

Abstract
Current pharmacotherapies for addiction represent opportunities for facilitating treatment and are forming a foundation for evaluating new medications. Furthermore, validated animal models of addiction and a surge in understanding of neurocircuitry and neuropharmacological mechanisms involved in the development and maintenance of addiction — such as the neuroadaptive changes that account for the transition to dependence and the vulnerability to relapse — have provided numerous potential therapeutic targets. Here, we emphasize a ‘Rosetta Stone approach’, whereby existing pharmacotherapies for addiction are used to validate and improve animal and human laboratory models to identify viable new treatment candidates. This approach will promote translational research and provide a heuristic framework for developing efficient and effective pharmacotherapies for addiction.
Drug addiction is a chronically relapsing disorder characterized by a compulsion to seek and take a drug, loss of control in limiting intake and emergence of a negative emotional state (for example, dysphoria, anxiety and irritability) when access to the drug is prevented1. An important goal of current neurobiological research is to understand the molecular, neuropharmacological and neurocircuitry changes that mediate the transition from occasional, controlled drug use to the loss of behavioural control over drug seeking and drug taking that defines chronic addiction. In this Review, we suggest that a combination of validated animal models for addiction, neurobiological targets derived from such models, and translation to and from the clinical domain provides a heuristic framework for the development of pharmacotherapies for addiction. Moreover, the application of known treatments for addiction to existing animal and human laboratory models can provide an evolving ‘Rosetta Stone approach’ for accelerating the translation of newly identified targets to pharmacotherapies for addiction (BOX 1; FIG. 1).
Box 1 Disease concept: addiction as a treatable disease.
Addiction is a brain disease, and is defined as a chronically relapsing disorder of compulsive drug use. Advances in our understanding of the neurobiology of addiction have given substantial support to the disease basis for addiction. Changes in specific neuronal and neurochemical circuits have been identified that correspond to different components of the addiction cycle. Perhaps more importantly, these changes are long lasting and in some cases can be permanent. One goal of medications development for addiction is to reverse or compensate for such pathological effects.
The concept of addiction as a disease is also supported by overwhelming evidence that addiction leads to brain pathology from a functional perspective, and this pathology is manifested by reversible, and possibly some irreversible, brain changes. In the United States alone, illicit-drug abuse and addiction costs society US$180.9 billion per year159; in addition, alcoholism costs $180 billion160 and tobacco addiction costs $167 billion161.
From the perspective of treatment, relapse rates for addiction with abstinence as a goal are high: generally 90% without treatment after 1 year. However, such relapse rates for addiction are similar to those for other chronic relapsing disorders, such as diabetes, hypertension and asthma162. Treatments for addiction have limited success, often only doubling the number of individuals that do not relapse after 1 year. However, even capturing 10% of subjects per year could afford considerable savings in human suffering and societal cost. Appropriately monitored replacement treatments, such as methadone and buprenorphine, have a relatively high success rate in terms of reducing or eliminating illicit use of opioids. Furthermore, recent successes with naltrexone, acamprosate, buprenorphine and varenicline hold promise for future medications development for addiction.
Figure 1. The ‘Rosetta Stone approach‘ to drug development.
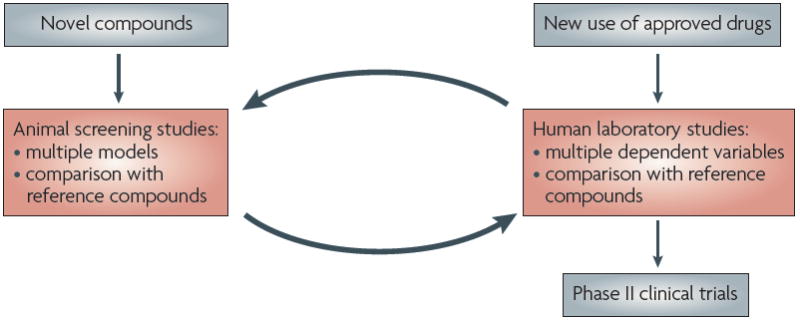
A crucial aspect of the proposed Rosetta Stone approach is the dynamic feedback from animal models and clinical data which can be used to identify treatments for drug addiction that are likely to succeed in clinical trials and to facilitate further development of animal and human models. These data may ultimately provide a rational basis for combination therapies such that multiple components of the addiction cycle can be treated by a given pharmacological strategy.
A key element of this approach will be to prevent predictions from animal models being limited by the constructs of the models themselves. Several aspects of the Rosetta Stone approach address this issue. First, because no single animal or human laboratory model exists for all of the aspects of addiction, various models are used to emulate different components of addiction, some of which are still evolving2. Second, there is a dynamic approach to the process of validation, with changes being discovered and implemented at both ends. New targets from neurobiology will feed forwards through the system and new medications will feed backwards, regardless of whether these medications are derived from the feed-forward process, clinical experience or serendipity. Third, symptoms or components of the addiction cycle provide a face-valid model that facilitates the creation of new animal and human laboratory models (for example, the animal model of compulsivity and drug seeking in the context of aversive consequences, or the human model of brain imaging in the context of cue-induced imagery3-5).
The addiction cycle
A useful psychiatry-based motivational framework that integrates well with animal models of addiction is the concept that drug addiction has aspects of both impulse control disorders (such as kleptomania) and compulsive disorders (such as obsessive–compulsive disorder). It has been suggested that, as an individual moves from an impulsive disorder to a compulsive disorder, a shift from positive reinforcement to negative reinforcement drives the motivated behaviour1 within a cycle comprising three stages: binge–intoxication, withdrawal–negative affect, and preoccupation–anticipation.
Animal models relevant to the addiction cycle
Animal models of addiction have outstanding face validity (for example, for intravenous self-administration) and recapitulate aspects of the condition in humans. They also have substantial construct validity (for example, deregulated stress responsivity during drug withdrawal), that is, explanatory power or functional equivalence for the condition in humans. Although no animal model fully reproduces addiction in humans, such models do permit investigation of elements of the drug addiction process that can be defined by models of addiction symptoms within the three stages of the addiction cycle. Different animal models for the study of the neurobiology of addiction can be superimposed on these three stages, collectively reproducing the pathological state known as addiction1 (TABLE 1).
Table 1.
Laboratory models of the stages of the addiction cycle
| Stage of addiction cycle | Animal models | Human laboratory models |
|---|---|---|
| Binge–intoxication | Drug or alcohol self-administration172 | Self-administration in dependent subjects120,176,177 |
| Conditioned place preference6 | Impulsivity178-180 | |
| Brain stimulation reward thresholds173 | ||
| Increased motivation for self-administration in dependent animals3,4,174,175 | ||
| Withdrawal–negative affect | Anxiety-like responses60,72,181 | Acute withdrawal125,188,189 |
| Conditioned place aversion182 | Self-medication190-192 | |
| Elevated reward thresholds183 | Mood induction57,149 | |
| Withdrawal-induced increases in drug self-administration175,184-187 | ||
| Preoccupation–anticipation | Drug-induced reinstatement6 | Drug reinstatement132 |
| Cue-induced reinstatement6 | Cue reactivity134,137,139 | |
| Stress-induced reinstatement6 | Emotional reactivity55 | |
| Stress-induced craving135,141-143 | ||
| Resistance to relapse150 | ||
| Cue-induced brain imaging responses5,151,152 |
Animal models for the binge–intoxication stage of the addiction cycle incorporate drug reinforcement and include drug and alcohol self-administration. For the withdrawal–negative affect stage, animal models exist for the somatic signs of withdrawal for almost all drugs of abuse. However, more relevant to addiction are the animal models of components of the motivational signs of withdrawal and the negative reinforcing effects of dependence, which are beginning to be used to explore how the nervous system is involved in motivation and adapts to drug use. These include anxiety-like responses, conditioned place aversion (a form of place conditioning), elevated reward thresholds and withdrawal-induced increases in drug self-administration. For the preoccupation–anticipation (craving) stage, models include drug-, cue- and stress-induced reinstatement of drug-seeking behaviour. Animal models of craving can also include the conditioned rewarding effects of drugs of abuse, measures of the conditioned aversive effects of withdrawal, and signs and symptoms of protracted abstinence6,7.
Neurobiological targets in addiction
Neurocircuitry of addiction
A crucial issue for the development of treatments for addiction is that selection of relevant targets should be informed by an empirical understanding of the neurobiology of addiction7. Three neurobiological circuits have been identified that have heuristic value for the study of the neurobiological changes associated with the development and persistence of drug dependence (FIG. 2).
Figure 2. Neural circuitry, current drugs and potential targets associated with the three stages of the addiction cycle.

In the binge–intoxication stage, reinforcing effects of drugs may engage associative mechanisms and neurotransmitters that signal reward in the shell (or medial portion) and core of the nucleus accumbens (Acb) and then engage stimulus response habits that depend on the dorsal striatum (DS). In the withdrawal–negative affect stage, the extended amygdala (AMG) may be activated. It consists of several basal forebrain structures, including the bed nucleus of the stria terminalis (BNST), the central nucleus of the amygdala (CeA), and a transition area in the shell of the nucleus accumbens. Neurons containing a key neurotransmitter in the extended amygdala, corticotropin-releasing factor (CRF), project to the brainstem, from which noradrenergic neurons provide a major reciprocal projection. In the preoccupation–anticipation (craving) stage, conditioned reinforcement is processed in the basolateral amygdala (BLA) and contextual information is processed in the hippocampus. Executive control depends on the prefrontal cortex and includes representation of contingencies, representation of outcomes, and their value and subjective states (that is, craving and feelings) associated with drugs. Functional imaging studies have shown that the subjective states, called drug craving in humans, involve activation of the orbital and anterior cingulate cortex and the temporal lobe, including the amygdala. For each stage of the addiction process, the existing medications and potential future medications for addiction treatment that are particularly relevant to that stage are shown. Dashed arrows represent output circuits. CRF1, CRF receptor 1; DA, dopamine; DGP, dorsal globus pallidus; GP, globus pallidus; mPFC (AC), medial prefrontal cortex (anterior cingulate); NA, noradrenaline; OFC, orbitofrontal cortex; SNc, substantia nigra pars compacta; VGP, ventral globus pallidus; VS, ventral striatum; VTA, ventral tegmental area. Figure is modified, with permission, from REF. 171 © (2008) Academic Press.
The circuitry related to the origin and terminal regions of the mesocorticolimbic dopamine system, which includes signalling by dopamine and opioid peptides, is a crucial mediator of the positive reinforcing effects of drugs associated with the binge–intoxication stage of the addiction cycle8. The preoccupation–anticipation (craving) stage involves key glutamatergic projections to the extended amygdala and nucleus accumbens from the prefrontal cortex (for drug-induced reinstatement of drug seeking) and from the basolateral amygdala (for cue-induced reinstatement of drug seeking)9. Compulsive drug-seeking behaviour is thought to engage ventral striatal–ventral pallidal–thalamic–cortical loops that could subsequently engage dorsal striatal–dorsal pallidal–thalamic–cortical loops10, both of which are exaggerated by concomitant decreased activity in reward circuits11,12. The neural substrates and neuropharmacological mechanisms for the negative motivational effects of the withdrawal–negative affect stage of the addiction cycle may involve not only disruption of the neural systems implicated in the positive reinforcing effects of drugs, but also recruitment of brain stress systems13. Common responses during acute withdrawal from the main drugs of abuse include decreased dopaminergic activity, an activated pituitary–adrenal stress response and an activated brain stress response with activated extrahypothalamic corticotropin-releasing factor (CRF) systems in the amygdala. However, repeated cycles of addiction lead to a blunted pituitary–adrenal response and a sensitized extrahypothalamic CRF stress system response in the amygdala13.
Molecular targets within the brain circuits associated with addiction
Molecular changes at the signal transduction, gene transcription or gene level are thought to provide insights into how the circuits described above become deregulated and maintain such deregulation, and provide several contributions to medications development. Drugs of abuse perturb intracellular signal transduction, leading to changes in nuclear function and rates of transcription of certain genes14. This leads to altered activity of the neurons in which such changes occur and ultimately to changes in the function of the associated neural circuits. To date, no medication targets have been identified from molecular targets but, in the future, molecular studies may provide the basis for future targets for pharmacotherapeutic approaches and a key to understanding the vulnerability to addiction.
Animal models of addiction: reverse validity
Several medications are currently on the market for the treatment of addiction (FIG. 2; TABLE 2). The validation procedure termed the Rosetta Stone or reverse validity approach uses drugs that are known to be effective in human clinical studies to validate animal models and human laboratory models, and can provide a means of refining such models. For more detailed information on the effects of drugs approved for the treatment of addiction on established animal models of addiction, see Supplementary information S1 (box).
Table 2.
Medications currently on the market for the treatment of drug addiction
| Name | Addiction | Year of FDA approval | Description |
|---|---|---|---|
| Disulfiram | Alcohol | 1954 |
|
| Methadone | Opiate | 1972 |
|
| Naltrexone | Alcohol | 1994 and 2005 (extended-release formulation) |
|
| Bupropion (Wellbutrin/Zyban; GlaxoSmithKline) | Nicotine | 1997 | |
| Buprenorphine (Subutex; Schering–Plough) | Opiate | 2002 |
|
| Acamprosate (Campral/Aotal; Merck–Serono/Forest Laboratories) | Alcohol | 2004 |
|
| Varenicline (Chantix/Champix; Pfizer) | Nicotine | 2006 |
|
| Nicotine replacement therapy | Nicotine |
|
FDA, US Food and Drug Administration; NMDA, N-methyl-d-aspartate.
New targets for medications development
The premise of this Review is that different components of the addiction cycle can be targeted by different medications, selected on the basis of information from three key sources. These comprise research into basic neurobiological mechanisms for the different stages of the addiction cycle, the effects of medications approved for the treatment of addiction on animal models of the different stages of the addiction cycle (Supplementary information S1 (box)) and clinical studies of medications approved for other indications that overlap with specific components of addiction (discussed below) (FIG. 2; TABLE 3).
Table 3.
