is frequently co-mutated with LKB1 in NSCLC (Makowski & Hayes 2008). To model these tumors, mice bearing a conditional activated allele of K-ras were crossed to those bearing a conditional inactivating allele of LKB1 and then mice were treated with an aerosolized adenovirus inducing deletion and hence simultaneous activation of K-ras and loss of LKB1 in isolated lung cells (Ji et al. 2007). The K-ras, LKB1 double mutant mice showed a dramatic increase in their tumor incidence and metastasis resulting in rapid acceleration of death (25 weeks for K-ras alone vs. 10 weeks for K-ras and LKB1 mutant). Immunohistochemical analysis of these tumors revealed that the LKB1-deficient tumors showed loss of AMPK signaling and even further increases in mTORC1 signaling beyond what was observed with K-ras alone (Ji et al. 2007). Combined with the human data, these data reinforce that the AMPK/ mTORC1 pathway may be relevant for tumorigenesis in this setting. Whether mTORC1 inhibitors as a single agent would show any utility against these tumors is unclear, although given that the tumors are not only mutant for LKB1, but also have all the K-ras dependent mitogenic signaling pathways stimulated, one might imagine a combination of mTORC1 and Erk, or mTORC1 and Akt inhibitors might demonstrate greater clinical efficacy.
Beyond Peutz-Jeghers syndrome and NSCLC patients, the fact that LKB1 is rate-limiting for tumor formation in a number of tissue settings in the mouse suggests that LKB1, and hence AMPK, may have a wider role in human cancers than currently appreciated. In particular, LKB1 deficiency in prostate, keratinocytes, uterine epithelium, pancreas, and bone all give rise to tumor-prone phenotypes (Hezel & Bardeesy 2008). Whether these tumors all possess elevations of mTORC1 remains to be determined. As AMPK likely has multiple substrates involved growth control, and LKB1 regulates 12 kinases in addition to AMPK, the relative importance of mTORC1 hyperactivation to the tumor phenotype in each of these contexts is an open question that remains to be answered.
Finally, therapeutics aimed at AMPK activation also hold potential for cancer. AMPK activation by metformin or AICAR inhibits the growth of tumor cell in culture as well as in xenograft models (Buzzai et al. 2007). Moreover, metformin treatment suppresses naturally-arising tumors in transgenic and carcinogen-treated rodent cancer models (Schneider et al. 2001, Anisimov et al. 2005, Huang et al. 2008). Given the known pharmacokinetics and widespread long-term clinical use of metformin, its utility as a potential chemotherapeutic modality deserves further attention. In addition, the biguanide phenformin which was removed from clinical use for type 2 diabetes due to incidence of fatalities from lactic acidosis, may find modern utility as a anti-cancer agent as the dosing and duration of its use would be quite distinct from that used for diabetes. Recently, the first study to directly compare the anti-tumoral efficacy of metformin, phenformin, and the small molecule compound Abbott A769662 was reported (Huang et al. 2008). The authors examined the ability of each of these AMPK activators to suppress tumors in Pten+/− mice, which are prone to spontaneously-arising lymphomas. While all three compounds resulted in delayed tumor onset, phenformin and Abbott A769662 suppressed tumors with much greater efficacy, which correlated with their ability to activate AMPK and suppress mTORC1 in a wide number of tissues in vivo, unlike metformin, which caused more transient and less robust changes on the signaling pathways in most tissues examined. Perhaps key to the success observed here is the fact that the tumors initiated from loss of Pten and hence activation of PI3-kinase, making mTORC1 hyperactivation of the biochemical initiating events for this tumor type and increasing the odds that mTORC1 signaling is required for the tumor. These data also suggest a possible therapeutic window for the use of AMPK agonists to treat tumors arising in patients with Tuberous Sclerosis Complex or for tumors exhibiting hyperactivation of mTOR by other genetic lesions. The fact that the targeted Abbott compound also performed well suggests that AMPK is in fact the key target of the biguanides in tumor reduction, though off-target effects of the Abbott compound have also been reported (Moreno et al. 2008).
