

Summary
Incorrectly specified or mis-specified cells often undergo cell death or are transformed to adopt a different cell fate during development. The underlying cause for this distinction is largely unknown. In many developmental mutants in Drosophila, large numbers of mis-specified cells die synchronously, providing a convenient model for analysis of this phenomenon. The maternal mutant bicoid is particularly useful model with which to address this issue because its mutant phenotype is a combination of both transformation of tissue (acron to telson) and cell death in the presumptive head and thorax regions. We show that a subset of these mis-specified cells die through an active gene-directed process involving transcriptional upregulation of the cell death inducer hid. Upregulation of hid also occurs in oskar mutants and other segmentation mutants. In hid bicoid double mutants, mis-specified cells in the presumptive head and thorax survive and continue to develop, but they are transformed to adopt a different cell fate. We provide evidence that the terminal torso signaling pathway protects the mis-specified telson tissue in bicoid mutants from hid-induced cell death, whereas mis-specified cells in the head and thorax die, presumably because equivalent survival signals are lacking. These data support a model whereby mis-specification can be tolerated if a survival pathway is provided, resulting in cellular transformation.
Keywords: Mis-specification, Cell death, Transformation, Bicoid, Oskar, Hid, Drosophila
Introduction
Programmed cell death or apoptosis is an important component of normal development and tissue homeostasis. It is required to remove cells that are no longer needed, cells that have been produced in excess and cells that have sustained genetic damage (Baehrecke, 2002). The molecular mechanisms that activate and execute the cell death program are conserved from worms to flies to mammals (reviewed by Danial and Korsmeyer, 2004). One common feature is the activation of caspases, a highly specialized class of cell death proteases. Produced as inactive zymogens, activation of caspases occurs through proteolytic cleavage, leading to cell death (Salvesen and Abrams, 2004). In Drosophila, the cell death-inducing genes head involution defective (hid; W –FlyBase), reaper and grim integrate a large number of cell death-inducing stimuli, culminating in activation of caspases (White et al., 1994; White et al., 1996; Grether et al., 1995; Chen et al., 1996). Caspases proteolytically cleave a large number of cellular proteins resulting in death and removal of the affected cell.
During abnormal development, cell death is also a contributing factor to the phenotypes of many mutants in Drosophila. This was first noted more that 35 years ago for mutants affecting imaginal development (Fristrom, 1968; Fristrom, 1969) (reviewed by Bonini and Fortini, 1999), and it was later also found in mutants affecting embryonic patterning, including maternal-effect genes, gap genes, pair-rule genes and segment-polarity genes (Lehmann et al., 1986; Tepass et al., 1994; Magrassi and Lawrence, 1988; Martinez-Arias and Ingham, 1985; Perrimon and Mahowald, 1987; Klingensmith et al., 1989; Bejsovec and Wieschaus, 1993; Pazdera et al., 1998; Hughes and Krause, 2001). In these mutants, entire regions of the developing organism are deleted (Nüsslein-Volhard and Wieschaus, 1980). The underlying cause for cell death in these mutants is unknown, but apparently cells are able to monitor their ability to specify and differentiate correctly. If a cell fails to complete its normal developmental program (from now on referred to as mis-specified cell), it undergoes cell death.
However, it is not always the case that mis-specified cells die. Instead, a transformation to adopt a different cell fate has often been observed. For example, in embryos obtained from bicoid mutant females, the acron is transformed to become a telson (see below). Other examples include sevenless (sev) and bride of sevenless (boss) mutations, which cause a transformation of the R7 photoreceptor to a cone cell during eye development in the fly (Tomlinson and Ready, 1986; Reinke and Zipursky, 1988). The factors influencing the choice between cell death and cell fate transformation in various mutants are not well understood. To address this issue, we initiated an analysis of the cell death and transformation phenotypes of the maternal effect mutants bicoid and oskar during embryogenesis.
During Drosophila development, the wild-type embryo generates five distinct regions along the anteroposterior axis that are visible in the larval cuticle as acron, head, thorax, abdomen and telson (Fig. 1A,D) (Nüsslein-Volhard et al., 1987). The maternal effect mutants bicoid and oskar severely disrupt anteroposterior patterning. bicoid mutant females produce embryos (from now on referred to as bicoid mutants) that lack head and thorax, and a duplicated telson replaces the acron at the anterior tip of the embryo (Fig. 1B,E) (Frohnhöfer and Nüsslein-Volhard, 1986; Frohnhöfer and Nüsslein-Volhard, 1987). oskar mutant females produce embryos (referred to as oskar mutants) that lack the entire abdomen, with the telson intact (Fig. 1C,F) (Lehmann and Nüsslein-Volhard, 1986). Development of acron and telson is independent of bicoid and oskar, and requires the torso signaling pathway (Klingler et al., 1988; Schüpbach and Wieschaus, 1986). However, bicoid specifies acron versus telson at the anterior tip of the embryo (Fig. 1B) (Frohnhöfer and Nüsslein-Volhard, 1986).
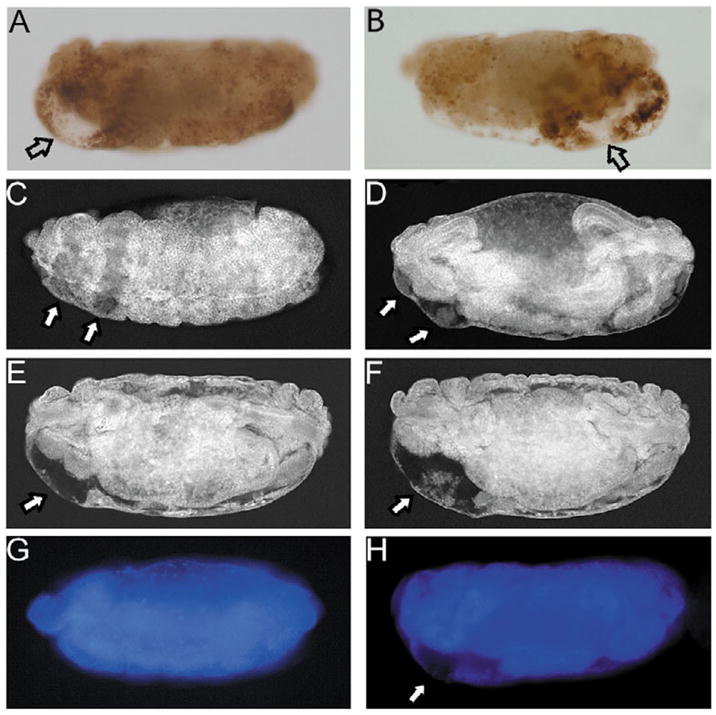
Fig. 1. Caspase-dependent cell death in bicoid and oskar mutants.

(A–C) Schematic illustration of the wild-type (A), bicoid (B) and oskar (C) phenotypes. In each panel, the embryonic fate maps are shown on the left, the differentiated larvae on the right. During development, wild-type embryos specify five distinct regions along the anteroposterior axis that are visible in the larval cuticle as Acron (Ac), Head (He), Thorax (Th), Abdomen (Ab) and Telson (Te). Arrows indicate the polarity of the tissues. T1-3 and A1-8 denote thoracic and abdominal segments, respectively. In bicoid and oskar mutants, this pattern is severely affected and some of the regions are missing. In addition, in bicoid mutants, the anterior acron is transformed into a telson (B). Modified, with permission, from Nüsslein-Volhard et al. (Nüsslein-Volhard et al., 1987). (D–F) Lateral views of larval cuticle preparations of wild-type (D), bicoid (E) and oskar (F) mutants. (G–I) Lateral views of TUNEL-labeled embryos of wild-type (G), bicoid (H) and oskar (I) mutants. (H,I) Brackets indicate areas of increased cell death; arrows indicate the presumptive telson (Te) areas, which are TUNEL negative. (J–L) CM1 labeling to detect active DrICE in wild-type (J), bicoid (K) and oskar (L) mutants. Lateral views. (K,L) Brackets highlight areas of increased caspase activation; arrows indicate the presumptive telson (Te) areas, which lack caspase activation. (M–O) Expression of the caspase inhibitor P35 blocks TUNEL-positive cell death in wild-type (M), bicoid (N) and oskar (O) mutants.