Potential pharmacotherapies derived from preclinical research
| Class | Candidates |
|---|---|
| Dopamine receptor partial agonists |
|
| Modulators of γ-aminobutyric acid (GABA) signalling |
|
| Modulators of brain stress systems |
|
| Modulators of glutamate signalling |
|
AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid; CRF1, corticotropin-releasing factor receptor 1; mGluR, metabotropic glutamate receptor; NMDA, N-methyl-d-aspartate.
The effects of selected compounds acting on specific neurobiological targets on models of the motivational components of the addiction cycle that are relevant for pharmacotherapies, using alcoholism as an example, are shown in TABLE 4. It compares novel approaches with two medications currently on the market (naltrexone, and acamprosate (Campral/Aotal; Merck–Serono/Forest Laboratories)). Different patterns of action on the animal models are thought to reflect actions in different stages of the addiction cycle. The Rosetta Stone approach places emphasis on elements of the withdrawal–negative affect stage (the ‘dark side’) of addiction. We propose that this framework is crucial for the neuroadaptations that lead to changes in motivation and thereby drive addiction to maintain an allostatic state12. There is compelling evidence that direct antagonism of the reinforcing effects of drugs of abuse, representing the binge–intoxication stage, produces compensatory increases in drug taking, motivational side effects that limit compliance, or a combination of both. Other reviews have focused on animal models of the preoccupation– anticipation (craving) state15. Here, we explore four neurotransmitter systems: dopamine, γ-aminobutyric acid (GABA), CRF and glutamate, all of which have targets that can restore deregulated reward systems, as in the withdrawal–negative affect stage, and in some cases affect the binge–intoxication stage (dopamine) and the preoccupation–anticipation stage (glutamate). Although GABA, glutamate and CRF are not new targets in the treatment of addiction2, targeting these and the dopamine system using the framework of validation described here has the potential to provide new medications.
Table 4.
Effects of drugs on animal models of the motivational components of the addiction cycle*
| Naltrexone | Acamprosate | CRF receptor antagonist | Noradrenaline receptor antagonist | GABA receptor modulator | Metabotropic glutamate receptor agonist | |
|---|---|---|---|---|---|---|
| Baseline drinking | ↓201 | No effect202 | No effect75 | ↓80 | ↓203 | ↓204 |
| Dependence-induced drinking | ↓98 | ↓205 | ↓75 | ↓80 | ↓56 | Not determined |
| cue-induced reinstatement | ↓206 | ↓207 | No effect206 | Not determined | ↓208 | ↓117 |
| Stress-induced reinstatement | No effect206 | Not determined | ↓206 | ↓209 | Not determined | ↓117 |
Using alcoholism as an example.
CRF, corticotropin-releasing factor; GABA, γ-aminobutyric acid.
Dopamine receptor partial agonists
The mesolimbic dopamine system projects from the ventral tegmental area to basal forebrain sites, the nucleus accumbens and the central nucleus of the amygdala, and has a key role in motivation. Activation of the mesolimbic dopamine system is thought to be important for directing behaviour towards salient rewarding stimuli16, but may not be necessary for hedonic experience17. Therefore, mesolimbic dopamine activity seems to be crucial for the reinforcing actions of indirect sympathomimetics, such as cocaine and amphetamines, and is involved in, but not essential for, the reinforcing actions of other drugs of abuse, such as opioids and alcohol.
More importantly for our dark side view, dopaminergic function is compromised during acute withdrawal from all major drugs of abuse18: levels of extracellular dopamine decrease in the nucleus accumbens following a binge of self-administered cocaine19; animals kept on a diet of alcohol show a decrease in extracellular levels of dopamine in the nucleus accumbens during withdrawal20. Withdrawal from most major drugs of abuse is also associated with decreased firing of dopaminergic neurons in the ventral tegmental area21.
Given the role of dopamine in the acute reinforcing effects of drugs and its deregulation during drug withdrawal, a dopamine partial agonist may be an effective treatment in different stages of the addiction cycle. A dopamine receptor partial agonist has antagonist properties in situations of high intrinsic activity and agonist properties in situations of low intrinsic activity. Because of its intermediate efficacy, a dopamine partial agonist acts as an agonist in the absence of dopamine and can act as an antagonist in the presence of dopamine23-25, and would hypothetically have less severe or fewer side effects than full agonists or antagonists22. Indeed, partial agonists of dopamine D2 receptors dose-dependently decrease the reinforcing effects of intravenous cocaine and amphetamine self-administration and oral alcohol self-administration in non-dependent rats26-29.
In a series of studies, D2 partial agonists have been shown to reverse psychostimulant withdrawal and block the increase in self-administration associated with extended access to the psychostimulant. A D2 partial agonist reversed the motivational deficit that occurs during amphetamine and methamphetamine withdrawal30,31. Animals with extended access to methamphetamine through intravenous self-administration show an increased intake of methamphetamine (escalation in intake)32. A notable effect of the D2 partial agonist aripiprazole on methamphetamine self-administration was a shift to the right of the dose–response function, with a greater effect in rats with higher methamphetamine intake associated with extended access. These data again suggest an increased sensitivity to the effects of the D2 partial agonists in dependent rats32.
Antagonists of the dopamine D3 receptor do not affect baseline cocaine self-administration, but block responses in a progressive-ratio schedule, a measure that is thought to reflect a compulsivity component of cocaine seeking. Additionally, D3 antagonists block cue-induced rein-statement of cocaine and alcohol self-administration33. Similarly, D3 partial agonists do not block baseline cocaine self-administration but block cue-induced reinstatement of self-administration34 and cue-induced drug seeking in a second-order schedule of reinforcement35. A D3 partial agonist also blocked amphetamine-induced conditioned place preference36.
D1 antagonists competitively block cocaine self-administration in rats37, but little work has been done on D1 partial agonists. One study has shown strain-dependent agonist and antagonist effects on cocaine self-administration with a D1 partial agonist in rats38. Together, these results suggest that deregulation of dopamine signalling contributes to the motivational effects of drug withdrawal and reinstatement, and dopamine partial agonists with the appropriate neuropharmacological and pharmacokinetic profile may be effective in treating certain aspects of addiction39.
GABAergic modulators
GABAA receptor antagonists and inverse agonists decrease alcohol self-administration40,41. However, their therapeutic actions are limited by potential side effects involving central nervous system hyperexcitability. By contrast, GABA receptor agonists or modulators can block drug-seeking behaviour through their actions on reward, dependence or both. GABA receptor modulators that increase GABAergic activity directly or indirectly decrease self-administration of cocaine, heroin, nicotine and alcohol in non-dependent rats42-45. GABA receptor agonists also block alcohol withdrawal in animals46 and humans, and decrease drinking and certain components of craving in humans with alcoholism47,48 (TABLE 4). GABAB receptor agonists also block the increased alcohol self-administration observed during acute withdrawal in dependent rats at lower doses than those that block alcohol self-administration in non-dependent rats, suggesting an increased sensitivity of this system during the development of dependence49. The GABAB agonist baclofen has been reported to reduce alcohol craving and intake in a preliminary double-blind, placebo-controlled trial47. However, GABAB agonists currently in therapeutic use for the relief of flexor spasms in multiple sclerosis have substantial sedative effects at therapeutic doses50. Thus, another approach is to explore the role of GABA receptor modulators that indirectly facilitate GABA release.
Gabapentin (Neurontin; Pfizer), an amino acid designed as a structural analogue of GABA51, is a novel anticonvulsant drug that is also used in the treatment of neuropathic pain. Gabapentin increases the concentration of GABA in the brain52 and GABA release from rat brain slices in vitro53. It also decreases synaptic transmission in the brain by selectively inhibiting Ca2+ influx through voltage-operated Ca2+ channels54 and may be an agonist of GABAB receptors55.
In animal models of alcohol dependence, gabapentin has strikingly different effects in non-dependent and alcohol-dependent rats, both cellularly and pharmacologically56. In non-dependent rats, gabapentin facilitated GABAergic transmission in the central nucleus of the amygdala but did not affect alcohol intake. However, in dependent rats, gabapentin decreased GABAergic transmission in the central nucleus of the amygdala and reduced excessive alcohol intake. Furthermore, gabapentin suppressed the anxiogenic-like effects of withdrawal from an acute alcohol injection. A possible explanation for these results is that, during the development of alcohol dependence, neuroadaptive changes occur in the GABAergic system, including a reduced sensitivity and/or a downregulation of presynaptic GABAB receptors56. Gabapentin has proved effective in human laboratory studies in decreasing craving, reversing physiological measures of protracted abstinence and reversing sleep deficits in protracted abstinence57, suggesting a key translation from animals to humans and supporting the translational approach suggested here.
Modulators of the brain stress system: CRF antagonists
Alcohol is a powerful activator of ‘stress systems’, which could have important implications for our understanding of the neurobiology of dependence and relapse. Alcohol-mediated activation of stress systems occurs through the hypothalamic–pituitary–adrenal axis and extensive extrahypothalamic, extraneuroendocrine CRF systems implicated in behavioural responses to stress58. Central administration of CRF mimics the hormonal, autonomic and behavioural response associated with activation and stress in rodents; competitive CRF receptor antagonists generally have the opposite effects13.
In animal models, a common response to acute withdrawal and protracted abstinence from all major drugs of abuse is the manifestation of anxiety-like responses. Withdrawal from repeated administration of cocaine, alcohol, nicotine and cannabinoids produces an anxiogenic-like response in the elevated plus maze and defensive burying test, and one or both of these effects are reversed by administration of either selective CRF receptor 1 (CRF1) antagonists or mixed CRF1–CRF2 antagonists60-66.
During alcohol withdrawal, extrahypothalamic CRF systems become hyperactive and there is an increase in extracellular CRF within the central nucleus of the amygdala and the bed nucleus of the stria terminalis in dependent rats67-69. Extracellular CRF is increased in the amygdala during withdrawal from chronic administration of nicotine64, cocaine70, opioids71 and cannabinoids66 in rats.
The ability of CRF antagonists to block the anxiogenic-like and aversive-like effects of drug withdrawal would predict the motivational effects of CRF antagonists in animal models of dependence40,62,63,72. A CRF1–CRF2 peptide antagonist that has no effect on alcohol self-administration in non-dependent rats eliminates excessive drinking in dependent rats during acute withdrawal and protracted abstinence73. Direct administration of a CRF1–CRF2 peptide antagonist into the central nucleus of the amygdala also eliminated excessive drinking during acute withdrawal67. Systemic injections of small-molecule CRF1 antagonists also blocked the increased alcohol intake during acute withdrawal and protracted abstinence in alcohol-dependent rats, but not in non-dependent rats74,75 (TABLE 4). These data suggest an important role for CRF, primarily in the central nucleus of the amygdala, in mediating the increased self-administration associated with alcohol dependence. CRF1 antagonists also selectively blocked the increased self-administration associated with extended access to cocaine76, nicotine64 and heroin77.
Several clinical trials that have been completed explored the effects of CRF1 antagonists on anxiety and depression, but no human laboratory studies or clinical trials have yet been initiated to investigate the effects of such drugs on alcohol dependence. In a trial terminated in Phase II because of increased levels of liver transaminases, the CRF1 antagonist R121919 caused a significant reduction in scores on the Hamilton Depression Inventory during the 30 day treatment period of the open trial78. A randomized, double-blind, placebo-controlled trial of another CRF1 antagonist, CP-316311, had no effect compared with the placebo, whereas the selective serotonin reuptake inhibitor sertraline produced a positive result79. CP-316311 had no adverse effects and in both trials the CRF1 antagonists were well tolerated, suggesting that CRF1 antagonists could be viable therapeutics for other addiction-related disorders.
Modulators of the brain stress systems: non-CRF targets
Preclinical data are emerging which suggest that other neurotransmitter systems and neuromodulators within the extended amygdala can be deregulated during the development of dependence on drugs of abuse13. Evidence for deregulation of noradrenaline in alcohol, cocaine and opioid dependence; dynorphin in cocaine and alcohol dependence; vasopressin in opioid and alcohol dependence; and orexin and substance P in cocaine, opioid and alcohol dependence are pertinent examples13. Administration of the noradrenergic α1-receptor antagonist prazosin decreases self-administration in rats that are dependent on alcohol, cocaine and opioids80-82 (TABLE 4). It has been proposed that noradrenergic–CRF interactions contribute to the brain stress activation associated with withdrawal from drugs of abuse13. In animal models of alcohol self-administration, the dramatic motivational effects of CRF in dependence can be observed in dependent animals.
Activation of the dynorphin–κ-opioid receptor system produces effects that are similar to those of most other opioid systems, but often opposite to those of μ-opioid receptors in the motivational domain83.κ-Opioid receptor agonists produce conditioned place aversion84, and depression and dysphoria in humans85. Substantial evidence suggests that expression of the gene encoding the dynorphin peptide and κ-opioid receptor activation are increased in the striatum and amygdala during acute and chronic administration of cocaine in rats86,87 and in humans88,89. The activation of the dynorphin system in the nucleus accumbens decreases activity in the dopamine system. It is therefore possible that the activation of the dynorphin system could contribute to the dysphoric syndrome associated with cocaine dependence90.
Dynorphin activity is also increased by stress91, suggesting a potential interaction with the CRF system. In mice, forced-swim stress and inescapable foot shock produced place aversions that were blocked by a κ-opioid receptor antagonist and dynorphin knockout92. Blockade of dynorphin activity, either by κ-opioid receptor antagonism or by disruption of the prodynorphin gene, blocked stress-induced reinstatement of cocaine-induced place preference in mice93 and blocked stress-induced reinstatement of cocaine-seeking behaviour in rats94, and CRF is thought to produce its aversive effect through activation of the dynorphin system92. There is also evidence that the link between reinstatement of drug-seeking behaviour and activation of κ-opioid receptors is mediated by CRF95. Thus, the dynorphin–κ-opioid system mimics stressor administration in animals in producing aversive effects and inducing drug-seeking behaviour. This aversive response could involve reciprocal interactions with dopamine in the nucleus accumbens and the extrahypothalamic brain CRF system.
Administration of a κ-opioid receptor antagonist had no effect on baseline self-administration of limited-access cocaine or heroin in primates96, but blunted the increased self-administration of cocaine in rats with extended access to the drug (S. Wee et al., in the press). A κ-opioid receptor antagonist also selectively blocked the increase in ethanol self-administration associated with withdrawal in alcohol-dependent rats97.