Importantly, compounds that activate AMPK will not only inhibit tumorigenesis via suppression of mTORC1 and lipogenic targets such as ACC, but perhaps also through alterations in organismal metabolism such as reducing blood glucose and insulin resistance, leading to lowered systemic blood insulin levels as well. Given the number of type 2 diabetics worldwide taking metformin daily (>100 million), epidemiologists have begun examing cancer incidence of those taking metformin. Initial studies have revealed that diabetic patients taking metformin exhibit a statistical reduction of tumor incidence (Evans et al. 2005, Bowker et al., 2006). Many additional epidemiological studies are required to determine whether there is indeed a clear effect of prolonged use of these compounds. Most important will be determining if any particular tumor types and any specific tumor genotypes best predict therapeutic efficacy of these compounds.
In fact, tumor cells lacking LKB1 are hypersensitive to apoptosis in culture following treatment with energy stress inducing agents, presumably originating from an inability to restore ATP levels due to AMPK deficiency (Shaw et al. 2004a, Carrereto et al. 2005, Memmott et al. 2008). However, in cells with functional AMPK signaling, the ability of AMPK to restore ATP levels results in cell survival effects in a number of cell types, which might result in increased tumor cell survival even if it caused a growth arrest of cells within the tumor. However, the magnitude of ATP depletion and effective level of energy stress different cell populations face in vivo induced by each combination of drugs, combined with the status of apoptotic and pro-survival signals in these cells is something that can only be modeled accurately in vivo, reinforcing the need for better mouse models of spontaneously arising tumors in their natural tissue setting.
Unanswered questions and future perspectives
The existence of a nutrient-regulated endogenous tumor suppressor pathway that couples cell growth to glucose and lipid metabolism results in a number of intriguing predictions and unanswered questions. For example, do environmental factors such as diet and exercise that contribute to physiological AMPK activation modulate tumorigenic risk through mTORC1 suppression? It is clear from a large number of epidemiology studies that cancer risk is elevated amongst individuals with metabolic syndrome, or obesity, or type 2 diabetes alone (Martinez et al. 2008). While a number of metabolic factors are deregulated in these individuals, not the least of which are increased insulin levels and commensurate changes in the insulin/IGF-1 signaling axis, these individuals also show hyperactivation of mTORC1 in many peripheral cell types which may contribute to increased cell proliferation of certain lineages. The identity of the cell types most sensitive to growth suppression effects of AMPK and LKB1 may reveal those lineages in which cell growth is most tightly coupled to dietary conditions. Conversely, exercise and caloric restriction, which both activate AMPK in some lineages, can lower overall cancer risk and improve cancer prognosis (McTiernan 2008). The mammalian cell types in which exercise and caloric restriction suppress cell growth and cancer risk also remain to be delineated. Though much remains to be done to examine whether AMPK mediates some of the beneficial effects of exercise and caloric restriction on cancer risk, a recent study revealed that AMPK was activated, and mTORC1 signaling was suppressed, in some rodent tissues in a dose-dependent manner by increasing amounts of dietary restriction (Jiang et al. 2008). Conversely, high fat diet was observed to increase mTOR and decrease AMPK activity in some mouse tissues (Moore et al., 2008).
Future studies are needed to further delineate the full set of tissue types and physiological conditions in which AMPK plays a prominent role in the regulation of overall mTOR activity. The investigation of mice deficient in AMPKα1 or AMPKα2, as well as mice bearing tissue-specific deletions of both will be critical for these studies, and will also be useful for defining tissues where loss of AMPK is sufficient to promote tumorigenesis. While AMPK regulation of mTOR is most likely to play a role in tissues that experience physiological or pathological energy stress, in other tissues and conditions, the activation of mTOR by nutrients and growth factors may play a more prominent role in dictating the level of mTOR activity.
In addition to cell proliferation, cell survival, and autophagy, inhibition of mTORC1 by AMPK will likely play a physiological role in other biological processes that mTORC1 is known to regulate including angiogenesis, mitochondrial metabolism, viral infection, and specific transcriptional responses. The existence of diverse biological and metabolic processes regulated by AMPK for which there are few known direct effectors suggests that there are many critical substrates yet to be identified. It will also be interesting to determine which of these will turn out to involve inhibition of mTORC1 as the as-of-yet unappreciated molecular mechanism.
In addition to clear roles in cell growth, the central role of AMPK and mTOR in autophagic control may be key to many differentiated non-dividing adult cells throughout the body in response to dietary and hormonal homeostasis fluctuations. The critical role that autophagy is beginning to be appreciated to play in the pathology of cancer, neurological diseases, and metabolic diseases, is just one more indication that this basic signaling pathway connecting AMPK to mTORC1 is likely to lie at the heart of many human maladies (Levine & Kroemer 2008). As the response to a shortage of environmental nutrients and resultant loss in cellular energy represents one of the most fundamental pathological events in all organisms, we anticipate that further investigation of the downstream targets of AMPK will provide great insight into the emerging nexus of cancer and metabolic disease controlled by this ancestral signaling pathway.