In wild-type, bicoid+ mRNA is maternally localized at the anterior tip of the embryo (Berleth et al., 1988). After fertilization, Bicoid protein forms an exponential concentration gradient along the anteroposterior axis with a maximum at the anterior tip, the source of its translation (Driever and Nüsslein-Volhard, 1988a) (reviewed by Ephrussi and St Johnston, 2004). Bicoid, which is a homeodomain-containing transcription factor, induces target gene expression in a dose-dependent manner, which is required for proper specification of head and thorax (Driever and Nüsslein-Volhard, 1988b; Driever and Nüsslein-Volhard, 1989; Struhl et al., 1989). Thus, loss of Bicoid results in failure to provide this specification, and the mutant embryos do not develop head and thorax (Fig. 1B,E). Similarly, oskar mRNA is localized at the posterior tip of the embryo where it is required to localize the posterior determinant nanos (Ephrussi et al., 1991; Kim-Ha et al., 1991). In the absence of oskar function, posterior development is disturbed, and the entire abdomen fails to develop (Fig. 1C,F).
The mechanisms that cause loss of embryonic tissue in bicoid and oskar mutants are unclear. In previous studies, these mutants were examined from fertilization to gastrulation, when the wild-type functions of bicoid and oskar are required for proper specification of cell fates along the anteroposterior axis. Hence, little is known about the events after gastrulation, when the bicoid and oskar mutant phenotypes, which result in significant tissue loss, are established. In principle, loss of tissue could result from decreased cell proliferation or increased cell death. Because nuclear divisions and cellularization are normal in bicoid and oskar mutants (Frohnhöfer and Nüsslein-Volhard, 1986; Frohnhöfer and Nüsslein-Volhard, 1987; Lehmann and Nüsslein-Volhard, 1986), defects in cell proliferation are unlikely to account for tissue loss observed in these mutants. Rather, increased cell death is an attractive mechanism to explain the bicoid and oskar mutant phenotypes.
Indeed, cell corpses in the abdomen of oskar mutants have been observed previously (Lehmann and Nüsslein-Volhard, 1986); however, the underlying cause has never been carefully examined. Here, we show that cell death is responsible for the loss of tissue in bicoid and oskar mutant embryos. Furthermore, our analysis implies that cellular mis-specification in these mutants triggers cell death through an active gene-directed pathway leading to expression of the cell death-inducing gene hid, resulting in caspase-dependent cell death. However, our data also show that if cell death is blocked either by removing hid or by providing a survival signaling pathway, mis-specification is tolerated and transformation can occur.
Materials and methods
Fly stocks and genetics
The following fly stocks were used: Canton S (as wild-type reference), bicoidE1, bicoidE2, bicoidGB, oskar88, oskar166, torso1, torso4, hidWR+X1, hidH99 and hidA329. The hidH99 bicoidE2 and hidA329 oskar88 double mutants were obtained by meiotic recombination. hid bicoid double mutants were maternally mutant for bicoid and zygotically mutant for hid, and were obtained from females of the genotype hidH99 bicoidE2/bicoidGB mated to hidWR+X1/+ males. hid oskar double mutants are maternally mutant for oskar and zygotically mutant for hid, and were obtained from hidA329 oskar88/oskar166 females mated to hidH99/+ males. To inhibit caspases by P35 expression, females of the maternal genotype UASp-P35, bicoidE1/bicoidGB and UASp-P35, oskar88/oskar166 were crossed with tub-GAL4 males.
For generation of UASp-P35 transgenic flies, primers GAGCTTGCGGCCGCAAAATGTGTGTAATTTTTCCGG and TTAGGCTCTAGATTTTAACATTTATTTAATTGTG were used to amplify the P35-coding region by PCR. The PCR product was treated with NotI and XbaI, and cloned into NotI/XbaI-treated pUASp vector (Rørth, 1998). Transgenic flies were generated by P-element-mediated transformation.
Immunohistochemistry
TUNEL assays, immunohistochemistry, in situ hybridization and Acridine Orange labeling of whole-mount embryos were performed in accordance with standard procedures (Goyal et al., 2000; Patel, 1994; Tautz and Pfeifle, 1989; Abrams et al., 1993). The CM1 antibody as well as monoclonal antibodies against Dlg (4F3), Abd-B (1A2E9) and Antp (8C11) were used at 1:1,000, 1:300, 1:20 and 1:100 dilutions, respectively. For cuticle preparations, differentiated embryos were embedded in Hoyer’s medium (van der Meer, 1977).
Results
Cell death and caspase activation in bicoid and oskar mutants
Reasoning that cell death might be an important contributor to the bicoid and oskar mutant phenotypes, we employed TUNEL as an assay to visualize cell death (Gavrieli et al., 1992). Compared with wild-type embryos, both bicoid and oskar mutants showed an increased number of cells undergoing TUNEL-positive cell death (Fig. 1G-I). In bicoid mutants, TUNEL labeling is elevated in two distinct stripes in the presumptive head and thorax region of the embryos, whereas almost the entire posterior half of oskar mutants is strongly TUNEL positive. These TUNEL-positive regions correspond to the missing tissues of the larvae (Fig. 1E,F). It is not clear why TUNEL-labeling in bicoid embryos starts out in two distinct stripes. However, later in development these stripes become wider and eventually fuse (see Fig. 2). Similar results were obtained by labeling embryos with Acridine Orange, an alternative method to labeling dying cells (see Fig. 4A). The presumptive telson regions in bicoid and oskar embryos are TUNEL negative (arrows in Fig. 1H,I). However, in torsoc mutants, acron and telson tissues also undergo TUNEL-positive cell death (data not shown).
Fig. 2.
Development of the areas of clearance. (A,B) CM1 labeling of stage 15 bicoid (A) and oskar (B) mutants. Open arrows indicate areas of clearance. (C–F) Confocal images of lateral views of stage 14 (C), stage 15 (D), stage 16 (E) and stage 17 (F) bicoid mutants stained with anti-Discs large (Dlg) antibody to visualize cell outline. Clearance of tissue is initially detectable in two distinct zones (see arrows in C and D), but becomes broader over time, fuses (E) and enlarges (F). Similar results were obtained for oskar (not shown). (G,H) DAPI staining of wild-type (G) and bicoid (H) mutants to visualize chromosomal DNA. The areas of clearance (arrow in H) do not contain DNA.
Fig. 4.
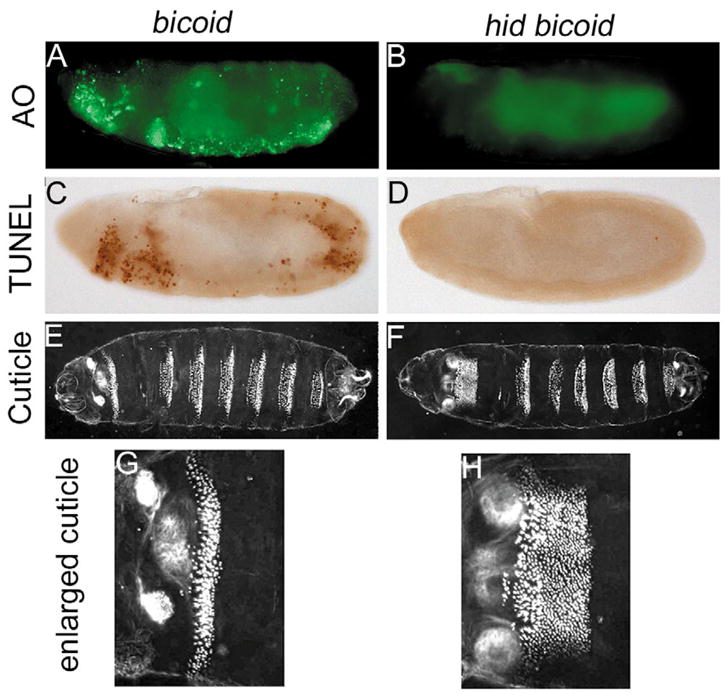
hid bicoid double mutant analysis. Acridine Orange (A,B) and TUNEL (C,D) labeling of dying cells in stage 10 bicoid mutant (A,C) and hid bicoid double mutant (B,D) embryos. (E,F) Ventral views of larval cuticle preparations of bicoid (E) and hid bicoid (F) mutants.