Neuromodulatory systems that oppose the actions of CRF in modulating stress and emotional behaviour could also be future targets for addiction treatment. These include neuropeptide Y and nociceptin systems, in which agonists reduce the excessive drinking associated with alcohol dependence98,99. Targeting the receptor system for substance P, known as the neurokinin 1 receptor (NK1R; also known as TACR1) system, which modulates emotional states, provides an example of a translational approach100. NK1R-knockout mice backcrossed onto a high-drinking strain of mice showed a major decrease in voluntary alcohol consumption, and recently detoxified subjects with alcohol dependence and treated with an NK1R antagonist showed decreased craving, blunted cortisol responses and decreased functional magnetic resonance imaging (fMRI) responses to affective stimuli. This suggests that NK1R may be a viable therapeutic target for the treatment of the dark side of addiction.
Glutamate modulators
Glutamate is thought to have several roles in the neurobiology of addiction, many of which provide potential targets for medications development. At low doses, ethanol may act as an NMDA (N-methyl-d-aspartate) receptor antagonist, which could contribute to the acute rewarding effects of ethanol102. Acute and protracted abstinence from ethanol described in alcohol dependence models seems to involve overactive glutamatergic systems, which are thought to be a target for the therapeutic effects of acamprosate39,102,103 (TABLE 4).
Glutamate has also long been associated with the neuroplasticity that is important for behavioural sensitization to drugs of abuse, particularly psychostimulants. AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) receptor antagonists and NMDA receptor antagonists block the development of locomotor sensitization to psychomotor stimulants104, and NMDA receptor antagonists also block the long-term potentiation and long-term depression associated with repeated administration of psychostimulants105. Some of these effects have been linked to increases in AMPA receptors that lack the subunit known as glutamate receptor 2 (GluR2)106.
One prominent hypothesis is that repeated self-administration of psychostimulants decreases basal release of glutamate in key brain circuits associated with the preoccupation–anticipation (craving) stage of the addiction cycle, but an exaggerated response of glutamate to activity in these circuits could confer susceptibility to relapse107. For example, drug-induced reinstatement of drug seeking seems to be mediated by a glutamatergic projection from the prefrontal cortex to the nucleus accumbens9. Cue-induced reinstatement of drug seeking involves a glutamatergic projection to the nucleus accumbens from both the basolateral amygdala and the ventral subiculum108,109. In addition, in cocaine-treated mice, there was less strengthening of synaptic transmission in ventral tegmental area slices from mice that lacked mGluR1 or the NMDA receptor subunit NR1 in dopamine neurons, suggesting a direct dopamine neuron target produced by glutamatergic plasticity110.
Additional evidence that AMPA or kainate receptors mediate glutamate modulation in addiction comes from a ‘top–down’ perspective. Clinical trials of topiramate, an anticonvulsant with some glutamate antagonist activity, have reported decreases in drinking behaviour in alcohol dependence and improvements in quality of life, but with significant adverse effects on memory and concentration111,112. In preclinical studies, topiramate decreased alcohol consumption and preference113 and decreased stress-induced increases in alcohol consumption and preference in mice114. However, topiramate also decreased saccharin preference, increased water intake and failed to block conditioned place preference to alcohol113,115. Together, these results suggest that topiramate can interact with both withdrawal-induced negative affective states and the rewarding effects of ethanol.
Pharmacological agents that modulate glutamate function may not only have a role in the basal hypoexcitability or hyperexcitability of glutamatergic systems during protracted abstinence, depending on the drug of abuse, but may also decrease drug-and cue-induced reinstatement of drug self-administration. To this end, antagonists of AMPA receptors, NMDA receptors and metabotropic glutamate receptor 5 (mGluR5), and agonists of mGluR2 and mGluR3, all of which decrease glutamate function, have been shown to block cue-induced reinstatement of drug self-administration116-119.
Given the side effects associated with direct glutamatergic antagonists, drugs that modulate the system may be more promising candidates for the treatment of addiction. Agents that can restore homeostasis to systems associated with both the withdrawal–negative affect (dark side) stage and the preoccupation–anticipation (craving) stage would be optimal. Studying the withdrawal–negative affect stage of the addiction cycle reveals numerous targets for pharmacotherapy development for addiction. A similar case can be made for the preoccupation–anticipation stage using the neurobiological targets identified here7. However, in both domains, a limiting step for clinical development is the progression from identifying preclinical targets to testing in humans.
Human laboratory studies
A crucial step in medications development is the submission of an investigational new drug (IND) application to the US Food and Drug Administration (FDA), or its equivalent in other countries, so that the drug can be tested in humans. See Supplementary information S2 (box) for further information regarding INDs for pharmacotherapies for the treatment of addiction.
Human laboratory studies provide a potentially powerful means of exploring treatment targets for specific components of the addiction cycle without the need for expensive double-blind, placebo-controlled trials. They can potentially predict efficacy measures of potential treatments for each stage of the addiction cycle (TABLES 1,4). Although the predictive validity of human laboratory models remains to be determined, ongoing studies with established medications for addiction can be used in a Rosetta Stone approach to evaluate the validity of animal and human models. Human laboratory models can also then serve as a springboard for the development of new pharmacotherapies.
The binge–intoxication stage
For the binge–intoxication stage of the addiction cycle, self-administration procedures for cocaine, heroin and marijuana in humans have been established largely using operant responding paradigms, in which participants who are dependent on a drug make a behavioural response such as pressing a key on a computer to receive the drug120. As in animal models, heroin self-administration is reduced by all three medications approved by the FDA to treat opioid dependence — methadone, naltrexone and buprenorphine (Subutex; Schering–Plough) — supporting the predictive validity of the animal models121,122. However, for cocaine, support for the validity of these models is less robust. A range of medications have been shown to reduce the subjective effects and craving associated with cocaine but do not decrease cocaine self-administration itself120. These results are consistent with clinical data showing that, of more than 60 medications tested, none has proved reliably effective in clinical trials123. To date, little or no work has been done on treatments for marijuana dependence.
Other measures, such as impulsivity, could be considered endophenotypes of the binge–intoxication stage and have some potential for predicting which pharmacotherapies could have efficacy for addiction treatment. Impulsivity can increase the probability that an individual will engage in initial drug intake, and the subsequent effects of the drug on impulsivity may increase impulsive behaviours that facilitate further drug use, prolonging the binge or even provoking relapse (discussed below). Various tasks have been used to assess impulsivity, including delayed discounting (that is, whether subjects show a relative preference for smaller, more immediate rewards over larger, more delayed rewards), behavioural inhibition (for example, the Stop Task124) and attentional measures (that is, whether subjects show increased variability in performance in a simple reaction time task that reflects ‘lapses in attention’).
The withdrawal–negative affect stage
For the withdrawal–negative affect stage, negative reinforcement mechanisms are in operation. Numerous human laboratory measures of acute withdrawal have proved to be sensitive to drug substitution125. In the laboratory, marijuana withdrawal is alleviated by marijuana smoking or by administration of oral Δ9-tetrahydrocannabinol (THC)126. Cognitive measures are sensitive to withdrawal effects of drug dependence during acute and protracted abstinence, and can be considered to be another endophenotype of the addiction process that could be used in medication screening127 (TABLE 1). Nicotine can improve cognitive processing and reduce negative affect128. The cascade of stress hormone interactions with drugs of abuse — which can facilitate the binge–intoxication stage, exaggerate the withdrawal–negative affect stage, and cause sensitization to stress-induced relapse — may all be amenable to human laboratory studies129.
The preoccupation–anticipation (craving) stage
For the preoccupation–anticipation stage, three main external factors (priming doses of drug, drug-associated cues and stressor exposure) and two internal factors (the malaise of protracted abstinence and an associated state of stress that contributes to malaise) are thought to contribute to relapse. Several human laboratory procedures have been developed to reflect these aspects of the preoccupation–anticipation stage. Drug reinstatement has been developed in human laboratory models, notably for alcohol and tobacco addiction. Priming-induced drinking in alcohol-dependent subjects in a bar-like setting was greater than in social drinkers and was selectively decreased in the alcohol-dependent groups by administration of opioid antagonists130. Similar results were observed in individuals who were dependent on alcohol, had a family history of alcohol dependence and received a priming dose of alcohol131, and in cigarette smokers primed with five cigarettes132.
Exposure to alcohol cues, such as the sight or smell of alcoholic beverages using the cue reactivity paradigm, reliably increases the urge to drink alcohol, salivation and attention to cues57,133,134. Furthermore, cue reactivity can predict treatment outcome135 and has been validated in some cases using medications that successfully treat alcohol dependence. For example, naltrexone, but not topiramate, blocked cue reactivity in subjects with alcohol dependence134,136, and nicotine replacement therapy decreased craving associated with smoking cues137. Other drugs not currently in therapeutic use for addiction have shown positive results with cue-induced reactivity paradigms, including carbamazepine (Tegretol; Novartis) for alcohol138 and amantadine for cocaine139.
Stress responses, including changes in the activities of the hypothalamic–pituitary–adrenal axis and extra-hypothalamic brain stress systems, affect all phases of the addiction cycle but may be particularly relevant to both the withdrawal–negative affect stage and the preoccupation–anticipation stage. Stress and stressors have also been associated with relapse and vulnerability to relapse129,140. Negative affect, stress or withdrawal-related distress also increases drug craving57,135,141,142. Both stress and drugs of abuse activate the hypothalamic–pituitary–adrenal axis, but the glucocorticoid response becomes blunted with chronic high-dose drug use. High glucocorticoid tone can, in turn, drive the brain stress systems in the amygdala129. Thus, drugs of abuse can trigger a cascade of stress hormone interactions that can facilitate the binge–intoxication stage and exacerbate the withdrawal–negative affect stage. They may also cause hypersensitivity of brain stress systems that contribute to maintaining the withdrawal–negative affect stage and sensitize the individual to stress-induced relapse. All of these changes may be amenable to study in humans in a laboratory setting.
Stress-related responses and stress-induced craving have been elicited in individuals with an addiction using a new model of stress-induced responsivity with an emotional imagery paradigm142 based on the early work of Lang and colleagues143. In this paradigm, individuals using the higher amounts of cocaine and alcohol and subjects recovering from an alcohol dependence showed greater craving and physiological responses to stressors than control social drinkers144. Perhaps of greatest importance in terms of paradigm validation, stress-induced cocaine craving in the laboratory could be used to accurately predict time to relapse145. Similar results have been observed for subjects dependent on alcohol or nicotine146,147. Preliminary results suggest that an α2-adrenoceptor agonist and an antagonist of sympathetic signalling, but not naltrexone, significantly decreased stress-induced opioid craving in subjects dependent on opioids148. These results support the construct and predictive validity of this laboratory model for stress-induced craving. Future studies crossreferencing pharmacological probes from the animal and human studies should provide an excellent basis for translational advances.
Exploration of the interaction of cue exposure with emotional states during protracted abstinence has provided a novel approach to the study of cue reactivity in alcohol craving57,149. A sample of non-treatment-seeking subjects with alcohol dependence was exposed to affective stimuli that had positive or negative valence (that is, they produced emotional responses that were positive or negative in nature) and then to a beverage cue but with no opportunity to self-administer alcohol. Cue reactivity was measured using subjective measures of craving, measures of emotional reactivity and psychophysiological measures. Alcohol exposure and both positive and negative emotional cues had the expected effects on subjective and emotional reactivity, and related effects on psychophysiological measures57. Gabapentin significantly decreased subjective craving and craving that was affectively evoked, and improved several measures of sleep quality57. These results suggest that affective priming, combined with alcohol cue exposure, could provide a powerful means to evaluate potential pharmacotherapies for addiction treatment.
Another novel approach involves measuring resistance to relapse in humans. The ‘smoking lapse behaviour’ model allows the measurement of two crucial features of relapse: the ability to resist the first cigarette and subsequent smoking behaviour150. Subjects with nicotine dependence are first exposed to precipitants of smoking relapse, such as alcohol, stress and nicotine deprivation, and then their ability to resist smoking when presented with their preferred brand of cigarettes is measured150. This model remains to be validated with existing anti-craving medications but provides an intriguing extension of cue reactivity studies that could be useful as an intermediary step between preclinical models and clinical trials.
An evolving area in human laboratory relapse models is the measurement of neural correlates of cues for relapse. Increased functional brain activation elicited by drug-associated cues, as measured in brain imaging studies, may correlate with an increased risk of relapse. Cue-induced functional activation of the brain can be assessed by measuring changes in cerebral blood flow with positron emission tomography or single photon emission computed tomography, or by combining blood flow measurements with fMRI. Core regions activated in most studies include the anterior cingulate, orbitofrontal cortex, basolateral amygdala, ventral striatum and dorsal striatum5. Strong cue-induced activation of similar regions, including the ventral striatum, dorsal striatum, medial prefrontal cortex and anterior cingulate, has been observed in subjects with alcohol dependence who have repeatedly suffered relapses151,152. Even more intriguingly, reduced functional activation of the ventral striatum in response to cues that signal non-drug rewards was observed in individuals with alcoholism, suggesting a shift in incentive salience to drug-related cues153. Imaging studies may therefore provide unique insights into subjects who exhibit the most dramatic functional activation in response to cues and, by extrapolation, the subjects who are more likely to relapse. Future studies will need to explore pharmacotherapeutic approaches to normalizing such cue-induced responses and whether such measures will predict therapeutic efficacy in treatment100.
Genetic variations and medications development
Widespread attempts are being made to identify genetic markers for addiction. However, a more exciting possibility is that certain single nucleotide polymorphisms may predict vulnerability to certain subtypes of excessive drinking syndromes and, of particular relevance to this Review, may predict responsiveness to pharmacotherapies in the treatment of alcoholism. Animal and human studies are beginning to realize this potential opportunity. Genetic association studies have focused on two pathways: one representing the reward side of addiction (the μ-opioid peptide system) and one representing the dark side of addiction (the CRF brain stress system). The human μ-opioid receptor is encoded by the OPRM1 gene and is a primary candidate for causing the pharmacogenetic variability of the clinical effects of opioid drugs and opioid receptor antagonists in the treatment of addiction. Mutations in OPRM1 have been found in the promoter, coding regions and introns of the gene. One mutation that has received considerable attention is the A118G single nucleotide polymorphism, which causes an amino acid substitution of asparagine with aspartate at position 40 of the μ-opioid receptor protein (Asn40Asp)154 (BOX 2).