Acknowledgements
The author would like to thank Katja Lamia for her editorial suggestions. Research in the author’s laboratory is generously supported by grants from the NIH (P01 CA120964, R01 DK080425), American Cancer Society, American Diabetes Association, V Foundation for Cancer Research, and Adler Foundation.
Footnotes
Conflict of Interest
There is no conflict of interest.
References
- Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Kovalenko IG, Poroshina TE, Semenchenko AV, Provinciali M, Re F, Franceschi C. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol. 2005;40:685–693. doi: 10.1016/j.exger.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Apfeld J, O'Connor G, McDonagh T, DiStefano PS, Curtis R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004;18:3004–3009. doi: 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsham AM, Neufeld TP. Thinking globally and acting locally with TOR. Curr Opin Cell Biol. 2006;18:589–597. doi: 10.1016/j.ceb.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Ashrafi K, Lin SS, Manchester JK, Gordon JI. Sip2p and its partner snf1p kinase affect aging in S. cerevisiae. Genes Dev. 2000;14:1872–1885. [PMC free article] [PubMed] [Google Scholar]
- Averous J, Fonseca BD, Proud CG. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene. 2008;27:1106–1113. doi: 10.1038/sj.onc.1210715. [DOI] [PubMed] [Google Scholar]
- Baena-Gonzalez E, Rolland F, Thevelein JM, Sheen J. A central integrator of transcription networks in plant stress and energy signalling. Nature. 2007;448:938–942. doi: 10.1038/nature06069. [DOI] [PubMed] [Google Scholar]
- Baugh LR, Sternberg PW. DAF-16/FOXO regulates transcription of cki-1/Cip/Kip and repression of lin-4 during C. elegans L1 arrest. Curr Biol. 2006;16:780–785. doi: 10.1016/j.cub.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Bertram PG, Choi JH, Carvalho J, Chan TF, Ai W, Zheng XF. Convergence of TOR-nitrogen and Snf1-glucose signaling pathways onto Gln3. Mol Cell Biol. 2002;22:1246–1252. doi: 10.1128/MCB.22.4.1246-1252.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdeaux R, Goebel N, Banaszynski L, Takemori H, Wandless T, Shelton GD, Montminy M. SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat Med. 2007;13:597–603. doi: 10.1038/nm1573. [DOI] [PubMed] [Google Scholar]
- Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J, Salisbury S, Franz DN. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokko PB, Francione L, Bandala-Sanchez E, Ahmed AU, Annesley SJ, Huang X, Khurana T, Kimmel AR, Fisher PR. Diverse cytopathologies in mitochondrial disease are caused by AMP-activated protein kinase signaling. Mol Biol Cell. 2007;18:1874–1886. doi: 10.1091/mbc.E06-09-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29:254–258. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–6752. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- Carretero J, Medina PP, Blanco R, Smit L, Tang M, Roncador G, Maestre L, Conde E, Lopez-Rios F, Clevers HC, Sanchez-Cespedes M. Dysfunctional AMPK activity, signalling through mTOR and survival in response to energetic stress in LKB1-deficient lung cancer. Oncogene. 2007;26:1616–1625. doi: 10.1038/sj.onc.1209951. [DOI] [PubMed] [Google Scholar]
- Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem. 2004;279:32771–32779. doi: 10.1074/jbc.M403528200. [DOI] [PubMed] [Google Scholar]
- Cheng SW, Fryer LG, Carling D, Shepherd PR. Thr2446 is a novel mammalian target of rapamycin (mTOR) phosphorylation site regulated by nutrient status. J Biol Chem. 2004;279:15719–15722. doi: 10.1074/jbc.C300534200. [DOI] [PubMed] [Google Scholar]
- Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, Clements M, Al-Qassab H, Heffron H, Xu AW, Speakman JR, Barsh GS, Viollet B, Vaulont S, Ashford ML, Carling D, Withers DJ. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest. 2007;117:2325–2336. doi: 10.1172/JCI31516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, Hsueh T, Chen Y, Wang W, Youngkin D, Liau L, Martin N, Becker D, Bergsneider M, Lai A, Green R, et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5:e8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corredetti MN, Inoki K, Bardeesy N, DePinho R, Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004;18:1533–1538. doi: 10.1101/gad.1199104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312:927–930. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- Curtis R, O'Connor G, DiStefano PS. Aging networks in Caenorhabditis elegans: AMP-activated protein kinase (aak-2) links multiple aging and metabolism pathways. Aging Cell. 2006;5:119–126. doi: 10.1111/j.1474-9726.2006.00205.x. [DOI] [PubMed] [Google Scholar]