(G,H) Enlargements of the first abdominal segment of bicoid (E) and hid bicoid (F) mutants, respectively.
In wild-type embryos, normal developmental cell death starts at embryonic stage 11 (~7 hours) (Abrams et al., 1993). However, TUNEL-positive cell death in bicoid and oskar mutants is already detectable at stage 10, about 1 hour before the onset of normal cell death in wild-type embryos, and remains detectable in the affected regions throughout development until shortening of the mutant embryos is visible (data not shown). These results suggest that cell death contributes to the loss of tissue in bicoid and oskar mutants.
We analyzed whether the increased cell death observed in bicoid and oskar mutants is caspase dependent. Cleavage and activation of caspases was determined immunohistochemically using the CM1 antibody (Srinivasan et al., 1998), which recognizes the active Drosophila caspase DrICE (Yu et al., 2002). Compared with wild-type embryos, increased CM1 labeling is detectable in bicoid and oskar mutants (Fig. 1J-L) in the regions that were identified by increased TUNEL labeling (Fig. 1H,I). The telson areas are spared from caspase activation. Similar to the onset of TUNEL-positive cell death, CM1 labeling is first detectable at stage 10, suggesting a direct correlation between caspase activation and cell death in these domains. Next, we analyzed whether caspase activation is required for increased cell death in bicoid and oskar mutants. The caspase inhibitor P35 has been shown to inhibit developmental cell death in Drosophila embryos (Fig. 1M) (Hay et al., 1993). Thus, we expressed P35 in bicoid and oskar mutants. TUNEL-positive cell death is strongly reduced in these embryos (Fig. 1N,O), suggesting that cell death in bicoid and oskar mutants is dependent on caspase activation.
Cell death causes clearance of tissue in bicoid and oskar mutants
CM1 labeling is present in the affected regions of the embryos until they are shortened. We detected an interesting intermediate phenotype in bicoid and oskar mutants, beginning at stage 14. Labeling with the CM1 antibody revealed large areas with no staining signal surrounded by immunopositive tissue (Fig. 2A,B, arrows). These areas also fail to give signals with antibodies recognizing ubiquitously expressed proteins such as Tubulin and Dronc, another Drosophila caspase (data not shown). We refer to these areas as ‘areas of clearance’.
Areas of clearance are not observed in wild-type embryos and in P35-expressing embryos (Fig. 1N,O), suggesting that they develop as a direct consequence of cell death in bicoid and oskar embryos. Hence, we characterized them in a time course using an antibody against the ubiquitously expressed membrane-associated Discs large (Dlg) protein (Parnas et al., 2001). Initially, in stage 14 bicoid embryos, beginning of clearance is visible in two distinct zones similar to TUNEL labeling and caspase activation (Fig. 2C). However, the two zones fuse over the next few stages and the areas of clearance enlarge significantly (Fig. 2D–F). Later these areas collapse, and the final phenotype of bicoid mutants is established. Similar data were obtained for oskar mutants (data not shown). These areas also do not contain DNA (Fig. 2G,H). Thus, DNA and proteins are cleared as a result of the apoptotic events in these tissues. Moreover, because Dlg is membrane associated, these data indicate that de-cellularization, the end result of the apoptotic process, has occurred. In Fig. 5 and Fig. S2 (see supplementary material), we are using the areas of clearance as markers of cell death in response to developmental mis-specification.
Fig. 5.
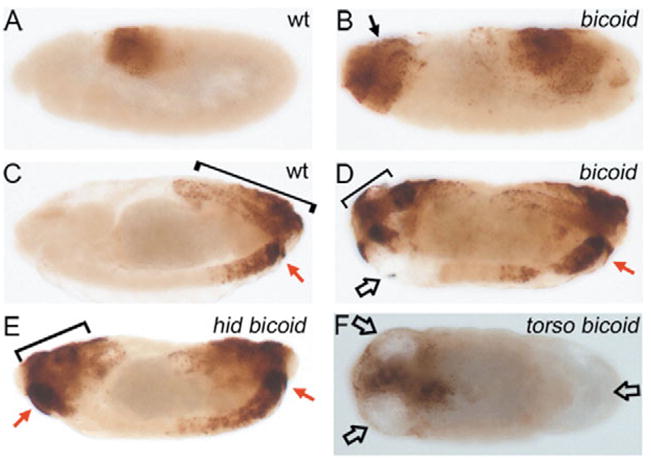
Abd-B distribution in wild-type, bicoid, hid bicoid and torso;bicoid embryos. Lateral views of stage 8 (A,B) and stage 15 (C–F) wild-type (A,C), bicoid (B,D), hid bicoid (E) and torso;bicoid (F) mutants labeled with anti-Abd-B antibodies. Wild-type embryos (A,C) contain Abd-B protein in the posterior region only. In bicoid mutant embryos (B,D), Abd-B protein is present at the posterior and at the anterior pole (see arrow in B). The brackets in C–E indicate the dorsally located component of Abd-B expression that develop into telson structures. Cell death clears part of the mis-specified area at the anterior (open arrow in D). Tissue clearance does not occur in hid bicoid double mutants (E). The red arrows in C–E indicate the relative position that specifies A8 segments in wild-type (C), bicoid (D) and hid bicoid (E) mutants. In torso; bicoid double mutants (F), the dorsally located component of anterior Abd B expression as well as the ventral and the posterior component are cleared (open arrows in F).
Expression of hid in bicoid and oskar embryos
During Drosophila embryogenesis, the genes reaper, hid and grim are essential for cell death through activation of caspases (White et al., 1994; White et al., 1996; Grether et al., 1995; Chen et al., 1996). We determined whether they are involved in the cell death response of bicoid and oskar mutants. Compared with wild-type embryos, hid expression is significantly elevated in those parts of bicoid and oskar embryos that showed high levels of TUNEL-positive cell death and activated DrICE (Fig. 3A–C). hid expression is not detectable in the presumptive telson regions. Expression of reaper and grim was not upregulated (data not shown). The increased expression of hid is first visible in stage 9 embryos, preceding cell death by ~1–2 hours, suggesting that transcriptional induction of hid triggers cell death in bicoid and oskar mutants. Expression of the caspase inhibitor P35 did not affect expression of hid (data not shown), demonstrating that caspase activation occurs downstream of hid. This is consistent with the proposed role of hid as caspase activator (Grether et al., 1995). Interestingly, hid expression in the affected parts of bicoid and oskar mutants is maintained for less than 2 hours, and then downregulated.
Fig. 3.
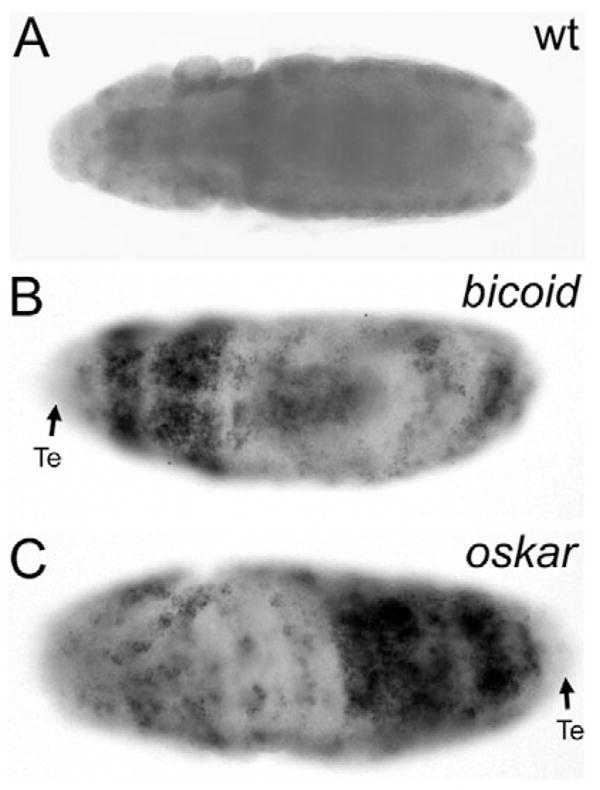
Expression of hid in bicoid and oskar mutants. Ventral views of stage 9 wild-type (A), bicoid (B) and oskar (C) mutant embryos, labeled for expression of hid by in situ hybridization. The arrows in B,C indicate the presumptive telsons (Te), which lack hid expression.