Box 2 Genetic association studies — two examples.
μ-opioid receptor
In transfected AV-12 cells, A118G substitution leads to increased receptor affinity for β-endorphin154
A118G substitution is associated with higher pain thresholds and greater requirements for opioid medication163
Subjects with A118G substitution show greater response to alcohol and opioid antagonists164,165
A118G substitution does not predict vulnerability to alcoholism166
Subjects with the A118G single nucleotide polymorphism had a significantly greater cortisol response to naloxone167
Corticotropin-releasing factor receptor 1 (CRF1)
Adolescent subjects homozygous for the C allele of R1876831 drank more alcohol per occasion and had higher lifetime rates of heavy drinking in response to negative life events than subjects carrying the T allele168
Increased expression of CRF1 is associated with higher intake of ethanol in animal studies169,170
CRF1 antagonists block increased ethanol intake associated with acute withdrawal and protracted abstinence in animal studies75,170
In a separate series of studies, an association was found between single nucleotide polymorphisms of the gene that encodes CRF1 and binge drinking in adolescents and in adults with an alcohol dependence155. One of these single nucleotide polymorphisms, R1876831, is located in an intron that could potentially influence transcription of the gene that encodes CRF1 (BOX 2).
Clinical trials: challenges and opportunities
Double-blind, placebo-controlled trials with random assignment to treatments are the accepted standard for determining pharmacotherapy efficacy. A number of unique features of clinical trials for medications to treat addiction need to be emphasized. These include a lack of consensus about clinically relevant outcome measures, admission criteria and methods for detecting relapse to use of substances between study visits. Additionally, general issues related to medication compliance, placebo response and dropout rates present challenges for the design of clinical trials in addiction. There are also safety and tolerability issues that are specific to addiction, including the potential interaction of alcohol or other substances of abuse with the pharmacotherapy under study.
Outcome measures
The choice of the outcome for which a pharmacotherapy is likely to show efficacy may be guided by results from preclinical models of addiction and human laboratory studies. For example, naltrexone is thought to reduce the rewarding effects of drinking such that the individual with an alcohol dependence is no longer motivated to drink heavily. An outcome of reduced heavy drinking, as opposed to complete abstinence, is well characterized for naltrexone in human laboratory models involving alcohol administration and in clinical trials with subjects that are non-abstinent. Conversely, medications such as acamprosate or gabapentin, which are thought to support abstinence from alcohol by normalizing activity in a brain pathway that has become chronically deregulated, have shown efficacy in human laboratory models of cue reactivity but not in alcohol administration models, and have primarily shown efficacy on abstinence outcomes in clinical trials (see below).
Historically, the rate of complete abstinence has been the primary outcome measure of pharmacotherapy efficacy. However, clinically relevant abstinence-oriented outcomes also include latency to first lapse, percentage of abstinence days over the study duration, longest duration of abstinence during the study, abstinence at the end of the study (as indicative of behaviour likely to continue after the study) and statistical modelling of the trajectory of abstinence over the course of the study. Depending on the pharmacokinetic properties of the medication (for example, time to achieve steady-state serum concentrations), it may be necessary to specify a grace period during which a lapse may occur that would not normally be considered in efficacy determinations. Alternatively, to ensure an abstinent starting point across all patients, detoxification and a brief abstinent interval (for example, 2–5 days) may be required before randomization.
Harm reduction and non-hazardous use of substances are alternative end points for patients that do not have abstinence as their treatment goal. Related outcomes may include time to first heavy use, percentage of heavy use days during the study or statistical models of the trajectory of heavy use days over the course of the study.
Admission criteria
Admission criteria related to pre-randomization substance use or abstinence vary as a function of the stage of the addiction cycle that a pharmacotherapy is thought to target. To show efficacy, pharmacotherapies that are thought to support abstinence through normalization of a brain pathway deregulated by discontinuation of the addictive substance require a study sample that has achieved a minimal period of abstinence before randomization (usually 2–5 days to avoid the effects of acute withdrawal). Conversely, pharmacotherapies that are predicted to decrease heavy or hazardous use may require a non-abstinent sample with a high pre-randomization rate of heavy use to show efficacy.
Safety considerations specific to a medication also influence admission criteria. For example, subjects with an addiction, especially those dependent on alcohol, frequently present for treatment with pathologically elevated liver function test values. Trials of pharmacotherapies that are associated with hepatotoxicity (for example, naltrexone) typically exclude patients with liver function test results that are more than three times the upper limit of the normal range. However, such necessary exclusion criteria may also limit the dependence severity in the study sample and the extent to which clinically relevant generalizations can be made. Although women with childbearing potential are often omitted from clinical trials, this group needs to be represented in clinical trials of substance dependence because gender can affect drug efficacy. For example, long-acting injectable naltrexone showed efficacy in males but not in females in a multicentre trial of alcohol dependence156.
Measures of substance use
Measures of substance use are another unique aspect of clinical trials for pharmacotherapy development for addiction. Urine dipsticks and hand-held analysers of alcohol or carbon monoxide in expired breath can provide an immediate indication of drug use, drinking and cigarette smoking, respectively, at the time of a clinical trial research visit. However, most substances of abuse and their active metabolites (if any) have short half-lives, and their use cannot be assessed in breath, urine or plasma during the interval between study visits. One exception is Δ9-THC, the primary metabolite of cannabis, which can be present in urine for 1 month after use, potentially resulting in an underestimate of abstinence if detected. Because of such a long half-life, urinary Δ9-THC levels can be normalized to the urinary creatinine concentration to reduce variability in drug measurement owing to urine dilution157; abstinence is reflected by a reduction in the Δ9-THC/creatinine ratio across visits.
Available biomarkers of substance use between study visits (for example, γ-glutamyl transpeptidase levels, mean corpuscular volume and carbohydrate-deficient transferrin levels in the case of alcohol) have sensitivity and/or specificity limitations that can contribute to inaccurate estimations of use. A standardized interview using a calendar format, typically a variation on the timeline followback interview158, uses prompts such as weekend versus weekdays, paydays or holidays to assess quantity and frequency of substance use between study visits. Periodic interviews with collateral informants such as close friends or relatives of the subject can also be used in conjunction with biological measures to validate the subject’s timeline followback interview self-report measures of substance use. If discrepancies between sources cannot be resolved, the most negative outcome is typically assumed to be accurate.
Medication compliance
Medication non-compliance is not a common factor that influences outcomes in clinical trials involving addiction. When it occurs, factors such as drug tolerability should be assessed if standard measures to ensure compliance have been implemented. Such measures can include packaging medication in blister cards with day and time of day indicated for each dose, identifying frequently missed doses by reviewing unused medication at every visit or using multiple event monitoring system cap data, linking missed dosing with an activity of daily living, such as meals or brushing teeth, and a suggestion to affirm a commitment to recovery with every dose.
Premature study termination
Part of the definition of substance dependence by the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM IV) is an increased risk of relapse, impaired impulse control and psychosocial difficulties. Not surprisingly, therefore, trials of substance dependence may involve higher dropout rates than clinical trials of treatments for other psychiatric disorders. Therefore, incorporating strategies to enhance study completion in order to adequately assess use, safety and efficacy of a pharmacotherapy is important. Such strategies can include a flat rate of monetary compensation to offset transportation expenses at each visit and a lump sum payment for trial completion that is sufficient to be motivating but not coercive, the offer of a ~US$5 value coupon at each visit or systematic acknowledgement of the gains the subject is making (for example, income saved on days of abstinence that was previously spent on substances or normalization of liver function tests). Obtaining comprehensive contact information is also prudent.
Conclusions and future directions
The US National Institutes of Health (NIH) has promoted many of the elements required for the development of pharmacotherapies for addiction. To our knowledge, equivalent programmes do not exist in Europe, and should be encouraged. Areas of success include tremendous breakthroughs in the basic neurobiology of addiction, the successful development and validation of behavioural and pharmacological treatments of addiction (such as buprenorphine, supported by the National Institute on Drug Abuse (NIDA), and naltrexone, supported by the National Institute on Alcohol Abuse and Alcoholism (NIAAA)), and improved infrastructure for some aspects of pharmacotherapy development. For example, NIDA has established an extensive clinical trials network (see Further information for a link to the NIDA clinical trials network website). However, tremendous resources have been devoted to the development of pharmacotherapies for cocaine addiction, with little or no success reported to date2. It is hoped that the burgeoning use of human laboratory studies, outlined above, and the Rosetta Stone approach linking human and animal studies promoted here will yield better results.
A relevant issue is therefore how the NIH can do more to help. Several suggestions can be provided on the basis of this Review. These include developing a more balanced portfolio at the preclinical and clinical stage regarding which drugs of abuse should be studied and expanding the development and validation of human laboratory studies for potential medications. A major bottleneck for testing new medications is obtaining IND approvals for new drugs for human laboratory studies and Phase II clinical trials. A core facility like that of the NIDA clinical trials network should be considered for pooling resources for IND development, not only by NIDA and NIAAA, but by all central nervous system-related NIH institutes. Of crucial importance is a scientifically determined and clinically relevant ‘decision tree’ of which drugs should go forward. Such a decision tree could be built on the framework elaborated here, by incorporating a Rosetta Stone-like validation into the strategic planning of the NIH to facilitate translational research.
Another key element is that the pharmaceutical industry must consider that pharmacotherapies to target addiction are potentially profitable. Recent success with acamprosate and varenicline (Chantix/Champix; Pfizer) should provide some indication that further investigations in this area of disease are merited. Increased interaction with NIH in general to pool resources could reduce some of the developmental costs for industry, and some mechanisms are clearly underway in the form of small business innovation research grants and small business technology transfer grants (see Further information for a link to the Small Business Innovation Research (SBIR) and Small Business Technology Transfer (STTR) programmes). However, more efforts could be made by industry to partner with the NIH for the development of medications for addiction treatment. Furthermore, the pharmaceutical industry should consider allowing compounds that have been granted IND status but which they are no longer developing to be used in proof-of-concept human laboratory studies of addiction.
Thus, there is substantial potential for the development of future pharmacotherapies for addiction. Medications currently on the market for this indication have not only provided information on the opportunities for facilitating treatment but are also forming a means to evaluate future medications. A combination of validated animal models of addiction and a surge in understanding the neurocircuits and neuropharmacological mechanisms involved in the development and maintenance of addiction through basic research has provided numerous viable targets for future medications. New neurobiological targets will be derived from this basic research on addiction, with a focus on the neuroadaptive changes that account for the transition to dependence and the vulnerability to relapse, possibly within a genetic context. We propose that the Rosetta Stone framework outlined here could provide a heuristic approach for efficient and effective development of pharmacotherapies for addiction, and promote the flourishing of translational research interactions among independent investigators, the NIH and private industry.
Acknowledgments
This is manuscript number 19,996 authored from The Scripps Research Institute. The authors thank M. Arends for his assistance with manuscript preparation. Preparation of this work was supported by the Pearson Center for Alcoholism and Addiction Research and National Institutes of Health grants AA12602, AA08459, (R37)AA014028 and AA06420 from the National Institute on Alcohol Abuse and Alcoholism, DA04043, DA04398, (P20)DA024194 and DA10072 from the National Institute on Drug Abuse, DK26741 from the National Institute of Diabetes and Digestive and Kidney Diseases, and 17RT-0095 from the Tobacco-Related Disease Research Program from the State of California. The opinions expressed in this article by G.K.L. are as an individual and not as an employee of Nereus Pharmaceuticals.
- Addiction
This term can be used interchangeably with substance dependence (as currently defined by the Diagnostic and Statistical Manual of Mental Disorders, 4th edition) to refer to a final stage of a usage process. Clinically, the occasional but limited use of a drug with the potential for abuse or dependence is distinct from the emergence of addiction
- Face-valid model
A model that looks or seems to be a valid representation of what it purports to measure
- Kleptomania
A classic impulse control disorder in which there is an increase in tension before stealing an object or objects that are not needed and relief after the act, but little or no regret or self-reproach
- Obsessive–compulsive disorder
A classic compulsive disorder is one in which obsessions of contamination or harm drive anxiety, the reduction of which requires repetitive compulsive acts to reduce the anxiety
- Binge
Any behaviour indulged to excess. In alcohol abuse, a binge is defined in the United States as four drinks for females and five drinks for males in a 2-hour period or reaching a blood alcohol level of 0.08 g per 100 ml
- Withdrawal
A collection of physiological signs and symptoms that present after the sudden cessation of drug intake, which can include shaking, sweating and anxiety, depending on the drug
- Place conditioning
A procedure for assessing the reinforcing efficacy of drugs using a classical or Pavlovian conditioning procedure. Animals typically show conditioned place preference for an environment associated with the common drugs of addiction in humans and avoid environments associated with aversive states of drug withdrawal (that is, they show conditioned place aversion)
- Corticotropin-releasing factor
A 41-amino-acid polypeptide with wide distribution throughout the brain and high concentrations in cell bodies in the paraventricular nucleus of the hypothalamus, the basal forebrain and notably the extended amygdala and brainstem
- GABAA receptor
A receptor that is coupled to Cl− channels and forms a receptor complex that includes recognition sites for convulsants, benzodiazepines, barbiturates and steroids
- GABAB receptor
A metabotropic receptor that regulates K+ and Ca2+ channels through a G protein-coupled mechanism. Both GABAA and GABAB receptors have an inhibitory action in the central nervous system and are thought to mediate the anxiety-decreasing, motor-uncoordinating, sedative and hypnotic effects of alcohol.
- Hamilton Depression Inventory
A validated scale to measure the severity of depressive symptoms
- Dynorphins
Opioid peptides derived from the prodynorphin precursor that contain the leucine–encephalin sequence at their amino termini. They are the presumed endogenous ligands of the κ-opioid receptor and have long been thought to mediate negative emotional states
- Behavioural sensitization
An increased drug-induced locomotor response or drug reward response with repeated administration
- Endophenotype
Measurable components, unseen by the unaided eye, along the pathway between disease and genotype
- Multiple event monitoring system cap
Caps on pill bottles with built-in microelectronics that record each date and time the cap is removed
Footnotes
Competing interests statement The authors declare competing financial interests: see web version for details.