- Davies DM, Johnson SR, Tattersfield AE, Kingswood JC, Cox JA, McCartney DL, Doyle T, Elmslie F, Saggar A, de Vries PJ, Sampson JR. Sirolimus therapy in tuberous sclerosis or sporadic lymphangioleiomyomatosis. N Engl J Med. 2008;358:200–203. doi: 10.1056/NEJMc072500. [DOI] [PubMed] [Google Scholar]
- Deshmukh AS, Treebak JT, Long YC, Viollet B, Wojtaszewski JF, Zierath JR. Role of adenosine 5'-monophosphate-activated protein kinase subunits in skeletal muscle mammalian target of rapamycin signaling. Mol Endocrinol. 2008;22:1105–1112. doi: 10.1210/me.2007-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Troya S, Perez-Perez ME, Florencio FJ, Crespo JL. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy. 2008;4:851–865. doi: 10.4161/auto.6555. [DOI] [PubMed] [Google Scholar]
- Dreyer HC, Fujita S, Cadenas JG, Chinkes DL, Volpi E, Rasmussen BB. Resistance exercise increases AMPK activity and reduces 4E-BP1 phosphorylation and protein synthesis in human skeletal muscle. J Physiol. 2006;576:613–624. doi: 10.1113/jphysiol.2006.113175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Chen Q, Takemori H, Xu H. SIK2 can be activated by deprivation of nutrition and it inhibits expression of lipogenic genes in adipocytes. Obesity. 2008;16:531–538. doi: 10.1038/oby.2007.98. [DOI] [PubMed] [Google Scholar]
- Dubouloz F, Deloche O, Wanke V, Cameroni E, De Virgilio C. The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol Cell. 2005;19:15–26. doi: 10.1016/j.molcel.2005.05.020. [DOI] [PubMed] [Google Scholar]
- Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. Bmj. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochem J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. 2005;280:32081–32089. doi: 10.1074/jbc.M502876200. [DOI] [PubMed] [Google Scholar]
- Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007;6:95–110. doi: 10.1111/j.1474-9726.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008;4:e24. doi: 10.1371/journal.pgen.0040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, Gout I, Downes CP, Lamb RF. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, Jarvinen H, Kristo P, Pelin K, Ridanpaa M, Salovaara R, Toro T, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- Hezel AF, Bardeesy N. LKB1; linking cell structure and tumor suppression. Oncogene. 2008;27:6908–6919. doi: 10.1038/onc.2008.342. [DOI] [PubMed] [Google Scholar]
- Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569–580. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Hong SP, Leiper FC, Woods A, Carling D, Carlson M. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci USA. 2003;100:8839–8843. doi: 10.1073/pnas.1533136100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SP, Carlson M. Regulation of snf1 protein kinase in response to environmental stress. J Biol Chem. 2007;282:16838–16845. doi: 10.1074/jbc.M700146200. [DOI] [PubMed] [Google Scholar]
- Hong YH, Varanasi US, Yang W, Leff T. AMP-activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem. 2003;278:27495–27501. doi: 10.1074/jbc.M304112200. [DOI] [PubMed] [Google Scholar]
- Hoyer-Hansen M, Jaattela M. AMP-activated protein kinase: a universal regulator of autophagy? Autophagy. 2007;3:381–383. doi: 10.4161/auto.4240. [DOI] [PubMed] [Google Scholar]
- Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, McBurnie W, Fleming S, Alessi DR. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008;412:211–221. doi: 10.1042/BJ20080557. [DOI] [PubMed] [Google Scholar]
- Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, Kovacevic Z, Lesovoy V, Schmidt-Wolf IG, Barbarash O, Gokmen E, O'Toole T, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–2281. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- Hutti JE, Jarrell ET, Chang JD, Abbott DW, Storz P, Toker A, Cantley LC, Turk BE. A rapid method for determining protein kinase phosphorylation specificity. Nat Methods. 2004;1:27–29. doi: 10.1038/nmeth708. [DOI] [PubMed] [Google Scholar]
- Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. Cell cycle regulation via p53 phosphorylation by a 5'-AMP activated protein kinase activator, 5-aminoimidazole- 4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem Biophys Res Commun. 2001;287:562–567. doi: 10.1006/bbrc.2001.5627. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, Macdougald OA, You M, Williams BO, Guan KL. TSC2 Integrates Wnt and Energy Signals via a Coordinated Phosphorylation by AMPK and GSK3 to Regulate Cell Growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- Inoue E, Yamauchi J. AMP-activated protein kinase regulates PEPCK gene expression by direct phosphorylation of a novel zinc finger transcription factor. Biochem Biophys Res Commun. 2006;351:793–799. doi: 10.1016/j.bbrc.2006.10.124. [DOI] [PubMed] [Google Scholar]
- Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaleel M, McBride A, Lizcano JM, Deak M, Toth R, Morrice NA, Alessi DR. Identification of the sucrose non-fermenting related kinase SNRK, as a novel LKB1 substrate. FEBS Lett. 2005;579:1417–1423. doi: 10.1016/j.febslet.2005.01.042. [DOI] [PubMed] [Google Scholar]
- Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, Liang MC, Cai D, Naumov GN, Bao L, Contreras CM, Li D, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–810. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131:3897–3906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- Jia K, Levine B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy. 2007;3:597–599. doi: 10.4161/auto.4989. [DOI] [PubMed] [Google Scholar]
- Jiang W, Zhu Z, Thompson HJ. Dietary energy restriction modulates the activity of AMP-activated protein kinase, Akt, and mammalian target of rapamycin in mammary carcinomas, mammary gland, and liver. Cancer Res. 2008;68:5492–5499. doi: 10.1158/0008-5472.CAN-07-6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen CM, Johnson BW, Williams SM, Chan AW, Reczek EE, Lynch RC, Rioth MJ, McClatchey A, Ryeom S, Cichowski K. TORC1 is essential for NF1-associated malignancies. Curr Biol. 2008;18:56–62. doi: 10.1016/j.cub.2007.11.066. [DOI] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, Powers RW, 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, Kennedy BK. Protein translation, 2008. Aging Cell. 2008;7:777–782. doi: 10.1111/j.1474-9726.2008.00439.x. [DOI] [PubMed] [Google Scholar]
- Kamada Y, Sekito T, Ohsumi Y. Autophagy in yeast: a TOR-mediated response to nutrient starvation. Curr Top Microbiol Immunol. 2004;279:73–84. doi: 10.1007/978-3-642-18930-2_5. [DOI] [PubMed] [Google Scholar]
- Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuman P, Gozani O, Odze RD, Zhou XC, Zhu H, Shaw R, Brien TP, Bozzuto CD, Ooi D, Cantley LC, Yuan J. The Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell death. Mol Cell. 2001;7:1307–1319. doi: 10.1016/s1097-2765(01)00258-1. [DOI] [PubMed] [Google Scholar]
- Kimball SR, Siegfried BA, Jefferson LS. Glucagon represses signaling through the mammalian target of rapamycin in rat liver by activating AMP-activated protein kinase. J Biol Chem. 2004;279:54103–54109. doi: 10.1074/jbc.M410755200. [DOI] [PubMed] [Google Scholar]
- Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, Takemori H, Montminy M. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- Lefebvre DL, Rosen CF. Regulation of SNARK activity in response to cellular stresses. Biochim Biophys Acta. 2005;1724:71–85. doi: 10.1016/j.bbagen.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Legembre P, Schickel R, Barnhart BC, Peter ME. Identification of SNF1/AMP kinase-related kinase as an NF-kappaB-regulated anti-apoptotic kinase involved in CD95-induced motility and invasiveness. J Biol Chem. 2004;279:46742–46747. doi: 10.1074/jbc.M404334200. [DOI] [PubMed] [Google Scholar]
- Levine AJ, Feng Z, Mak TW, You H, Jin S. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev. 2006;20:267–275. doi: 10.1101/gad.1363206. [DOI] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM, Mills GB. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- Lippai M, Csikos G, Maroy P, Lukacsovich T, Juhasz G, Sass M. SNF4Agamma, the Drosophila AMPK gamma subunit is required for regulation of developmental and stress-induced autophagy. Autophagy. 2008;4:476–486. doi: 10.4161/auto.5719. [DOI] [PubMed] [Google Scholar]
- Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21:521–531. doi: 10.1016/j.molcel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, Alessi DR. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. Embo J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, Loda M, Lane HA, Sellers WR. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10:594–601. doi: 10.1038/nm1052. [DOI] [PubMed] [Google Scholar]
- Makowski L, Hayes DN. Role of LKB1 in lung cancer development. Br J Cancer. 2008;99:683–688. doi: 10.1038/sj.bjc.6604515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal S, Guptan P, Owusu-Ansah E, Banerjee U. Mitochondrial regulation of cell cycle progression during development as revealed by the tenured mutation in Drosophila. Dev Cell. 2005;9:843–854. doi: 10.1016/j.devcel.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol. 2004;167:399–403. doi: 10.1083/jcb.200408161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez ME, Marshall JR, Giovannucci E. Diet and cancer prevention: the roles of observation and experimentation. Nat Rev Cancer. 2008 doi: 10.1038/nrc2441. [epub in print] [DOI] [PubMed] [Google Scholar]
- Mayordomo I, Estruch F, Sanz P. Convergence of the target of rapamycin and the Snf1 protein kinase pathways in the regulation of the subcellular localization of Msn2, a transcriptional activator of STRE (Stress Response Element)-regulated genes. J Biol Chem. 2002;277:35650–35656. doi: 10.1074/jbc.M204198200. [DOI] [PubMed] [Google Scholar]
- McGee SL, Mustard KJ, Hardie DG, Baar K. Normal hypertrophy accompanied by phosphoryation and activation of AMP-activated protein kinase alpha1 following overload in LKB1 knockout mice. J Physiol. 2008;586:1731–1741. doi: 10.1113/jphysiol.2007.143685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTiernan A. Mechanisms linking physical activity with cancer. Nat Rev Cancer. 2008;8:205–211. doi: 10.1038/nrc2325. [DOI] [PubMed] [Google Scholar]
- Meijer AJ, Codogno P. AMP-activated protein kinase and autophagy. Autophagy. 2007;3:238–240. doi: 10.4161/auto.3710. [DOI] [PubMed] [Google Scholar]
- Memmott RM, Gills JJ, Hollingshead M, Powers MC, Chen Z, Kemp B, Kozikowski A, Dennis PA. Phosphatidylinositol ether lipid analogues induce AMP-activated protein kinase-dependent death in LKB1-mutant non small cell lung cancer cells. Cancer Res. 2008;68:580–588. doi: 10.1158/0008-5472.CAN-07-3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore T, Beltran L, Carbajal S, Strom S, Traag J, Hursting SD, DiGiovanni J. Dietary energy balance modulates signaling through the Akt/mammalian target of rapamycin pathways in multiple epithelial tissues. Cancer Prev Res. 2008;1:65–76. doi: 10.1158/1940-6207.CAPR-08-0022. [DOI] [PubMed] [Google Scholar]
- Moreno D, Knecht E, Viollet B, Sanz P. A769662, a novel activator of AMP-activated protein kinase, inhibits non-proteolytic components of the 26S proteasome by an AMPK-independent mechanism. FEBS Lett. 2008;582:2650–2654. doi: 10.1016/j.febslet.2008.06.044. [DOI] [PubMed] [Google Scholar]
- Mu J, Brozinick JT, Jr, Valladares O, Bucan M, Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell. 2001;7:1085–1094. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- Narbonne P, Roy R. Inhibition of germline proliferation during C. elegans dauer development requires PTEN, LKB1 and AMPK signalling. Development. 2006;133:611–619. doi: 10.1242/dev.02232. [DOI] [PubMed] [Google Scholar]
- Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, Kang H, Shaw RJ, Evans RM. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- Noga AA, Soltys CL, Barr AJ, Kovacic S, Lopaschuk GD, Dyck JR. Expression of an active LKB1 complex in cardiac myocytes results in decreased protein synthesis associated with phenylephrine-induced hypertrophy. Am J Physiol Heart Circ Physiol. 2007;292:H1460–H1469. doi: 10.1152/ajpheart.01133.2006. [DOI] [PubMed] [Google Scholar]
- Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K, Hara K, Tanaka N, Avruch J, Yonezawa K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003;278:15461–15464. doi: 10.1074/jbc.C200665200. [DOI] [PubMed] [Google Scholar]
- Orlova M, Kanter E, Krakovich D, Kuchin S. Nitrogen availability and TOR regulate the Snf1 protein kinase in Saccharomyces cerevisiae. Eukaryot Cell. 2006;5:1831–1837. doi: 10.1128/EC.00110-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, Kapahi P. Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell. 2007;6:111–119. doi: 10.1111/j.1474-9726.2006.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A. PHA-4/Foxa mediates dietrestriction-induced longevity of C. elegans. Nature. 2007;447:550–555. doi: 10.1038/nature05837. [DOI] [PubMed] [Google Scholar]
- Podsypanina K, Lee RT, Politis C, Hennessy I, Crane A, Puc J, Neshat M, Wang H, Yang L, Gibbons J, Frost P, Dreisbach V, Blenis J, Gaciong Z, Fisher P, Sawyers C, et al. An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase activity in Pten+/− mice. Proc Natl Acad Sci USA. 2001;98:10320–10325. doi: 10.1073/pnas.171060098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8:224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruznak AM, Kazi AA, Frost RA, Vary TC, Lang CH. Activation of AMP-activated protein kinase by 5-aminoimidazole-4-carboxamide-1-beta-D-ribonucleoside prevents leucine-stimulated protein synthesis in rat skeletal muscle. J Nutr. 2008;138:1887–1894. doi: 10.1093/jn/138.10.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3:179–192. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Sanchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM, Westra WH, Herman JG, Sidransky D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002;62:3659–3662. [PubMed] [Google Scholar]
- Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403:139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalm SS, Fingar DC, Sabatini DM, Blenis J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr Biol. 2003;13:797–806. doi: 10.1016/s0960-9822(03)00329-4. [DOI] [PubMed] [Google Scholar]
- Schneider MB, Matsuzaki H, Haorah J, Ulrich A, Standop J, Ding XZ, Adrian TE, Pour PM. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology. 2001;120:1263–1270. doi: 10.1053/gast.2001.23258. [DOI] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose Restriction Extends Caenorhabditis elegans Life Span by Inducing Mitochondrial Respiration and Increasing Oxidative Stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Scott JW, Norman DG, Hawley SA, Kontogiannis L, Hardie DG. Protein kinase substrate recognition studied using the recombinant catalytic domain of AMP-activated protein kinase and a model substrate. J Mol Biol. 2002;317:309–323. doi: 10.1006/jmbi.2001.5316. [DOI] [PubMed] [Google Scholar]
- Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650–1656. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004a;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004b;6:91–99. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Cantley LC. Ras, PI(3)K, and mTOR signaling controls tumor cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- Sheaffer KL, Updike DL, Mango SE. The Target of Rapamycin pathway antagonizes pha-4/FoxA to control development and aging. Curr Biol. 2008;18:1355–1364. doi: 10.1016/j.cub.2008.07.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuck BJ, Lenski M, Bohm M, Laufs U. Metabolic Switch and Hypertrophy of Cardiomyocytes following Treatment with Angiotensin II Are Prevented by AMP-activated Protein Kinase. J Biol Chem. 2008;283:32562–32569. doi: 10.1074/jbc.M801904200. [DOI] [PubMed] [Google Scholar]
- Steffen KK, MacKay VL, Kerr EO, Tsuchiya M, Hu D, Fox LA, Dang N, Johnston ED, Oakes JA, Tchao BN, Pak DN, Fields S, Kennedy BK, Kaeberlein M. Yeast life span extension by depletion of 60s ribosomal subunits is mediated by Gcn4. Cell. 2008;133:292–302. doi: 10.1016/j.cell.2008.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan JS, Herman PK. The regulation of autophagy in eukaryotic cells: do all roads pass through Atg1? Autophagy. 2006;2:146–148. doi: 10.4161/auto.2.2.2485. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Kusakai G, Kishimoto A, Minegichi Y, Ogura T, Esumi H. Induction of cell-cell detachment during glucose starvation through F-actin conversion by SNARK, the fourth member of the AMP-activated protein kinase catalytic subunit family. Biochem Biophys Res Commun. 2003;311:156–161. doi: 10.1016/j.bbrc.2003.09.184. [DOI] [PubMed] [Google Scholar]