To determine whether cell death in bicoid mutants requires hid, embryos double mutant for hid and bicoid (see Materials and methods) were analyzed by TUNEL and Acridine Orange labeling. Cell death was not detectable in stage 10 hid bicoid double mutant embryos (Fig. 4B,D), suggesting that hid is genetically required for increased cell death in bicoid embryos. In summary, this analysis highlights the fact that cells in the affected regions of bicoid and oskar mutants do not simply die by a passive mechanism because of a lack of appropriate developmental information. Instead, these cells induce a transcriptional response leading to expression of hid. Thus, they die by an active gene-directed process.
The ability to block cell death in hid bicoid double mutants allowed us to address the fates of cells that normally undergo cell death in bicoid single mutants. Cuticle preparations of hid bicoid double mutant larvae revealed that the patterning defect (loss of head and thorax, and transformation of acron into telson, see Fig. 1B) is not rescued, as expected, because the wild-type function of bicoid is still missing (Fig. 4F). However, the anterior-most abdominal segment is considerably expanded in size, whereas the remaining abdominal segments are of normal size (Fig. 4F, see enlargement in Fig. 4H). This expansion is not present in either bicoid (Fig. 4E,G) or hid single mutants (data not shown). Hence, we conclude that this expansion is derived from cells which would normally die in bicoid mutants. Significantly, the rescued cells secrete cuticle elements, suggesting that they are terminally differentiated. Similarly, it has also been observed in C. elegans that cells that are programmed to die are able to differentiate if cell death is blocked (Avery and Horvitz, 1988).
Rescued mis-specified cells in hid bicoid double mutants have posterior identity
It has previously been proposed that mis-specified cells have at least two options: they either die or they survive and are transformed to adopt a different fate (reviewed by Bonini and Fortini, 1999). The underlying cause for this distinction is unclear. The bicoid mutant phenotype combines both transformation of tissue (acron to telson, Fig. 1B) and cell death in the presumptive head and thorax region (Fig. 1H). Thus, we asked why some mis-specified cells in bicoid mutants die, while others survive. To address this, we closely examined the identity of the expanded first abdominal segment in hid bicoid double mutants. Interestingly, the polarity of the denticles and the width of the segment resemble segment A8, the posterior-most segment (Fig. 1A), suggesting that anterior tissue may have been transformed toward a posterior identity in bicoid mutants. As mentioned above, transformation of acron into telson in bicoid mutants has previously been reported (Frohnhöfer and Nüsslein-Volhard, 1986) (Fig. 1B). However, the hid bicoid double mutant analysis revealed that the transformation of anterior tissue into posterior identity expands beyond the telson, and that this expansion undergoes hid-induced cell death in bicoid single mutants.
To verify this interpretation, we analyzed the posterior identity of anterior tissue in more detail using an antibody raised against the Abdominal-B (Abd-B) protein as posterior marker. In wild-type embryos, Abd-B protein is present in the posterior part of the embryo (Fig. 5A,C) (Celniker et al., 1989). However, in bicoid mutants it is found in both posterior and anterior poles of the embryo (Fig. 5B,D), consistent with our morphological observation. Thus, in bicoid mutants, anterior cells are incorrectly specified because they receive a developmental signal that in wild-type embryos is present only posteriorly.
To determine whether mis-specified cells induce hid expression and cell death in bicoid mutants, we analyzed Abd-B protein distribution in wild-type, bicoid single and hid bicoid double mutant embryos. In stage 15 wild-type embryos, Abd-B protein is detectable in two populations of cells at the posterior pole, a dorsally located component that gives rise to the telson (Fig. 5C, bracket), and a ventrally located component which specifies segment A8 (Fig. 5C, red arrow). In bicoid mutants, Abd-B protein distribution is mirror-imaged at the anterior pole (Fig. 5D). Significantly, however, part of the Abd-B-expressing ventral tissue in bicoid mutants is cleared as a consequence of cell death (Fig. 5D, open arrow; compare with Fig. 2). Consistently, in hid bicoid double mutants, tissue clearance does not occur and Abd-B protein persists (Fig. 5E, red arrow). Thus, the mis-specified ventral tissue expressing Abd-B survives in the double mutant. Similar data were obtained using a different abdominal marker, abd-A (see Fig. S1 in the supplementary material). Taken together, these findings suggest that mis-specified cells in bicoid mutants can induce hid expression and undergo cell death. Furthermore, the red arrows in Fig. 5C–E indicate the highest expression levels of Abd-B specifying abdominal segment A8 in wild-type, bicoid mutants and hid bicoid double mutants. Thus, the rescued cells in hid bicoid double mutants experience highest levels of Abd-B and develop into a segment with A8 identity (Fig. 4F). We also determined that cell death in oskar mutants largely affects mis-specified tissue, and that hid oskar double mutants rescue this tissue (see Fig. S2 in the supplementary material). Thus, similar to bicoid, mis-specified cells in oskar mutants induce hid to undergo cell death.
In torso;bicoid double mutants all Abd-B-expressing tissue is removed
From the analysis presented above, it is also clear that not all mis-specified cells at the anterior of bicoid mutants undergo cell death. The tissue containing the dorsally located component of Abd-B (bracket in Fig. 5D) is spared from tissue clearance, and thus cell death. This tissue undergoes transformation from acron to telson in bicoid mutants (Fig. 1B), for which it requires torso signaling (Nüsslein-Volhard et al., 1987; Frohnhöfer and Nüsslein-Volhard, 1986; Klingler et al., 1988; Schüpbach and Wieschaus, 1986). To determine whether torso signaling protects this mis-specified tissue from cell death, we performed a double mutant analysis. In stage 15 torso;bicoid double mutants, areas of clearance are detectable in both the ventral and dorsal components of Abd-B-expressing tissue at the mis-specified anterior tip, as well as at the posterior tip, which is also affected in torso mutants (Fig. 5F) (Klingler et al., 1988; Schüpbach and Wieschaus, 1986). Consistently, expression of hid is also upregulated in the presumptive acron and telson regions of torso mutants (data not shown). In summary, this analysis suggests that torso+ protects the mis-specified dorsal component at the anterior tip of bicoid embryos from hid-induced cell death, providing an explanation of why these cells survive. This analysis suggests that if an alternative survival signaling pathway is provided, such as torso+ in this example, mis-specification is tolerated and transformation can occur (see Discussion). Thus, the posterior to anterior transformation in bicoid embryos affects cells differently with respect to survival depending on their relative location. The telson survives because the torso signaling pathway is intact in bicoid embryos. However, the anterior tissue with posterior identity next to the telson undergoes cell death in bicoid embryos suggesting that it lacks an ‘anterior’ survival signal or is otherwise unable to inhibit expression of hid.