DATABASES
Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
OPRM1
UniProtKB: http://www.uniprot.org
κ-opioid receptor | μ-opioid receptor | CRF1 | D2 | D3 | NK1R
FURTHER INFORMATION
NIDA clinical trials network: http://www.nida.nih.gov/ctn/
Small Business Innovation Research (SBIR) and Small Business Technology Transfer (STTR) programmes: http://grants.nih.gov/grants/funding/sbirsttr_programs.htm
Supplementary Material
References
- 1.Koob GF, Le Moal M. Drug abuse: hedonic homeostatic dysregulation. Science. 1997;278:52–58. doi: 10.1126/science.278.5335.52. [DOI] [PubMed] [Google Scholar]
- 2.Kreek MJ, LaForge KS, Butelman E. Pharmacotherapy of addictions. Nature Rev Drug Discov. 2002;1:710–726. doi: 10.1038/nrd897. erratum 1, 926 (2002) [DOI] [PubMed] [Google Scholar]
- 3.Deroche-Gamonet V, Belin D, Piazza PV. Evidence for addiction-like behavior in the rat. Science. 2004;305:1014–1017. doi: 10.1126/science.1099020. [DOI] [PubMed] [Google Scholar]
- 4.Vanderschuren LJ, Everitt BJ. Drug seeking becomes compulsive after prolonged cocaine self-administration. Science. 2004;305:1017–1019. doi: 10.1126/science.1098975. [DOI] [PubMed] [Google Scholar]
- 5.Heinz A, Beck A, Grüsser SM, Grace AA, Wrase J. Identifying the neural circuitry of alcohol craving and relapse vulnerability. Addict Biol. 2009;14:108–118. doi: 10.1111/j.1369-1600.2008.00136.x.. This study showed a positive correlation between functional brain activation elicited by alcohol-related cues and risk of relapse, suggesting a novel imaging approach in humans.
- 6.Sanchis-Segura C, Spanagel R. Behavioural assessment of drug reinforcement and addictive features in rodents: an overview. Addict Biol. 2006;11:2–38. doi: 10.1111/j.1369-1600.2006.00012.x. [DOI] [PubMed] [Google Scholar]
- 7.Koob GF, Le Moal M. Neurobiology of Addiction. Elsevier; London: 2006. [Google Scholar]
- 8.Nestler EJ. Is there a common molecular pathway for addiction? Nature Neurosci. 2005;8:1445–1449. doi: 10.1038/nn1578.. The authors showed that drugs of abuse have very different acute mechanisms of action but converge on the reward pathways of the brain in the ventral tegmental area and nucleus accumbens to produce common functional effects.
- 9.Kalivas PW, McFarland K. Brain circuitry and the reinstatement of cocaine-seeking behavior. Psychopharmacology. 2003;168:44–56. doi: 10.1007/s00213-003-1393-2. [DOI] [PubMed] [Google Scholar]
- 10.Vanderschuren LJ, Everitt BJ. Behavioral and neural mechanisms of compulsive drug seeking. Eur J Pharmacol. 2005;526:77–88. doi: 10.1016/j.ejphar.2005.09.037. [DOI] [PubMed] [Google Scholar]
- 11.Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nature Neurosci. 2005;8:1442–1444. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- 12.Koob GF, Le Moal M. Addiction and the brain antireward system. Annu Rev Psychol. 2008;59:29–53. doi: 10.1146/annurev.psych.59.103006.093548.. A neurobiological model of the brain emotional systems was proposed to explain the persistent changes in motivation that are associated with vulnerability to dependence in addiction.
- 13.Koob GF. A role for brain stress systems in addiction. Neuron. 2008;59:11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nestler EJ. Molecular neurobiology of addiction. Am J Addict. 2001;10:201–217. doi: 10.1080/105504901750532094. [DOI] [PubMed] [Google Scholar]
- 15.Shaham Y, Shalev U, Lu L, de Wit H, Stewart J. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology. 2003;168:3–20. doi: 10.1007/s00213-002-1224-x.. This review elegantly summarizes the neuronal events that mediate reinstatement of heroin-, cocaine-and alcohol-seeking following acute priming injections of drugs, drug-associated cues and environmental stressors.
- 16.Schultz W. Multiple dopamine functions at different time courses. Annu Rev Neurosci. 2007;30:259–288. doi: 10.1146/annurev.neuro.28.061604.135722. [DOI] [PubMed] [Google Scholar]
- 17.Berridge KC. The debate over dopamine’s role in reward: the case for incentive salience. Psychopharmacology. 2007;191:391–431. doi: 10.1007/s00213-006-0578-x. [DOI] [PubMed] [Google Scholar]
- 18.Weiss F, Koob GF. Drug addiction: functional neurotoxicity of the brain reward systems. Neurotox Res. 2000;3:145–156. doi: 10.1007/BF03033235. [DOI] [PubMed] [Google Scholar]
- 19.Weiss F, Markou A, Lorang MT, Koob GF. Basal extracellular dopamine levels in the nucleus accumbens are decreased during cocaine withdrawal after unlimited-access self-administration. Brain Res. 1992;593:314–318. doi: 10.1016/0006-8993(92)91327-b. [DOI] [PubMed] [Google Scholar]
- 20.Weiss F, et al. Ethanol self-administration restores withdrawal-associated deficiencies in accumbal dopamine and 5-hydroxytryptamine release in dependent rats. J Neurosci. 1996;16:3474–3485. doi: 10.1523/JNEUROSCI.16-10-03474.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melis M, Spiga S, Diana M. The dopamine hypothesis of drug addiction: hypodopaminergic state. Int Rev Neurobiol. 2005;63:101–154. doi: 10.1016/S0074-7742(05)63005-X.. The authors report that tonic mesolimbic dopamine transmission seems to be drastically reduced in animal models of drug addiction and human subjects with the disease, and suggest that restoring dopamine transmission (not necessarily with classic receptor-oriented drugs) may reveal new treatment options.
- 22.Pulvirenti L, Koob GF. Being partial to psychostimulant addiction therapy. Trends Pharmacol Sci. 2002;23:151–153. doi: 10.1016/s0165-6147(00)01991-x. [DOI] [PubMed] [Google Scholar]
- 23.Clark D, et al. Behavioural profile of partial D2 dopamine receptor agonists: 1 Atypical inhibition of d-amphetamine-induced locomotor hyperactivity and stereotypy. Psychopharmacology. 1991;105:381–392. doi: 10.1007/BF02244434. [DOI] [PubMed] [Google Scholar]
- 24.Pulvirenti L, Koob GF. Dopamine receptor agonists, partial agonists and psychostimulant addiction. Trends Pharmacol Sci. 1994;15:374–379. doi: 10.1016/0165-6147(94)90158-9. [DOI] [PubMed] [Google Scholar]
- 25.Svensson K, et al. Effects of the partial dopamine receptor agonists SDZ 208-911, SDZ 208-912 and terguride on central monoamine receptors: a behavioral, biochemical and electrophysiological study. Naunyn Schmiedebergs Arch Pharmacol. 1991;344:263–274. doi: 10.1007/BF00182999. [DOI] [PubMed] [Google Scholar]
- 26.Pulvirenti L, Smith D, Koob GF. SDZ 208-911, an amino-ergoline with partial dopamine agonistic properties, dose dependently increases cocaine self-administration in the rat. Psychopharmacology. 1994;113:518–520. doi: 10.1007/BF02245232. [DOI] [PubMed] [Google Scholar]
- 27.Pulvirenti L, Balducci C, Piercy M, Koob GF. Characterization of the effects of the partial dopamine agonist terguride on cocaine self-administration in the rat. J Pharmacol Exp Ther. 1998;286:1231–1238. [PubMed] [Google Scholar]
- 28.Izzo E, Orsini C, Koob GF, Pulvirenti L. A dopamine partial agonist and antagonist block amphetamine self-administration in a progressive ratio schedule. Pharmacol Biochem Behav. 2001;68:701–708. doi: 10.1016/s0091-3057(01)00472-5. [DOI] [PubMed] [Google Scholar]
- 29.Bono G, Balducci C, Richelmi P, Koob GF, Pulvirenti L. Dopamine partial receptor agonists reduce ethanol intake in the rat. Eur J Pharmacol. 1996;296:233–238. doi: 10.1016/0014-2999(95)00592-7. [DOI] [PubMed] [Google Scholar]
- 30.Orsini C, Koob GF, Pulvirenti L. Dopamine partial agonist reverses amphetamine withdrawal in rats. Neuropsychopharmacology. 2001;25:789–792. doi: 10.1016/S0893-133X(01)00270-6. [DOI] [PubMed] [Google Scholar]
- 31.Hoefer ME, Voskanian SJ, Koob GF, Pulvirenti L. Effects of terguride, ropinirole, and acetyl-l-carnitine on methamphetamine withdrawal in the rat. Pharmacol Biochem Behav. 2006;83:403–409. doi: 10.1016/j.pbb.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 32.Wee S, Wang Z, Woolverton WL, Pulvirenti L, Koob GF. Effect of aripiprazole, a partial D2 receptor agonist, on increased rate of methamphetamine self-administration in rats with prolonged access. Neuropsychopharmacology. 2007;32:2238–2247. doi: 10.1038/sj.npp.1301353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heidbreder CA, et al. The role of central dopamine D3 receptors in drug addiction: a review of pharmacological evidence. Brain Res Rev. 2005;49:77–105. doi: 10.1016/j.brainresrev.2004.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gyertyan I, et al. Effects of RGH-237 (N-(4-(4-(3-amin ocarbonyl-phenyl)-piperazin-1-yl)-butyl)-4-bromo-benzamide), an orally active, selective dopamine D3 receptor partial agonist in animal models of cocaine abuse. J Pharmacol Exp Ther. 2007;320:1268–1278. doi: 10.1124/jpet.106.107920. [DOI] [PubMed] [Google Scholar]
- 35.Pilla M, et al. Selective inhibition of cocaine-seeking behaviour by a partial dopamine D3 receptor agonist. Nature. 1999;400:371–375. doi: 10.1038/22560. [DOI] [PubMed] [Google Scholar]
- 36.Aujla H, Beninger RJ. The dopamine D3 receptor-preferring partial agonist BP 897 dose-dependently attenuates the expression of amphetamine-conditioned place preference in rats. Behav Pharmacol. 2005;16:181–186. doi: 10.1097/00008877-200505000-00007. [DOI] [PubMed] [Google Scholar]
- 37.Caine SB, Koob GF. Effects of mesolimbic dopamine depletion on responding maintained by cocaine and food. J Exp Anal Behav. 1994;61:213–221. doi: 10.1901/jeab.1994.61-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haile CN, Kosten TA. Differential effects of D1-and D2-like compounds on cocaine self-administration in Lewis and Fischer 344 inbred rats. J Pharmacol Exp Ther. 2001;299:509–518. [PubMed] [Google Scholar]
- 39.Spanagel R, Kiefer F. Drugs for relapse prevention of alcoholism: ten years of progress. Trends Pharmacol Sci. 2008;29:109–115. doi: 10.1016/j.tips.2007.12.005.. This review summarizes the many neurochemical pathways involved in mediating craving and relapse to alcohol, and focuses on new targets in these domains for the treatment of alcoholism
- 40.Rassnick S, D’Amico E, Riley E, Koob GF. GABA antagonist and benzodiazepine partial inverse agonist reduce motivated responding for ethanol. Alcohol Clin Exp Res. 1993;17:124–130. doi: 10.1111/j.1530-0277.1993.tb00736.x. [DOI] [PubMed] [Google Scholar]
- 41.Hyytia P, Koob GF. GABAA receptor antagonism in the extended amygdala decreases ethanol self-administration in rats. Eur J Pharmacol. 1995;283:151–159. doi: 10.1016/0014-2999(95)00314-b. [DOI] [PubMed] [Google Scholar]
- 42.Weerts EM, Froestl W, Griffiths RR. Effects of GABAergic modulators on food and cocaine self-administration in baboons. Drug Alcohol Depend. 2005;80:369–376. doi: 10.1016/j.drugalcdep.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 43.Paterson NE, et al. Positive modulation of GABAB receptors decreased nicotine self-administration and counteracted nicotine-induced enhancement of brain reward function in rats. J Pharmacol Exp Ther. 2008;326:306–314. doi: 10.1124/jpet.108.139204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Colombo G, et al. Baclofen suppresses motivation to consume alcohol in rats. Psychopharmacology. 2003;167:221–224. doi: 10.1007/s00213-003-1397-y. [DOI] [PubMed] [Google Scholar]
- 45.Xi ZX, Stein EA. Baclofen inhibits heroin self-administration behavior and mesolimbic dopamine release. J Pharmacol Exp Ther. 1999;290:1369–1374. [PubMed] [Google Scholar]
- 46.Frye GD, McCown TJ, Breese GR. Differential sensitivity of ethanol withdrawal signs in the rat to gamma-aminobutyric acid (GABA) mimetics: blockade of audiogenic seizures but not forelimb tremors. J Pharmacol Exp Ther. 1983;226:720–725. [PubMed] [Google Scholar]
- 47.Addolorato G, et al. Baclofen efficacy in reducing alcohol craving and intake: a preliminary double-blind randomized controlled study. Alcohol Alcohol. 2002;37:504–508. doi: 10.1093/alcalc/37.5.504. [DOI] [PubMed] [Google Scholar]
- 48.Addolorato G, et al. Rapid suppression of alcohol withdrawal syndrome by baclofen. Am J Med. 2002;112:226–229. doi: 10.1016/s0002-9343(01)01088-9. [DOI] [PubMed] [Google Scholar]
- 49.Walker BM, Koob GF. The γ-aminobutyric acid-B receptor agonist baclofen attenuates responding for ethanol in ethanol-dependent rats. Alcohol Clin Exp Res. 2007;31:11–18. doi: 10.1111/j.1530-0277.2006.00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahmadi-Abhari SA, et al. Baclofen versus clonidine in the treatment of opiates withdrawal, side-effects aspect: a double-blind randomized controlled trial. J Clin Pharmacol Ther. 2001;26:67–71. doi: 10.1046/j.1365-2710.2001.00325.x. [DOI] [PubMed] [Google Scholar]
- 51.Sills GJ. The mechanisms of action of gabapentin and pregabalin. Curr Opin Pharmacol. 2006;6:108–113. doi: 10.1016/j.coph.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 52.Taylor CP, et al. A summary of mechanistic hypotheses of gabapentin pharmacology. Epilepsy Res. 1998;29:233–249. doi: 10.1016/s0920-1211(97)00084-3. [DOI] [PubMed] [Google Scholar]
- 53.Gotz E, Feuerstein TJ, Lais A, Meyer DK. Effects of gabapentin on release of gamma-aminobutyric acid from slices of rat neostriatum. Arzneimittelforschung. 1993;43:636–638. [PubMed] [Google Scholar]