- Sutherland CM, Hawley SA, McCartney RR, Leech A, Stark MJ, Schmidt MC, Hardie DG. Elm1p is one of three upstream kinases for the Saccharomyces cerevisiae SNF1 complex. Curr Biol. 2003;13:1299–1305. doi: 10.1016/s0960-9822(03)00459-7. [DOI] [PubMed] [Google Scholar]
- Syntichaki P, Troulinaki K, Tavernarakis N. eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature. 2007;445:922–926. doi: 10.1038/nature05603. [DOI] [PubMed] [Google Scholar]
- Thelander M, Olsson T, Ronne H. Snf1-related protein kinase 1 is needed for growth in a normal day-night light cycle. Embo J. 2004;23:1900–1910. doi: 10.1038/sj.emboj.7600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson DM, Porter BB, Tall JH, Kim HJ, Barrow JR, Winder WW. Skeletal muscle and heart LKB1 deficiency causes decreased voluntary running and reduced muscle mitochondrial marker enzyme expression in mice. Am J Physiol Endocrinol Metab. 2007;292:E196–E202. doi: 10.1152/ajpendo.00366.2006. [DOI] [PubMed] [Google Scholar]
- Thomson DM, Fick CA, Gordon SE. AMPK activation attenuates S6K1, 4E-BP1, and eEF2 signaling responses to high-frequency electrically stimulated skeletal muscle contractions. J Appl Physiol. 2008;104:625–632. doi: 10.1152/japplphysiol.00915.2007. [DOI] [PubMed] [Google Scholar]
- Tiainen M, Vaahtomeri K, Ylikorkala A, Makela TP. Growth arrest by the LKB1 tumor suppressor: induction of p21(WAF1/CIP1) Hum Mol Genet. 2002;11:1497–1504. doi: 10.1093/hmg/11.13.1497. [DOI] [PubMed] [Google Scholar]
- Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100:328–341. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–323. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426:620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- Wang C, Mao X, Wang L, Liu M, Wetzel MD, Guan KL, Dong LQ, Liu F. Adiponectin sensitizes insulin signaling by reducing p70 S6 kinase-mediated serine phosphorylation of IRS-1. J Biol Chem. 2007;282:7991–7996. doi: 10.1074/jbc.M700098200. [DOI] [PubMed] [Google Scholar]
- Wei C, Amos CI, Zhang N, Wang X, Rashid A, Walker CL, Behringer RR, Frazier ML. Suppression of Peutz-Jeghers polyposis by targeting mammalian target of rapamycin signaling. Clin Cancer Res. 2008;14:1167–1171. doi: 10.1158/1078-0432.CCR-07-4007. [DOI] [PubMed] [Google Scholar]
- Wendel HG, Malina A, Zhao Z, Zender L, Kogan SC, Cordon-Cardo C, Pelletier J, Lowe SW. Determinants of sensitivity and resistance to rapamycin-chemotherapy drug combinations in vivo. Cancer Res. 2006;66:7639–7646. doi: 10.1158/0008-5472.CAN-06-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods SC, Seeley RJ, Cota D. Regulation of food intake through hypothalamic signaling networks involving mTOR. Annu Rev Nutr. 2008;28:295–311. doi: 10.1146/annurev.nutr.28.061807.155505. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Yang W, Hong YH, Shen XQ, Frankowski C, Camp HS, Leff T. Regulation of transcription by AMP-activated protein kinase: phosphorylation of p300 blocks its interaction with nuclear receptors. J Biol Chem. 2001;276:38341–38344. doi: 10.1074/jbc.C100316200. [DOI] [PubMed] [Google Scholar]
- Zang M, Zuccollo A, Hou X, Nagata D, Walsh K, Herscovitz H, Brecher P, Ruderman NB, Cohen RA. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J Biol Chem. 2004;279:47898–47905. doi: 10.1074/jbc.M408149200. [DOI] [PubMed] [Google Scholar]
- Zarrinpashneh E, Beauloye C, Ginion A, Pouleur AC, Havaux X, Hue L, Viollet B, Vanoverschelde JL, Bertrand L. AMPKalpha2 counteracts the development of cardiac hypertrophy induced by isoproterenol. Biochem Biophys Res Commun. 2008;376:677–681. doi: 10.1016/j.bbrc.2008.09.057. [DOI] [PubMed] [Google Scholar]
- Zhang P, Hu X, Xu X, Fassett J, Zhu G, Viollet B, Xu W, Wiczer B, Bernlohr DA, Bache RJ, Chen Y. AMP activated protein kinase-alpha2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension. 2008;52:918–924. doi: 10.1161/HYPERTENSIONAHA.108.114702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]