Expression of hid in segmentation mutants
Because hid is transcriptionally upregulated in dying mis-specified cells of bicoid and oskar mutants, we determined whether induction of hid expression represents a broader mechanism that also applies to mis-specified cells in other developmental mutants. We analyzed mutants of gap genes [knirps (kni)], pair-rule genes [odd-skipped (odd)] and segment-polarity genes [wingless (wg)]. These mutants are characterized by loss of tissue (Nüsslein-Volhard and Wieschaus, 1980), and ectopic cell death has been reported for these mutants (Tepass et al., 1994; Perrimon and Mahowald, 1987; Klingensmith et al., 1989; Bejsovec and Wieschaus, 1993; Pazdera et al., 1998; Hughes and Krause, 2001). As in the case of bicoid and oskar mutants, hid expression is upregulated during stage 9 of embryogenesis in the regions of the mutant embryos that are later deleted in the larvae (Fig. 6). This is most evident for odd, which lacks every other segment in the larvae. Correspondingly, we detected upregulation of hid in every other segment (Fig. 6C, arrowheads). In kni mutants, upregulation of hid is detectable in the posterior part of the embryo (Fig. 6B), where kni+ function is required (Nauber et al., 1988; Pankratz et al., 1992). In wg mutants, hid expression is detectable in every segment (Fig. 6D), consistent with the notion that the segment polarity phenotype is the result of regional mis-specification and subsequently cell death (Klingensmith et al., 1989; Perrimon and Mahowald, 1987). Similar data were also obtained for additional mutants, including hunchback, Krüppel, fushi-tarazu, hedgehog and engrailed (data not shown). Thus, these data support the notion that upregulation of hid in mis-specified cells is a common feature in many developmental mutants.
Fig. 6.
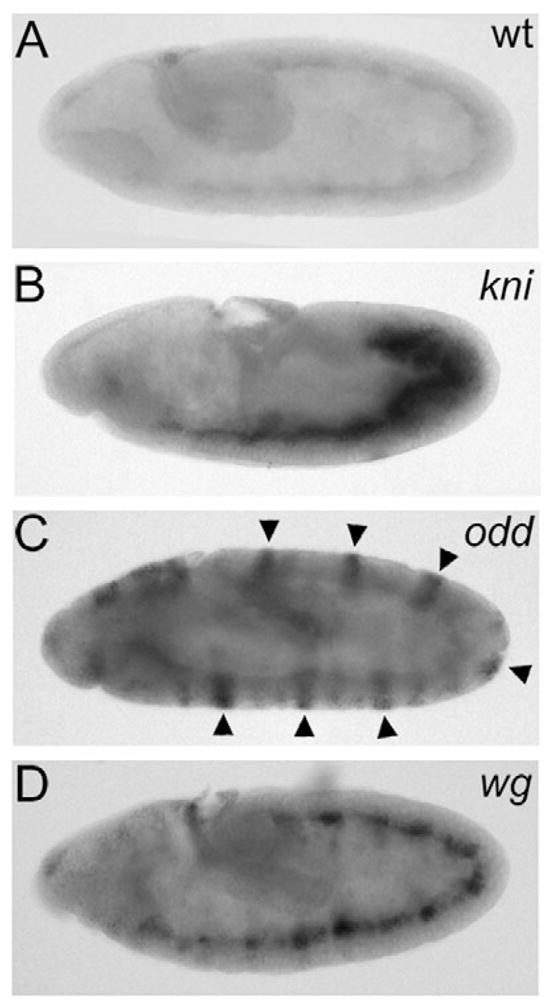
hid expression in segmentation mutants. Lateral views of stage 9 wild-type (A), kni (B), odd (C) and wg (D) embryos. Arrowheads in C indicate the pair-rule expression of hid in odd mutants.
Discussion
The events leading to cell death and tissue loss in response to mis-specification in bicoid and oskar mutants are summarized in Fig. 7. These mutants do not contain the correct developmental information for proper specification of the anterior and posterior parts of the embryos, respectively. Subsequently, the affected cells induce a transcriptional response leading to hid expression, which triggers caspase activation and cell death. Finally, clearance of the dying tissue occurs, and the embryos become visibly shortened.
Fig. 7.

Time course of the establishment of the bicoid and oskar mutant phenotypes. The developmental stages and approximate age of the embryos are indicated. The events that occur as development proceeds are summarized. See text for details.
Interestingly, it takes approximately 6 hours from caspase activation (stage 10; 7 hours of age) to the onset of cellular clearance (stage 14; 13 hours of age). During this time, the contents of the affected cells, including protein and DNA, are completely degraded and the cells are removed. This analysis highlights that developmental cell death occurs very rapidly, and that the bicoid and oskar mutants provide a convenient model with which to analyze the individual events of cell death with high temporal and spatial resolution because many cells die almost synchronously in these mutants.
Mis-specified cells in patterning mutants induce hid expression
Although we largely focus our analysis on the maternal effect mutants bicoid and oskar, it is likely that the principles we have uncovered are of broader significance. Segmentation mutants acting downstream of bicoid and oskar, including mutants of gap genes (Krüppel, knirps), pair-rule genes (odd, fushi-tarazu) and segment polarity genes (wg, hedgehog, engrailed) induce expression of hid (Fig. 6). These mutants are characterized by loss of larval tissue (Nüsslein-Volhard and Wieschaus, 1980), and for some of them cell death has been documented (Tepass et al., 1994; Perrimon and Mahowald, 1987; Magrassi and Lawrence, 1988; Klingensmith et al., 1989; Bejsovec and Wieschaus, 1993; Pazdera et al., 1998; Hughes and Krause, 2001). As in the case of bicoid and oskar, hid expression is upregulated during stage 9 of embryogenesis in the regions of the mutant embryos that are later deleted in the larvae. In addition, hid mutants rescue the cuticle phenotype of armadillo mutants (Cox et al., 2000). Finally, we also found hid expression accompanied by TUNEL-positive cell death in dorsal and Toll10b mutants which cause dorsalizing and ventralizing phenotypes, respectively, along the dorsoventral axis of Drosophila embryos (data not shown). Thus, these data support the notion that upregulation of hid appears to be a common trigger for a caspase-dependent cell death program in mis-specified cells of patterning mutants.
Furthermore, mutations affecting imaginal disc development result in loss of the adult appendage due to inappropriate cell death (Fristrom, 1968; Fristrom, 1969) (reviewed by Bonini and Fortini, 1999). We are currently determining whether these mutants also require hid expression to develop the final phenotypes. Moreover, many gene disruptions in mice result in inappropriate cell death in the tissue that requires the function of the disrupted gene (Rossel and Capecchi, 1999; McKay et al., 1994; Swiatek and Gridley, 1993), suggesting that similar mechanisms might exist in mammalian development. Finally, cell death may be an important contributing factor to human congenital birth defects. Thus, an understanding of the underlying mechanisms is of general interest.
Interestingly, not all segment polarity mutants analyzed induce hid expression and cell death. Embryos mutant for patched, which encodes the hedgehog receptor, were not found to express hid and do not contain increased cell death (data not shown), although hedgehog mutants both upregulate hid and contain increased amounts of cell death. The reasons for these differences are not known, but partial redundancy might account for lack of hid expression in patched mutants. The Drosophila genome encodes another patched homolog, patched-related, which might provide the survival requirement for mis-specified cells in patched mutants.
Mis-specified cells in bicoid and oskar mutants induce expression of hid. We did not observe increased reaper or grim expression in these mutants. However, expression of reaper has been reported in crumbs mutants, which affect epithelial integrity (Nordstrom et al., 1996). X-ray-treated embryos also preferentially respond by upregulation of reaper (Nordstrom et al., 1996; Brodsky et al., 2000; Ollmann et al., 2000), rather than hid (M. E. Grether, PhD thesis, MIT, 1994). Although we have not analyzed crumbs mutants for hid expression, it appears that cells contain several developmental checkpoints, which activate different cell death-inducing regulators depending on the type of abnormal cellular development.