- 54.Fink K, Meder W, Dooley DJ, Göthert M. Inhibition of neuronal Ca2+ influx by gabapentin and subsequent reduction of neurotransmitter release from rat neocortical slices. Brit J Pharmacol. 2000;130:900–906. doi: 10.1038/sj.bjp.0703380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bertrand S, et al. The anticonvulsant, antihyperalgesic agent gabapentin is an agonist at brain γ-aminobutyric acid type B receptors negatively coupled to voltage-dependent calcium channels. J Pharmacol Exp Ther. 2001;298:15–24. [PubMed] [Google Scholar]
- 56.Roberto M, et al. Cellular and behavioral interactions of gabapentin with alcohol dependence. J Neurosci. 2008;28:5762–5771. doi: 10.1523/JNEUROSCI.0575-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mason BJ, Light JM, Williams LD, Drobes DJ. Proof-of-concept human laboratory study for protracted abstinence in alcohol dependence: effects of gabapentin. Addict Biol. 2009;14:73–83. doi: 10.1111/j.1369-1600.2008.00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koob GF, Heinrichs SC, Menzaghi F, Pich EM, Britton KT. Corticotropin releasing factor, stress and behavior. Semin Neurosci. 1994;6:221–229. [Google Scholar]
- 59.Swanson LW, Sawchenko PE, Rivier J, Vale W. The organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36:165–186. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- 60.Sarnyai Z, et al. Brain corticotropin-releasing factor mediates “anxiety-like” behavior induced by cocaine withdrawal in rats. Brain Res. 1995;675:89–97. doi: 10.1016/0006-8993(95)00043-p. [DOI] [PubMed] [Google Scholar]
- 61.Basso AM, Spina M, Rivier J, Vale W, Koob GF. Corticotropin-releasing factor antagonist attenuates the “anxiogenic-like” effect in the defensive burying paradigm but not in the elevated plus-maze following chronic cocaine in rats. Psychopharmacology. 1999;145:21–30. doi: 10.1007/s002130051028. [DOI] [PubMed] [Google Scholar]
- 62.Knapp DJ, Overstreet DH, Moy SS, Breese GR. SB242084, flumazenil, and CRA1000 block ethanol withdrawal-induced anxiety in rats. Alcohol. 2004;32:101–111. doi: 10.1016/j.alcohol.2003.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Overstreet DH, Knapp DJ, Breese GR. Modulation of multiple ethanol withdrawal-induced anxiety-like behavior by CRF and CRF1 receptors. Pharmacol Biochem Behav. 2004;77:405–413. doi: 10.1016/j.pbb.2003.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.George O, et al. CRF–CRF1 system activation mediates withdrawal-induced increases in nicotine self-administration in nicotine-dependent rats. Proc Natl Acad Sci USA. 2007;104:17198–17203. doi: 10.1073/pnas.0707585104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stinus L, Cador M, Zorrilla EP, Koob GF. Buprenorphine and a CRF1 antagonist block the acquisition of opiate withdrawal-induced conditioned place aversion in rats. Neuropsychopharmacology. 2005;30:90–98. doi: 10.1038/sj.npp.1300487. [DOI] [PubMed] [Google Scholar]
- 66.Rodriguez de Fonseca F, Carrera MRA, Navarro M, Koob GF, Weiss F. Activation of corticotropin-releasing factor in the limbic system during cannabinoid withdrawal. Science. 1997;276:2050–2054. doi: 10.1126/science.276.5321.2050. [DOI] [PubMed] [Google Scholar]
- 67.Funk CK, O’Dell LE, Crawford EF, Koob GF. Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats. J Neurosci. 2006;26:11324–11332. doi: 10.1523/JNEUROSCI.3096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Merlo-Pich E, et al. Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci. 1995;15:5439–5447. doi: 10.1523/JNEUROSCI.15-08-05439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Olive MF, Koenig HN, Nannini MA, Hodge CW. Elevated extracellular CRF levels in the bed nucleus of the stria terminalis during ethanol withdrawal and reduction by subsequent ethanol intake. Pharmacol Biochem Behav. 2002;72:213–220. doi: 10.1016/s0091-3057(01)00748-1. [DOI] [PubMed] [Google Scholar]
- 70.Richter RM, Weiss F. In vivo CRF release in rat amygdala is increased during cocaine withdrawal in self-administering rats. Synapse. 1999;32:254–261. doi: 10.1002/(SICI)1098-2396(19990615)32:4<254::AID-SYN2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 71.Weiss F, et al. Compulsive drug-seeking behavior and relapse: neuroadaptation, stress, and conditioning factors. Ann NY Acad Sci. 2001;937:1–26. doi: 10.1111/j.1749-6632.2001.tb03556.x. [DOI] [PubMed] [Google Scholar]
- 72.Baldwin HA, Rassnick S, Rivier J, Koob GF, Britton KT. CRF antagonist reverses the “anxiogenic” response to ethanol withdrawal in the rat. Psychopharmacology. 1991;103:227–232. doi: 10.1007/BF02244208. [DOI] [PubMed] [Google Scholar]
- 73.Valdez GR, et al. Increased ethanol self-administration and anxiety-like behavior during acute withdrawal and protracted abstinence: regulation by corticotropin-releasing factor. Alcohol Clin Exp Res. 2002;26:1494–1501. doi: 10.1097/01.ALC.0000033120.51856.F0. [DOI] [PubMed] [Google Scholar]
- 74.Gehlert DR, et al. 3-(4-chloro-2-morpholin-4-yl-thiaz ol-5-yl)-8-(1-ethylpropyl)-2,6-dimethyl-imidazo(1,2-b) pyridazine: a novel brain-penetrant, orally available corticotropin-releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. J Neurosci. 2007;27:2718–2726. doi: 10.1523/JNEUROSCI.4985-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Funk CK, Zorrilla EP, Lee MJ, Rice KC, Koob GF. Corticotropin-releasing factor 1 antagonists selectively reduce ethanol self-administration in ethanol-dependent rats. Biol Psychol. 2007;61:78–86. doi: 10.1016/j.biopsych.2006.03.063.. This study showed that CRF1 receptors play an important part in mediating excessive alcohol self-administration in dependent rats, but not in non-dependent rats, and that CRF1 receptor antagonists could be new pharmacotherapeutic targets for the treatment of alcoholism in humans.
- 76.Specio SE, et al. CRF1 receptor antagonists attenuate escalated cocaine self-administration in rats. Psychopharmacology. 2008;196:473–482. doi: 10.1007/s00213-007-0983-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Greenwell TN, et al. Corticotropin-releasing factor-1 receptor antagonists decrease heroin self-administration in long-, but not short-access rats. Addict Biol. 2009;14:130–143. doi: 10.1111/j.1369-1600.2008.00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zobel AW, et al. Effects of the high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major depression: the first 20 patients treated. J Psychiatr Res. 2000;34:171–181. doi: 10.1016/s0022-3956(00)00016-9. [DOI] [PubMed] [Google Scholar]
- 79.Binneman B, et al. A 6-week randomized, placebo-controlled trial of CP-316311 (a selective CRH1 antagonist in the treatment of major depression) Am J Psychiatry. 2008;165:617–620. doi: 10.1176/appi.ajp.2008.07071199. [DOI] [PubMed] [Google Scholar]
- 80.Walker BM, Rasmussen DD, Raskind MA, Koob GF. α1-Noradrenergic receptor antagonism blocks dependence-induced increases in responding for ethanol. Alcohol. 2008;42:91–97. doi: 10.1016/j.alcohol.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Greenwell TN, Walker BM, Cottone P, Zorrilla EP, Koob GF. The adrenergic α1 receptor antagonist prazosin reduces heroin self-administration in rats with extended access to heroin administration. Pharmacol Biochem Behav. 2009;91:295–302. doi: 10.1016/j.pbb.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wee S, Mandyam CD, Lekic DM, Koob GF. α1-Noradrenergic system role in increased motivation for cocaine intake in rats with prolonged access. Eur Neuropsychopharmacol. 2008;18:303–311. doi: 10.1016/j.euroneuro.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shippenberg TS, Zapata A, Chefer VI. Dynorphin and the pathophysiology of drug addiction. Pharmacol Ther. 2007;116:306–321. doi: 10.1016/j.pharmthera.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mucha RF, Herz A. Motivational properties of kappa and mu opioid receptor agonists studied with place and taste preference conditioning. Psychopharmacology. 1985;86:274–280. doi: 10.1007/BF00432213. [DOI] [PubMed] [Google Scholar]
- 85.Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by kappa opiate receptors. Science. 1986;233:774–776. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- 86.Sivam SP. Cocaine selectively increases striatonigral dynorphin levels by a dopaminergic mechanism. J Pharmacol Exp Ther. 1989;250:818–824. [PubMed] [Google Scholar]
- 87.Spangler R, Unterwald EM, Kreek MJ. “Binge” cocaine administration induces a sustained increase of prodynorphin mRNA in rat caudate-putamen. Mol Brain Res. 1993;19:323–327. doi: 10.1016/0169-328x(93)90133-a.. This study showed a substantial increase in the concentration of prodynorphin mRNA in caudate putamen extracts of rats injected with cocaine, using a binge administration pattern designed to mimic cocaine abuse in humans.
- 88.Hurd YL, Herkenham M. Molecular alterations in the neostriatum of human cocaine addicts. Synapse. 1993;13:357–369. doi: 10.1002/syn.890130408. [DOI] [PubMed] [Google Scholar]
- 89.Unterwald EM, Rubenfeld JM, Kreek MJ. Repeated cocaine administration upregulates kappa and mu, but not delta, opioid receptors. Neuroreport. 1994;5:1613–1616. doi: 10.1097/00001756-199408150-00018. [DOI] [PubMed] [Google Scholar]
- 90.Nestler EJ. Historical review: molecular and cellular mechanisms of opiate and cocaine addiction. Trends Pharmacol Sci. 2004;25:210–218. doi: 10.1016/j.tips.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 91.Shirayama Y, et al. Stress increases dynorphin immunoreactivity in limbic brain regions and dynorphin antagonism produces antidepressant-like effects. J Neurochem. 2004;90:1258–1268. doi: 10.1111/j.1471-4159.2004.02589.x. [DOI] [PubMed] [Google Scholar]
- 92.Land BB, et al. The dysphoric component of stress is encoded by activation of the dynorphin κ-opioid system. J Neurosci. 2008;28:407–414. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Beardsley PM, Howard JL, Shelton KL, Carroll FI. Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine-seeking induced by footshock stressors vs cocaine primes and its antidepressant-like effects in rats. Psychopharmacology. 2005;183:118–126. doi: 10.1007/s00213-005-0167-4. [DOI] [PubMed] [Google Scholar]
- 95.Valdez GR, Platt DM, Rowlett JK, Rüedi-Bettschen D, Spealman RD. Kappa agonist-induced reinstatement of cocaine seeking in squirrel monkeys: a role for opioid and stress-related mechanisms. J Pharmacol Exp Ther. 2007;323:525–533. doi: 10.1124/jpet.107.125484. [DOI] [PubMed] [Google Scholar]
- 96.Negus SS, et al. Effect of antagonists selective for mu, delta and kappa opioid receptors on the reinforcing effects of heroin in rats. J Pharmacol Exp Ther. 1993;265:1245–1252. [PubMed] [Google Scholar]
- 97.Walker BM, Koob GF. Pharmacological evidence for a motivational role of κ-opioid systems in ethanol dependence. Neuropsychopharmacology. 2008;33:643–652. doi: 10.1038/sj.npp.1301438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Heilig M, Koob GF, Ekman R, Britton KT. Corticotropin-releasing factor and neuropeptide Y: role in emotional integration. Trends Neurosci. 1994;17:80–85. doi: 10.1016/0166-2236(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 99.Ciccocioppo R, Angeletti S, Panocka I, Massi M. Nociceptin–orphanin FQ and drugs of abuse. Peptides. 2000;21:1071–1080. doi: 10.1016/s0196-9781(00)00245-x. [DOI] [PubMed] [Google Scholar]
- 100.George DT, et al. Neurokinin 1 receptor antagonism as a possible therapy for alcoholism. Science. 2008;319:1536–1539. doi: 10.1126/science.1153813. [DOI] [PubMed] [Google Scholar]
- 101.Hoffman PL, Rabe CS, Moses F, Tabakoff B. N-methyl-d-aspartate receptors and ethanol: inhibition of calcium flux and cyclic GMP production. J Neurochem. 1989;52:1937–1940. doi: 10.1111/j.1471-4159.1989.tb07280.x. [DOI] [PubMed] [Google Scholar]
- 102.de Witte P, Littleton J, Parot P, Koob G. Neuroprotective and abstinence-promoting effects of acamprosate: elucidating the mechanism of action. CNS Drugs. 2005;19:517–537. doi: 10.2165/00023210-200519060-00004. [DOI] [PubMed] [Google Scholar]
- 103.Littleton JM. Acamprosate in alcohol dependence: implications of a unique mechanism of action. J Addict Med. 2007;1:115–125. doi: 10.1097/ADM.0b013e318156c26f. [DOI] [PubMed] [Google Scholar]
- 104.Li Y, Vartanian AJ, White FJ, Xue CJ, Wolf ME. Effects of the AMPA receptor antagonist NBQX on the development and expression of behavioral sensitization to cocaine and amphetamine. Psychopharmacology. 1997;134:266–276. doi: 10.1007/s002130050449. [DOI] [PubMed] [Google Scholar]
- 105.Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–387. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- 106.Conrad KL, et al. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995.. This paper proposed that increased levels of AMPA receptors lacking the GluR2 subunit causes increased reactivity of nucleus accumbens neurons to cocaine-related cues and leads to an intensification of drug craving and relapse.
- 107.Kalivas PW, et al. Glutamate transmission and addiction to cocaine. Ann NY Acad Sci. 2003;1003:169–175. doi: 10.1196/annals.1300.009. [DOI] [PubMed] [Google Scholar]
- 108.Everitt BJ, Wolf ME. Psychomotor stimulant addiction: a neural systems perspective. J Neurosci. 2002;22:3312–3320. doi: 10.1523/JNEUROSCI.22-09-03312.2002. Erratum 22, 1a(2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vorel SR, Liu X, Hayes RJ, Spector JA, Gardner EL. Relapse to cocaine-seeking after hippocampal theta burst stimulation. Science. 2001;292:1175–1178. doi: 10.1126/science.1058043. [DOI] [PubMed] [Google Scholar]
- 110.Engblom D, et al. Glutamate receptors on dopamine neurons control the persistence of cocaine seeking. Neuron. 2008;59:497–508. doi: 10.1016/j.neuron.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 111.Stringer S, Rueve M, Mossman D. Topiramate as treatment for alcohol dependence. J Am Med Assoc. 2008;299:405–406. doi: 10.1001/jama.299.4.405-c. [DOI] [PubMed] [Google Scholar]
- 112.Olmsted CL, Kockler DR. Topiramate for alcohol dependence. Ann Pharmacother. 2008;42:1475–1480. doi: 10.1345/aph.1L157. [DOI] [PubMed] [Google Scholar]
- 113.Gabriel KI, Cunningham CL. Effects of topiramate on ethanol and saccharin consumption and preferences in C57BL/6J mice. Alcohol Clin Exp Res. 2005;29:75–80. doi: 10.1097/01.alc.0000150014.79657.64. [DOI] [PubMed] [Google Scholar]
- 114.Farook JM, Lewis B, Littleton JM, Barron S. Topiramate attenuates the stress-induced increase in alcohol consumption and preference in male C57BL/6J mice. Physiol Behav. 2009;96:189–193. doi: 10.1016/j.physbeh.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 115.Gremel CM, Gabriel KI, Cunningham CL. Topiramate does not affect the acquisition or expression of ethanol conditioned place preference in DBA/2J or C57BL/6J mice. Alcohol Clin Exp Res. 2006;30:783–790. doi: 10.1111/j.1530-0277.2006.00091.x. [DOI] [PubMed] [Google Scholar]
- 116.Di Ciano P, Everitt BJ. Dissociable effects of antagonism of NMDA and AMPA/KA receptors in the nucleus accumbens core and shell on cocaine-seeking behavior. Neuropsychopharmacology. 2001;25:341–360. doi: 10.1016/S0893-133X(01)00235-4. [DOI] [PubMed] [Google Scholar]
- 117.Zhao Y, et al. Activation of group II metabotropic glutamate receptors attenuates both stress and cue-induced ethanol-seeking and modulates c-fos expression in the hippocampus and amygdala. J Neurosci. 2006;26:9967–9974. doi: 10.1523/JNEUROSCI.2384-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schroeder JP, et al. Cue-induced reinstatement of alcohol-seeking behavior is associated with increased ERK1/2 phosphorylation in specific limbic brain regions: blockade by the mGluR5 antagonist MPEP. Neuropharmacology. 2008;55:546–554. doi: 10.1016/j.neuropharm.2008.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Backstrom P, Hyytia P. Ionotropic and metabotropic glutamate receptor antagonism attenuates cue-induced cocaine seeking. Neuropsychopharmacology. 2006;31:778–786. doi: 10.1038/sj.npp.1300845. [DOI] [PubMed] [Google Scholar]
- 120.Haney M. Self-administration of cocaine, cannabis and heroin in the human laboratory: benefits and pitfalls. Addict Biol. 2009;14:9–21. doi: 10.1111/j.1369-1600.2008.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mello NK, Mendelson JH, Kuehnle JC, Sellers MS. Operant analysis of human heroin self-administration and the effects of naltrexone. J Pharmacol Exp Ther. 1981;216:45–54. [PubMed] [Google Scholar]
- 122.Comer SD, Collins ED, Fischman MW. Choice between money and intranasal heroin in morphine-maintained humans. Behav Pharmacol. 1997;8:677–690. doi: 10.1097/00008877-199712000-00002. [DOI] [PubMed] [Google Scholar]
- 123.Vocci FJ, Elkashef A. Pharmacotherapy and other treatments for cocaine abuse and dependence. Curr Opin Psychiatry. 2005;18:265–270. doi: 10.1097/01.yco.0000165596.98552.02. [DOI] [PubMed] [Google Scholar]
- 124.Logan GD. In: Inhibitory Processes in Attention, Memory and Language. Dagenbach D, Carr TH, editors. Academic Press; San Diego: 1994. pp. 189–236. [Google Scholar]
- 125.Dole VP. Implications of methadone maintenance for theories of narcotic addiction. J Am Med Assoc. 1988;260:3025–3029. [PubMed] [Google Scholar]
- 126.Haney M. Marijuana withdrawal in humans: effects of oral THC or divalproex. Neuropsychopharmacology. 2004;29:158–170. doi: 10.1038/sj.npp.1300310. [DOI] [PubMed] [Google Scholar]
- 127.Evans DE, Drobes DJ. Nicotine self-medication of cognitive-attentional processing. Addict Biol. 2009;14:32–42. doi: 10.1111/j.1369-1600.2008.00130.x. [DOI] [PubMed] [Google Scholar]
- 128.Kassel JD, Unrod M. Smoking, anxiety, and attention: support for the role of nicotine in attentionally mediated anxiolysis. J Abnorm Psychol. 2000;109:161–166. doi: 10.1037//0021-843x.109.1.161. [DOI] [PubMed] [Google Scholar]
- 129.Koob GF, Kreek MJ. Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry. 2007;164:1149–1159. doi: 10.1176/appi.ajp.2007.05030503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Drobes DJ, Anton RF, Thomas SE, Voronin K. A clinical laboratory paradigm for evaluating medication effects on alcohol consumption: naltrexone and nalmefene. Neuropsychopharmacology. 2003;28:755–764. doi: 10.1038/sj.npp.1300101. [DOI] [PubMed] [Google Scholar]
- 131.Krishnan-Sarin S, Krystal JH, Shi J, Pittman B, O’Malley SS. Family history of alcoholism influences naltrexone-induced reduction in alcohol drinking. Biol Psychiatry. 2007;62:694–697. doi: 10.1016/j.biopsych.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 132.Chornock WM, Stitzer ML, Gross J, Leischow S. Experimental model of smoking re-exposure: effects on relapse. Psychopharmacology. 1992;108:495–500. doi: 10.1007/BF02247427. [DOI] [PubMed] [Google Scholar]
- 133.Carter BL, Tiffany ST. Meta-analysis of cue-reactivity in addiction research. Addiction. 1999;94:327–340. [PubMed] [Google Scholar]
- 134.Monti PM, et al. Naltrexone’s effect on cue-elicited craving among alcoholics in treatment. Alcohol Clin Exp Res. 1999;23:1386–1394. [PubMed] [Google Scholar]
- 135.Cooney NL, Litt MD, Morse PA, Bauer LO, Gaupp L. Alcohol cue reactivity, negative-mood reactivity, and relapse in treated alcoholic men. J Abnorm Psychol. 1997;106:243–250. doi: 10.1037//0021-843x.106.2.243. [DOI] [PubMed] [Google Scholar]
- 136.Miranda R., Jr Effects of topiramate on urge to drink and the subjective effects of alcohol: a preliminary laboratory study. Alcohol Clin Exp Res. 2008;32:489–497. doi: 10.1111/j.1530-0277.2007.00592.x. [DOI] [PubMed] [Google Scholar]
- 137.Shiffman S, et al. Efficacy of acute administration of nicotine gum in relief of cue-provoked cigarette craving. Psychopharmacology. 2003;166:343–350. doi: 10.1007/s00213-002-1338-1. [DOI] [PubMed] [Google Scholar]
- 138.Hersh D, Bauer LO, Kranzler HR. Carbamazepine and cocaine-cue reactivity. Drug Alcohol Depend. 1995;39:213–221. doi: 10.1016/0376-8716(95)01165-3. [DOI] [PubMed] [Google Scholar]
- 139.Robbins SJ, Ehrman RN, Childress AR, O’Brien CP. Using cue reactivity to screen medications for cocaine abuse: a test of amantadine hydrochloride. Addict Behav. 1992;17:491–499. doi: 10.1016/0306-4603(92)90009-k. [DOI] [PubMed] [Google Scholar]
- 140.Marlatt G, Gordon J. In: Behavioral Medicine: Changing Health Lifestyles. Davidson P, Davidson S, editors. Brunner/Mazel; New York: 1980. pp. 410–452. [Google Scholar]
- 141.Childress AR, et al. Can induced moods trigger drug-related responses in opiate abuse patients? J Subst Abuse Treat. 1994;11:17–23. doi: 10.1016/0740-5472(94)90060-4. [DOI] [PubMed] [Google Scholar]
- 142.Sinha R, Fuse T, Aubin LR, O’Malley SS. Psychological stress, drug-related cues and cocaine craving. Psychopharmacology. 2000;152:140–148. doi: 10.1007/s002130000499. [DOI] [PubMed] [Google Scholar]
- 143.Lang PJ, Kozak MJ, Miller GA, Levin DN, McLean A., Jr Emotional imagery: conceptual structure and pattern of somato-visceral response. Psychophysiology. 1980;17:179–192. doi: 10.1111/j.1469-8986.1980.tb00133.x. [DOI] [PubMed] [Google Scholar]
- 144.Fox HC, Bergquist KL, Hong KI, Sinha R. Stress-induced and alcohol cue-induced craving in recently abstinent alcohol-dependent individual. Alcohol Clin Exp Res. 2007;31:395–403. doi: 10.1111/j.1530-0277.2006.00320.x. [DOI] [PubMed] [Google Scholar]
- 145.Sinha R, Garcia M, Paliwal P, Kreek MJ, Rounsaville BJ. Stress-induced cocaine craving and hypothalamic-pituitary-adrenal responses are predictive of cocaine relapse outcomes. Arch Gen Psychiatry. 2006;63:324–331. doi: 10.1001/archpsyc.63.3.324. [DOI] [PubMed] [Google Scholar]
- 146.Breese GR, Overstreet DH, Knapp DJ, Navarro M. Prior multiple ethanol withdrawals enhance stress-induced anxiety-like behavior: inhibition by CRF1-and benzodiazepine-receptor antagonists and a 5-HT1a-receptor agonist. Neuropsychopharmacology. 2005;30:1662–1669. doi: 10.1038/sj.npp.1300706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.al’Absi M, Hatsukami D, Davis GL. Attenuated adrenocorticotropic responses to psychological stress are associated with early smoking relapse. Psychopharmacology. 2005;181:107–117. doi: 10.1007/s00213-005-2225-3. [DOI] [PubMed] [Google Scholar]
- 148.Sinha R, Kimmerling A, Doebrick C, Kosten TR. Effects of lofexidine on stress-induced and cue-induced opioid craving and opioid abstinence rates: preliminary findings. Psychopharmacology. 2007;190:569–574. doi: 10.1007/s00213-006-0640-8. [DOI] [PubMed] [Google Scholar]
- 149.Mason BJ, Light JM, Escher T, Drobes DJ. Effect of positive and negative affective stimuli and beverage cues on measures of craving in non treatment-seeking alcoholics. Psychopharmacology. 2008;200:141–150. doi: 10.1007/s00213-008-1192-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.McKee SA. Developing human laboratory models of smoking lapse behavior for medication screening. Addict Biol. 2009;14:99–107. doi: 10.1111/j.1369-1600.2008.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Braus DF, et al. Alcohol-associated stimuli activate the ventral striatum in abstinent alcoholics. J Neural Transm. 2001;108:887–894. doi: 10.1007/s007020170038. [DOI] [PubMed] [Google Scholar]
- 152.Grusser SM, et al. Cue-induced activation of the striatum and medial prefrontal cortex is associated with subsequent relapse in abstinent alcoholics. Psychopharmacology. 2004;175:296–302. doi: 10.1007/s00213-004-1828-4. [DOI] [PubMed] [Google Scholar]
- 153.Wrase J, et al. Dysfunction of reward processing correlates with alcohol craving in detoxified alcoholics. Neuroimage. 2007;35:787–794. doi: 10.1016/j.neuroimage.2006.11.043. [DOI] [PubMed] [Google Scholar]
- 154.Bond C, et al. Single-nucleotide polymorphism in the human mu opioid receptor gene alters β-endorphin binding and activity: possible implications for opiate addiction. Proc Natl Acad Sci USA. 1998;95:9608–9613. doi: 10.1073/pnas.95.16.9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Treutlein J, et al. Genetic association of the human corticotropin releasing hormone receptor 1 (CRHR1) with binge drinking and alcohol intake patterns in two independent samples. Mol Psychiatry. 2006;11:594–602. doi: 10.1038/sj.mp.4001813. [DOI] [PubMed] [Google Scholar]
- 156.Garbutt JC, et al. Efficacy and tolerability of long-acting injectable naltrexone for alcohol dependence: a randomized controlled trial. J Am Med Assoc. 2005;293:1617–1625. doi: 10.1001/jama.293.13.1617. errata 293,1978, 2864 (2005) [DOI] [PubMed] [Google Scholar]
- 157.Huestis MA, Cone EJ. Differentiating new marijuana use from residual drug excretion in occasional marijuana users. J Anal Toxicol. 1998;22:445–454. doi: 10.1093/jat/22.6.445. [DOI] [PubMed] [Google Scholar]
- 158.Sobell LC, Sobell MB. In: Measuring Alcohol Consumption: Psychosocial and Biochemical Methods. Litten RZ, Allen JP, editors. Human Press; Totowa: 1992. pp. 41–72. [Google Scholar]
- 159.Office of National Drug Control Policy. The Economic Costs of Drug Abuse in the United States: 1992–2002. Office of National Drug Control Policy; Washington DC: 2004. [Google Scholar]
- 160.National Institute on Alcohol Abuse and Alcoholism. 10th special report to the US Congress on alcohol and health: highlights from current research. National Institute on Alcohol Abuse and Alcoholism; Bethesda: 2000. [Google Scholar]