Cell death versus cell fate transformation
Mis-specified cells can survive if an alternative survival pathway is provided. The example presented here is the acron into telson transformation in bicoid mutants, which is mediated by the torso signaling pathway. Although the cells giving rise to telson structures at the anterior tip are mis-specified based on Abd-B-labeling experiments, they survive because they receive a survival signal from the torso signaling system. In this case, transformation rather than cell death is favored. It has previously been shown that activation of the Ras/Mapk pathway protects cells from hid-induced apoptosis, both by transcriptional repression of hid (Kurada and White, 1998) and by phosphorylation of Hid protein by Mapk (Bergmann et al., 1998). Because Torso, which encodes a receptor tyrosine kinase (RTK) (Sprenger et al., 1989), is known to activate Ras and Mapk (Ghiglione et al., 1999; Gabay et al., 1997), we tested whether manipulation of active Mapk levels using a gain-of-function allele, MapkSem, can suppress hid expression and cell death in bicoid mutants. However, this was found not to be the case (data not shown). Thus, torso appears to protect mis-specified cells independently of Mapk activation.
The hid bicoid double mutant analysis reveals that the transformation of anterior into posterior identity expands beyond the telson, and that this expansion undergoes hid-induced cell death in bicoid single mutants. The rescued cells secrete larval cuticle elements, suggesting that mis-specified cells have the developmental capacity to terminally differentiate. However, in hid+ background, they instead die, presumably because equivalent survival signals are lacking. We propose that mis-specified cells undergo cell death if no alternative survival pathway is provided to protect them.
An alternative survival mechanism might also operate in other developmental mutants where transformation rather than cell death occurs. Mutations in the sev RTK and its ligand boss result in transformation of the R7 photoreceptor cell into a non-neuronal cone cell (Tomlinson and Ready, 1986; Reinke and Zipursky, 1988). Survival of this cell could be mediated by the Drosophila Egf receptor (Egfr), another RTK, which is required to maintain cell survival in the developing eye disk (Baker and Yu, 2001). Accordingly, activation of the Ras/Mapk pathway by Egfr would inhibit hid expression and support survival of the presumptive R7 photoreceptor cell. This interpretation is also consistent with observations that egfr− clones are small and undergo cell death (Diaz-Benjumea and Garcia-Bellido, 1990; Xu and Rubin, 1993), and that this death can be suppressed in hid mutants (Yu et al., 2002). Thus, transformation of the R7 photoreceptor to a cone cell rather than R7 cell death in sev and boss mutants could occur because of survival signaling by the Egfr.
Why do mis-specified cells die?
The hid bicoid double mutant analysis suggests that mis-specified cells can continue to develop and differentiate. Yet, they die. Presumably, this cell death protects the organism from potentially dangerous cells. For example, it is conceivable that in mammals, surviving mis-specified cells might lie dormant in the host organism for years. During this time, they might acquire additional genetic alterations that could drive the progressive transformation of these cells into malignant cancer. In wild-type embryos, mis-specification probably occurs in cells in isolation, and elimination of these cells does not interfere with development and survival of the organism. Only in extreme situations, such as the patterning mutants analyzed here, is the mis-specification caused by aberrant development so severe that the affected organism dies.
What is the nature of the mechanism that recognizes mis-specified cells?
The cause of mis-specification in each segmentation mutant is different. Usually, the expression of other segmentation genes is shifted and expanded, resulting in flattened gradients (e.g. Rivera-Pomar and Jäckle, 1996; Mullen and DiNardo, 1995). Yet, irrespective of the cause of mis-specification, most of these mutants have in common that they induce hid expression. It is currently unknown how the mis-specified fate of cells is recognized, and how hid expression is induced. One possibility might be that the protein gradients established by bicoid+ and oskar+, as well as other segmentation genes (Driever and Nüsslein-Volhard, 1988a; Ephrussi et al., 1991; Dahanukar and Wharton, 1996; Rivera-Pomar and Jäckle, 1996; Mullen and DiNardo, 1995) are used as readout for proper cellular specification. The steepness of protein gradients as a means to determine life or death decisions has recently been proposed (Moreno and Basler, 2004; de la Cova et al., 2004). Such a model would imply that cells are able to determine their position in a graded field and compare this readout with their neighbors. Because in bicoid and oskar mutants these gradients do not form, the concentration difference between neighboring cells would be zero. If the concentration difference between two neighboring cells is below a crucial threshold, they induce the expression of hid and undergo cell death. This model could also explain embryonic pattern repair, which was described in embryos that express six copies of the bicoid gene (Namba et al., 1997). In these embryos, the head and thorax primordia are expanded because of the presence of six copies of bicoid. However, this expansion is corrected for by induction of cell death, and relatively normal larvae develop (Namba et al., 1997). In this case, the Bicoid protein gradient does form, but would be flatter compared with wild type. Thus, the concentration difference between neighboring cells would be below a critical threshold, sufficient to induce hid-dependent cell death. However, it is largely unknown how cells compare their position in a graded field with those of their neighbors. It has been proposed that short-range cell interactions mediated via the cell-surface proteins Capricious and Tartan provide cues that support cell survival during wing development (Milan et al., 2002). Cells unable to participate in these interactions are eliminated by cell death. It is unclear, however, whether short-range interactions are sufficient to explain the cell death phenotype in bicoid and oskar mutants.
Irrespective of the underlying mechanism for sensing mis-specification, our results highlight the role of an active gene-directed process that removes mis-specified cells during development. However, if a survival mechanism is provided, mis-specified cells can survive and adopt a different fate. In wild-type embryos, mis-specification probably occurs in cells in isolation, and hence is difficult to study. However, in bicoid and oskar mutants, large regions of neighboring cells are mis-specified and undergo cell death simultaneously, providing a unique opportunity to clarify the signals that initiate cell death in situations where cells are developmentally mis-specified.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank the Bloomington stock center and Iris Koch for fly stocks; Hermann Steller, Georg Halder and the Developmental Studies Hybridoma Bank/The University of Iowa for antibodies; Eric Wieschaus and Amy Bejsovec for stimulating discussions; and Randy Johnson, Mary Ellen Lane, Robert Schulz, Richard Behringer and Georg Halder for comments on the manuscript. This work was supported by a Basil O’Connor Starter Scholar Research Award to A.B., The Robert A. Welch Foundation (G-1496) and The M.D. Anderson Research Trust.