- 161.Centers for Disease Control and Prevention. Annual smoking-attributable mortality, years of potential life lost, and productivity losses: United States, 1997–2001. Morb Mort Wkly Rep. 2005;54:625–628. [PubMed] [Google Scholar]
- 162.O’Brien CP, McLellan AT. Myths about the treatment of addiction. Lancet. 1996;347:237–240. doi: 10.1016/s0140-6736(96)90409-2. [DOI] [PubMed] [Google Scholar]
- 163.Lotsch J, Geisslinger G. Are μ-opioid receptor polymorphisms important for clinical opioid therapy? Trends Mol Med. 2005;11:82–89. doi: 10.1016/j.molmed.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 164.Ray LA, Hutchison KE. A polymorphism of the mu-opioid receptor gene (OPRM1) and sensitivity to the effects of alcohol in humans. Alcohol Clin Exp Res. 2004;28:1789–1795. doi: 10.1097/01.alc.0000148114.34000.b9. [DOI] [PubMed] [Google Scholar]
- 165.Anton RF, et al. An evaluation of μ-opioid receptor (OPRM1) as a predictor of naltrexone response in the treatment of alcohol dependence: results from the Combined Pharmacotherapies and Behavioral Interventions for Alcohol Dependence (COMBINE study) Arch Gen Psychiatry. 2008;65:135–144. doi: 10.1001/archpsyc.65.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.van der Zwaluw CS, et al. Polymorphisms in the μ-opioid receptor gene (OPRM1) and the implications for alcohol dependence in humans. Pharmacogenomics. 2007;8:1427–1436. doi: 10.2217/14622416.8.10.1427. [DOI] [PubMed] [Google Scholar]
- 167.Hernandez-Avila CA, Wand G, Luo X, Gelernter J, Kranzler HR. Association between the cortisol response to opioid blockade and the Asn40Asp polymorphism at the mu-opioid receptor locus (OPRM1) Am J Med Gen B Neuropsych Genet. 2003;118B:60–65. doi: 10.1002/ajmg.b.10054. [DOI] [PubMed] [Google Scholar]
- 168.Blomeyer D, et al. Interaction between CRHR1 gene and stressful life events predicts adolescent heavy alcohol use. Biol Psychiatry. 2008;63:146–151. doi: 10.1016/j.biopsych.2007.04.026. [DOI] [PubMed] [Google Scholar]
- 169.Hansson AC, et al. Variation at the rat Crhr1 locus and sensitivity to relapse into alcohol seeking induced by environmental stress. Proc Natl Acad Sci USA. 2006;103:15236–15241. doi: 10.1073/pnas.0604419103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sommer WH, et al. Upregulation of voluntary alcohol intake, behavioral sensitivity to stress, and amygdala crhr1 expression following a history of dependence. Biol Psychiatry. 2008;63:139–145. doi: 10.1016/j.biopsych.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 171.Koob GF, Everitt BJ, Robbins TW. In: Fundamental Neuroscience. 3. Squire LG, et al., editors. Academic Press; Amsterdam: 2008. pp. 987–1016. [Google Scholar]
- 172.Collins RJ, Weeks JR, Cooper MM, Good PI, Russell RR. Prediction of abuse liability of drugs using IV self-administration by rats. Psychopharmacology. 1984;82:6–13. doi: 10.1007/BF00426372. [DOI] [PubMed] [Google Scholar]
- 173.Kornetsky C, Bain G. In: Modern Methods in Pharmacology: Testing and Evaluation of Drugs of Abuse. Adler MW, Cowan A, editors. Vol. 6. Wiley-Liss; New York: 1990. pp. 211–231. [Google Scholar]
- 174.Tornatzky W, Miczek KA. Cocaine self-administration “binges”: transition from behavioral and autonomic regulation toward homeostatic dysregulation in rats. Psychopharmacology. 2000;148:289–298. doi: 10.1007/s002130050053. [DOI] [PubMed] [Google Scholar]
- 175.Ahmed SH, Koob GF. Transition from moderate to excessive drug intake: change in hedonic set point. Science. 1998;282:298–300. doi: 10.1126/science.282.5387.298. [DOI] [PubMed] [Google Scholar]
- 176.Kelly TH, Foltin RW, Emurian CS, Fischman MW. Are choice and self-administration of marijuana related to Δ9-THC content? Exp Clin Psychopharmacol. 1997;5:74–82. doi: 10.1037//1064-1297.5.1.74. [DOI] [PubMed] [Google Scholar]
- 177.Haney M, Foltin RW, Fischman MW. Effects of pergolide on intravenous cocaine self-administration in men and women. Psychopharmacology. 1998;137:15–24. doi: 10.1007/s002130050588. [DOI] [PubMed] [Google Scholar]
- 178.de Wit H. Impulsivity as a determinant and consequence of drug use: a review of underlying processes. Addict Biol. 2009;14:22–31. doi: 10.1111/j.1369-1600.2008.00129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Rachlin H, Green L. Commitment, choice and self-control. J Exp Anal Behav. 1972;17:15–22. doi: 10.1901/jeab.1972.17-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Leth-Steensen C, Elbaz ZK, Douglas VI. Mean response times, variability, and skew in the responding of ADHD children: a response time distributional approach. Acta Psychol. 2000;104:167–190. doi: 10.1016/s0001-6918(00)00019-6. [DOI] [PubMed] [Google Scholar]
- 181.Schulteis G, Yackey M, Risbrough V, Koob GF. Anxiogenic-like effects of spontaneous and naloxone-precipitated opiate withdrawal in the elevated plus-maze. Pharmacol Biochem Behav. 1998;60:727–731. doi: 10.1016/s0091-3057(98)00034-3. [DOI] [PubMed] [Google Scholar]
- 182.Tzschentke TM. Measuring reward with the conditioned place preference paradigm: a comprehensive review of drug effects, recent progress and new issues. Prog Neurobiol. 1998;56:613–672. doi: 10.1016/s0301-0082(98)00060-4. [DOI] [PubMed] [Google Scholar]
- 183.Markou A, Kosten TR, Koob GF. Neurobiological similarities in depression and drug dependence: a self-medication hypothesis. Neuropsychopharmacology. 1998;18:135–174. doi: 10.1016/S0893-133X(97)00113-9. [DOI] [PubMed] [Google Scholar]
- 184.Ahmed SH, Walker JR, Koob GF. Persistent increase in the motivation to take heroin in rats with a history of drug escalation. Neuropsychopharmacology. 2000;22:413–421. doi: 10.1016/S0893-133X(99)00133-5. [DOI] [PubMed] [Google Scholar]
- 185.Roberts AJ, Heyser CJ, Cole M, Griffin P, Koob GF. Excessive ethanol drinking following a history of dependence: animal model of allostasis. Neuropsychopharmacology. 2000;22:581–594. doi: 10.1016/S0893-133X(99)00167-0. [DOI] [PubMed] [Google Scholar]
- 186.Kitamura O, Wee S, Specio SE, Koob GF, Pulvirenti L. Escalation of methamphetamine self-administration in rats: a dose-effect function. Psychopharmacology. 2006;186:48–53. doi: 10.1007/s00213-006-0353-z. [DOI] [PubMed] [Google Scholar]
- 187.O’Dell LE, Koob GF. “Nicotine deprivation effect” in rats with intermittent 23-hour access to intravenous nicotine self-administration. Pharmacol Biochem Behav. 2007;86:346–353. doi: 10.1016/j.pbb.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hjalmarson AI. Effect of nicotine chewing gum in smoking cessation: a randomized, placebo-controlled, double-blind study. J Am Med Assoc. 1984;252:2835–2838. [PubMed] [Google Scholar]
- 189.Haney M. The marijuana withdrawal syndrome: diagnosis and treatment. Curr Psychiatry Rep. 2005;7:360–366. doi: 10.1007/s11920-005-0036-1. [DOI] [PubMed] [Google Scholar]
- 190.Rusted JM, Caulfield D, King L, Goode A. Moving out of the laboratory: does nicotine improve everyday attention? Behav Pharmacol. 2000;11:621–629. doi: 10.1097/00008877-200011000-00009. [DOI] [PubMed] [Google Scholar]
- 191.Lawrence NS, Ross TJ, Stein EA. Cognitive mechanisms of nicotine on visual attention. Neuron. 2002;36:539–548. doi: 10.1016/s0896-6273(02)01004-8. [DOI] [PubMed] [Google Scholar]
- 192.Giessing C, Thiel CM, Rösler F, Fink GR. The modulatory effects of nicotine on parietal cortex activity in a cued target detection task depend on cue reliability. Neuroscience. 2006;137:853–864. doi: 10.1016/j.neuroscience.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 193.Christensen JK, Moller IW, Ronsted P, Angelo HR, Johansson B. Dose-effect relationship of disulfiram in human volunteers. I: Clinical studies. Pharmacol Toxicol. 1991;68:163–165. doi: 10.1111/j.1600-0773.1991.tb01215.x. [DOI] [PubMed] [Google Scholar]
- 194.Goedde HW, Agarwal DP, Harada S. In: Isozymes: Current Topics in Biological and Medical Research: Cellular Localization, Metabolism, and Physiology. Rattazzi MC, Scandalios JG, Whitt GS, editors. Vol. 8. Liss; New York: 1983. pp. 175–193. [Google Scholar]
- 195.Volpicelli JR, Alterman AI, Hayashida M, O’Brien CP. Naltrexone in the treatment of alcohol dependence. Arch Gen Psychiatry. 1992;49:876–880. doi: 10.1001/archpsyc.1992.01820110040006. [DOI] [PubMed] [Google Scholar]
- 196.O’Malley SS, et al. Naltrexone and coping skills therapy for alcohol dependence: a controlled study. Arch Gen Psychiatry. 1992;49:881–887. doi: 10.1001/archpsyc.1992.01820110045007. [DOI] [PubMed] [Google Scholar]
- 197.Hurt RD, et al. A comparison of sustained-release bupropion and placebo for smoking cessation. N Engl J Med. 1997;337:1195–1202. doi: 10.1056/NEJM199710233371703. [DOI] [PubMed] [Google Scholar]
- 198.Mason BJ. Acamprosate and naltrexone treatment for alcohol dependence: an evidence-based risk-benefits assessment. Eur Neuropsychopharmacol. 2003;13:469–475. doi: 10.1016/j.euroneuro.2003.08.009.. This paper provides an evidence-based risk–benefit assessment of acamprosate and naltrexone in the treatment of alcohol dependence. The safety of the two drugs in combination is supported by two independent double-blind studies, suggesting that combination treatment is a realistic goal.
- 199.Mason BJ, Goodman AM, Chabac S, Lehert P. Effect of oral acamprosate on abstinence in patients with alcohol dependence in a double-blind, placebo-controlled trial: the role of patient motivation. J Psychiatr Res. 2006;40:383–393. doi: 10.1016/j.jpsychires.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 200.Jorenby DE, et al. Efficacy of varenicline, an α4β2 nicotinic acetylcholine receptor partial agonist, vs placebo or sustained-release bupropion for smoking cessation: a randomized controlled trial. J Am Med Assoc. 2006;296:56–63. doi: 10.1001/jama.296.1.56. erratum 296,1355 (2006) [DOI] [PubMed] [Google Scholar]
- 201.Altshuler HL, Phillips PE, Feinhandler DA. Alteration of ethanol self-administration by naltrexone. Life Sci. 1980;26:679–688. doi: 10.1016/0024-3205(80)90257-x. [DOI] [PubMed] [Google Scholar]
- 202.Heyser CJ, Moc K, Koob GF. Effects of naltrexone alone and in combination with acamprosate on the alcohol deprivation effect in rats. Neuropsychopharmacology. 2003;28:1463–1471. doi: 10.1038/sj.npp.1300175. [DOI] [PubMed] [Google Scholar]
- 203.Colombo G, et al. Ability of baclofen in reducing alcohol intake and withdrawal severity: I. Preclinical evidence. Alcohol Clin Exp Res. 2000;24:58–66. [PubMed] [Google Scholar]
- 204.Backstrom P, Hyytia P. Suppression of alcohol self-administration and cue-induced reinstatement of alcohol seeking by the mGlu2/3 receptor agonist LY379268 and the mGlu8 receptor agonist (S)-3, 4-DCPG. Eur J Pharmacol. 2005;528:110–118. doi: 10.1016/j.ejphar.2005.10.051. [DOI] [PubMed] [Google Scholar]
- 205.Le Magnen J, Tran G, Durlach J, Martin C. Dose-dependent suppression of the high alcohol intake of chronically intoxicated rats by Ca-acetyl homotaurinate. Alcohol. 1987;4:97–102. doi: 10.1016/0741-8329(87)90005-x. [DOI] [PubMed] [Google Scholar]
- 206.Liu X, Weiss F. Additive effect of stress and drug cues on reinstatement of ethanol seeking: exacerbation by history of dependence and role of concurrent activation of corticotropin-releasing factor and opioid mechanisms. J Neurosci. 2002;22:7856–7861. doi: 10.1523/JNEUROSCI.22-18-07856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Bachteler D, Economidou D, Danysz W, Ciccocioppo R, Spanagel R. The effects of acamprosate and neramexane on cue-induced reinstatement of ethanol-seeking behavior in rat. Neuropsychopharmacology. 2005;30:1104–1110. doi: 10.1038/sj.npp.1300657. [DOI] [PubMed] [Google Scholar]
- 208.Maccioni P, Bienkowski P, Carai MA, Gessa GL, Colombo G. Baclofen attenuates cue-induced reinstatement of alcohol-seeking behavior in Sardinian alcohol-preferring (sP rats) Drug Alcohol Depend. 2008;95:284–287. doi: 10.1016/j.drugalcdep.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 209.Le AD, Harding S, Juzytsch W, Funk D, Shaham Y. Role of alpha-2 adrenoceptors in stress-induced reinstatement of alcohol seeking and alcohol self-administration in rats. Psychopharmacology. 2005;179:366–373. doi: 10.1007/s00213-004-2036-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.