Footnotes
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/cgi/content/full/132/24/5343/DC1
References
- Abrams JM, White K, Fessler LI, Steller H. Programmed cell death during Drosophila embryogenesis. Development. 1993;117:29–43. doi: 10.1242/dev.117.1.29. [DOI] [PubMed] [Google Scholar]
- Avery L, Horvitz HR. A cell that dies during wild-type C. elegans development can function as a neuron in a ced-3 mutant. Cell. 1987;51:1071–1078. doi: 10.1016/0092-8674(87)90593-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehrecke EH. How death shapes life during development. Nat Rev Mol Cell Biol. 2002;3:779–787. doi: 10.1038/nrm931. [DOI] [PubMed] [Google Scholar]
- Baker NE, Yu SY. The EGF receptor defines domains of cell cycle progression and survival to regulate cell number in the developing Drosophila eye. Cell. 2001;104:699–708. doi: 10.1016/s0092-8674(01)00266-5. [DOI] [PubMed] [Google Scholar]
- Bejsovec A, Wieschaus E. Segment polarity gene interactions modulate epidermal patterning in Drosophila embryos. Development. 1993;119:501–517. doi: 10.1242/dev.119.2.501. [DOI] [PubMed] [Google Scholar]
- Bergmann A, Agapite J, McCall KA, Steller H. The Drosophila gene hid is a direct molecular target of Ras-dependent survival signaling. Cell. 1998;95:331–341. doi: 10.1016/s0092-8674(00)81765-1. [DOI] [PubMed] [Google Scholar]
- Berleth T, Burri M, Thoma G, Bopp D, Richstein S, Frigerio G, Noll M, Nüsslein-Volhard C. The role of localization of bicoid RNA in organizing the anterior pattern of the Drosophila embryo. EMBO J. 1988;7:1749–1756. doi: 10.1002/j.1460-2075.1988.tb03004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonini NM, Fortini ME. Surviving Drosophila eye development: integrating cell death with differentiation during formation of a neural structure. BioEssays. 1999;21:991–1003. doi: 10.1002/(SICI)1521-1878(199912)22:1<991::AID-BIES3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Brodsky MH, Nordstrom W, Tsang G, Kwan E, Rubin GM, Abrams JM. Drosophila p53 binds a damage response element at the reaper locus. Cell. 2000;101:103–113. doi: 10.1016/S0092-8674(00)80627-3. [DOI] [PubMed] [Google Scholar]
- Celniker SE, Keelan DJ, Lewis EB. The molecular genetics of the bithorax complex of Drosophila: characterization of the products of the Abdominal-B domain. Genes Dev. 1989;3:1424–1436. doi: 10.1101/gad.3.9.1424. [DOI] [PubMed] [Google Scholar]
- Chen P, Nordstrom W, Gish B, Abrams JM. grim, a novel cell death gene in Drosophila. Genes Dev. 1996;10:1773–1782. doi: 10.1101/gad.10.14.1773. [DOI] [PubMed] [Google Scholar]
- Condie JM, Mustard JA, Brower DL. Generation of anti-Antennapedia monoclonal antibodies and Antennapedia protein expression in imaginal discs. Dros Inf Service. 1990;70:52–54. [Google Scholar]
- Cox RT, McEwen DG, Myster DL, Duronio RJ, Loureiro J, Peifer M. A screen for mutations that suppress the phenotype of Drosophila armadillo, the beta-catenin homolog. Genetics. 2000;155:1725–1740. doi: 10.1093/genetics/155.4.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahanukar A, Wharton RP. The Nanos gradient in Drosophila embryos is generated by translational regulation. Genes Dev. 1996;10:2610–2620. doi: 10.1101/gad.10.20.2610. [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila myc regulates organ size by inducing cell competition. Cell. 2004;117:107–116. doi: 10.1016/s0092-8674(04)00214-4. [DOI] [PubMed] [Google Scholar]
- Diaz-Benjumea FJ, Garcia-Bellido A. Behaviour of cells mutant for an EGF receptor homologue of Drosophila in genetic mosaics. Proc Biol Sci. 1990;242:36–44. doi: 10.1098/rspb.1990.0100. [DOI] [PubMed] [Google Scholar]
- Driever W, Nüsslein-Volhard C. A gradient of bicoid protein in Drosophila embryos. Cell. 1988a;54:83–93. doi: 10.1016/0092-8674(88)90182-1. [DOI] [PubMed] [Google Scholar]
- Driever W, Nüsslein-Volhard C. The bicoid protein determines position in the Drosophila embryo in a concentration-dependent manner. Cell. 1988b;54:95–104. doi: 10.1016/0092-8674(88)90183-3. [DOI] [PubMed] [Google Scholar]
- Driever W, Nüsslein-Volhard C. The bicoid protein is a positive regulator of hunchback transcription in the early Drosophila embryo. Nature. 1989;337:138–143. doi: 10.1038/337138a0. [DOI] [PubMed] [Google Scholar]
- Ephrussi A, St Johnston D. Seeing is believing: the bicoid morphogen gradient matures. Cell. 2004;116:143–152. doi: 10.1016/s0092-8674(04)00037-6. [DOI] [PubMed] [Google Scholar]
- Ephrussi A, Dickinson LK, Lehmann R. Oskar organizes the germ plasm and directs localization of the posterior determinant nanos. Cell. 1991;66:37–50. doi: 10.1016/0092-8674(91)90137-n. [DOI] [PubMed] [Google Scholar]
- Fristrom D. Cellular degeneration in wing development of the mutant vestigial of Drosophila melanogaster. J Cell Biol. 1968;39:488–491. doi: 10.1083/jcb.39.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fristrom D. Cellular degeneration in the production of some mutant phenotypes in Drosophila melanogaster. Mol Gen Genet. 1969;103:363–379. doi: 10.1007/BF00383486. [DOI] [PubMed] [Google Scholar]
- Frohnhöfer HG, Nüsslein-Volhard C. Organization of anterior pattern in the Drosophila embryo by the maternal gene bicoid. Nature. 1986;324:120–125. [Google Scholar]
- Frohnhöfer HG, Nüsslein-Volhard C. Maternal genes required for the anterior localization of bicoid activity in the embryo of Drosophila. Genes Dev. 1987;1:880–890. [Google Scholar]
- Gabay L, Seger R, Shilo BZ. MAP kinase in situ activation atlas during Drosophila embryogenesis. Development. 1997;124:3535–3541. doi: 10.1242/dev.124.18.3535. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiglione C, Perrimon N, Perkins LA. Quantitative variations in the level of MAPK activity control patterning of the embryonic termini in Drosophila. Dev Biol. 1999;205:181–193. doi: 10.1006/dbio.1998.9102. [DOI] [PubMed] [Google Scholar]
- Goyal L, McCall K, Agapite J, Hartwieg E, Steller H. Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J. 2000;19:589–597. doi: 10.1093/emboj/19.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grether ME, Abrams JM, Agapite J, White K, Steller H. The head involution defective gene of Drosophila melanogaster functions in Programmed Cell Death. Genes Dev. 1995;9:1694–1708. doi: 10.1101/gad.9.14.1694. [DOI] [PubMed] [Google Scholar]
- Hay BA, Wolff T, Rubin GM. Expression of baculovirus P35 prevents cell death in Drosophila. Development. 1993;120:2121–2129. doi: 10.1242/dev.120.8.2121. [DOI] [PubMed] [Google Scholar]
- Hughes SC, Krause HM. Establishment and maintenance of parasegmental compartments. Development. 2001;128:1109–1118. doi: 10.1242/dev.128.7.1109. [DOI] [PubMed] [Google Scholar]
- Karch F, Bender W, Weiffenbach B. abdA expression in Drosophila embryos. Genes Dev. 1990;4:1573–1587. doi: 10.1101/gad.4.9.1573. [DOI] [PubMed] [Google Scholar]
- Kim-Ha J, Smith JL, Macdonald PM. oskar mRNA is localized to the posterior pole of the Drosophila oocyte. Cell. 1991;66:23–35. doi: 10.1016/0092-8674(91)90136-m. [DOI] [PubMed] [Google Scholar]
- Klingensmith J, Noll E, Perrimon N. The segment polarity phenotype of Drosophila involves differential tendencies toward transformation and cell death. Dev Biol. 1989;134:130–145. doi: 10.1016/0012-1606(89)90084-5. [DOI] [PubMed] [Google Scholar]
- Klingler M, Erdelyi M, Szabad J, Nüsslein-Volhard C. Function of torso in determining the terminal anlagen of the Drosophila embryo. Nature. 1988;335:275–277. doi: 10.1038/335275a0. [DOI] [PubMed] [Google Scholar]
- Kurada P, White K. Ras promotes cell survival in Drosophila by downregulating hid expression. Cell. 1998;95:319–329. doi: 10.1016/s0092-8674(00)81764-x. [DOI] [PubMed] [Google Scholar]
- Lehmann R, Nüsslein-Volhard C. Abdominal segmentation, pole cell formation, and embryonic polarity require the localized activity of oskar, a maternal gene in Drosophila. Cell. 1986;47:141–152. doi: 10.1016/0092-8674(86)90375-2. [DOI] [PubMed] [Google Scholar]
- Macias A, Casanova J, Morata G. Expression and regulation of the abd-A gene of Drosophila. Development. 1990;110:1197–1207. doi: 10.1242/dev.110.4.1197. [DOI] [PubMed] [Google Scholar]
- Magrassi L, Lawrence PA. The pattern of cell death in fushi tarazu, a segmentation gene of Drosophila. Development. 1988;104:447–451. doi: 10.1242/dev.104.3.447. [DOI] [PubMed] [Google Scholar]
- Martinez-Arias A, Ingham PW. The origin of pattern duplications in segment polarity mutants of Drosophila melanogaster. J Embryol Exp Morphol. 1985;87:129–135. [PubMed] [Google Scholar]
- McKay IJ, Muchamore I, Krumlauf R, Maden M, Lumsden A, Lewis J. The kreisler mouse: a hindbrain segmentation mutant that lacks two rhombomeres. Development. 1994;120:2199–2211. doi: 10.1242/dev.120.8.2199. [DOI] [PubMed] [Google Scholar]
- Milan M, Perez L, Cohen SM. Short-range cell interactions and cell survival in the Drosophila wing. Dev Cell. 2002;2:797–805. doi: 10.1016/s1534-5807(02)00169-7. [DOI] [PubMed] [Google Scholar]
- Moreno E, Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117:117–129. doi: 10.1016/s0092-8674(04)00262-4. [DOI] [PubMed] [Google Scholar]
- Mullen JR, DiNardo S. Establishing parasegments in Drosophila embryos: roles of the odd-skipped and naked genes. Dev Biol. 1995;169:295–308. doi: 10.1006/dbio.1995.1145. [DOI] [PubMed] [Google Scholar]
- Namba R, Pazdera TM, Cerrone R, Minden JS. Drosophila embryonic pattern repair: how embryos respond to bicoid dosage alteration. Development. 1997;124:1393–1403. doi: 10.1242/dev.124.7.1393. [DOI] [PubMed] [Google Scholar]
- Nauber U, Pankratz MJ, Kienlin A, Seifert E, Klemm U, Jäckle H. Abdominal segmentation of the Drosophila embryo requires a hormone receptor-like protein encoded by the gap gene knirps. Nature. 1988;336:489–492. doi: 10.1038/336489a0. [DOI] [PubMed] [Google Scholar]
- Nordstrom W, Chen P, Steller H, Abrams JM. Activation of the reaper gene during ectopic cell killing in Drosophila. Dev Biol. 1996;180:213–226. doi: 10.1006/dbio.1996.0296. [DOI] [PubMed] [Google Scholar]
- Nüsslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- Nüsslein-Volhard C, Frohnhöfer HG, Lehmann R. Determination of anteroposterior polarity in Drosophila. Science. 1987;238:1675–1681. doi: 10.1126/science.3686007. [DOI] [PubMed] [Google Scholar]
- Ollmann M, Young LM, Di Como CJ, Karim F, Belvin M, Robertson S, Whittaker K, Demsky M, Fisher WW, Buchman A, et al. Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell. 2000;101:91–101. doi: 10.1016/S0092-8674(00)80626-1. [DOI] [PubMed] [Google Scholar]
- Pankratz MJ, Busch M, Hoch M, Seifert E, Jäckle H. Spatial control of the gap gene knirps in the Drosophila embryo by posterior morphogen system. Science. 1992;255:986–989. doi: 10.1126/science.1546296. [DOI] [PubMed] [Google Scholar]
- Parnas DA, Haghighi P, Fetter RD, Kim SW, Goodman CS. Regulation of postsynaptic structure and protein localization by the Rho-Type guanine nucleotide exchange factor dPix. Neuron. 2001;32:415–424. doi: 10.1016/s0896-6273(01)00485-8. [DOI] [PubMed] [Google Scholar]
- Patel NH. Imaging neuronal subsets and other cell types in whole-mount Drosophila embryos and larvae using antibody probes. Methods Cell Biol. 1994;44:445–487. doi: 10.1016/s0091-679x(08)60927-9. [DOI] [PubMed] [Google Scholar]
- Pazdera TM, Janardhan P, Minden JS. Patterned epidermal cell death in wild-type and segment polarity mutant Drosophila embryos. Development. 1998;125:3427–3436. doi: 10.1242/dev.125.17.3427. [DOI] [PubMed] [Google Scholar]
- Perrimon N, Mahowald AP. Multiple functions of segment polarity genes in Drosophila. Dev Biol. 1987;119:587–600. doi: 10.1016/0012-1606(87)90061-3. [DOI] [PubMed] [Google Scholar]
- Reinke R, Zipursky SL. Cell-cell interaction in the Drosophila retina: the bride of sevenless gene is required in photoreceptor cell R8 for R7 cell development. Cell. 1988;55:321–330. doi: 10.1016/0092-8674(88)90055-4. [DOI] [PubMed] [Google Scholar]
- Rivera-Pomar R, Jäckle H. From gradients to stripes in Drosophila embryogenesis: filling in the gaps. Trends Genet. 1996;12:478–483. doi: 10.1016/0168-9525(96)10044-5. [DOI] [PubMed] [Google Scholar]
- Rørth P. Gal4 in the Drosophila female germline. Mech Dev. 1998;78:113–118. doi: 10.1016/s0925-4773(98)00157-9. [DOI] [PubMed] [Google Scholar]
- Rossel M, Capecchi MR. Mice mutant for both Hoxa1 and Hoxb1 show extensive remodeling of the hindbrain and defects in craniofacial development. Development. 1999;126:5027–5040. doi: 10.1242/dev.126.22.5027. [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Abrams JM. Caspase activation – stepping on the gas or releasing the brakes? Lessons from humans and flies. Oncogene. 2004;23:2774–2784. doi: 10.1038/sj.onc.1207522. [DOI] [PubMed] [Google Scholar]
- Schüpbach T, Wieschaus E. Germline autonomy of maternal-effect mutations altering the embryonic body pattern of Drosophila. Dev Biol. 1986;113:443–448. doi: 10.1016/0012-1606(86)90179-x. [DOI] [PubMed] [Google Scholar]
- Sprenger F, Stevens LM, Nüsslein-Volhard C. The Drosophila gene torso encodes a putative receptor tyrosine kinase. Nature. 1989;338:478–483. doi: 10.1038/338478a0. [DOI] [PubMed] [Google Scholar]
- Srinivasan A, Roth KA, Sayers RO, Shindler KS, Wong AM, Fritz LC, Tomaselli KJ. In situ immunodetection of activated caspase-3 in apoptotic neurons in the developing nervous system. Cell Death Differ. 1998;5:1004–1016. doi: 10.1038/sj.cdd.4400449. [DOI] [PubMed] [Google Scholar]
- Struhl G, Struhl K, Macdonald PM. The gradient morphogen bicoid is a concentration-dependent transcriptional activator. Cell. 1989;57:1259–1273. doi: 10.1016/0092-8674(89)90062-7. [DOI] [PubMed] [Google Scholar]
- Swiatek PJ, Gridley T. Perinatal lethality and defects in hindbrain development in mice homozygous for a targeted mutation of the zinc finger gene Krox20. Genes Dev. 1993;7:2071–2084. doi: 10.1101/gad.7.11.2071. [DOI] [PubMed] [Google Scholar]
- Tautz D, Pfeifle C. A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma. 1989;98:81–85. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- Tepass U, Fessler LI, Aziz A, Hartenstein V. Embryonic origin of hemocytes and their relationship to cell death Drosophila. Development. 1994;120:1829–1837. doi: 10.1242/dev.120.7.1829. [DOI] [PubMed] [Google Scholar]
- Tomlinson A, Ready DF. Sevenless: A cell specific homeotic mutation of the Drosophila eye. Science. 1986;231:400–402. doi: 10.1126/science.231.4736.400. [DOI] [PubMed] [Google Scholar]
- Van der Meer S. Optical clean and permanent whole mount preparation for phase contrast microscopy of cuticular structures of insect larvae. Dros Inf Service. 1977;52:160. [Google Scholar]
- White K, Grether ME, Abrams JM, Young L, Farrell K, Steller H. Genetic control of cell death in Drosophila. Science. 1994;264:677–683. doi: 10.1126/science.8171319. [DOI] [PubMed] [Google Scholar]
- White K, Tahaoglu E, Steller H. Cell killing by Drosophila reaper. Science. 1996;271:805–807. doi: 10.1126/science.271.5250.805. [DOI] [PubMed] [Google Scholar]
- Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- Yu SY, Yoo SJ, Yang L, Zapata C, Srinivasan A, Hay BA, Baker NE. A pathway of signals regulating effector and initiator caspases in the developing Drosophila eye. Development. 2002;129:3269–3278. doi: 10.1242/dev.129.13.3269. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.