

Abstract
The human disease von Recklinghausen's neurofibromatosis (Nf1) is one of the most common genetic disorders. It is caused by mutations in the NF1 tumor suppressor gene, which encodes a GTPase activating protein (GAP) that negatively regulates p21-RAS signaling. Dermal and plexiform neurofibromas as well as malignant peripheral nerve sheath tumors and other malignant tumors, are significant complications in Nf1. Neurofibromas are complex tumors and composed mainly of abnormal local cells including Schwann cells, endothelial cells, fibroblasts and additionally a large number of infiltrating inflammatory mast cells. Recent work has indicated a role for the microenvironment in plexiform neurofibroma genesis. The emerging evidence points to mast cells as crucial contributors to neurofibroma tumorigenesis. Therefore, further understanding of the molecular interactions between Schwann cells and their environment will provide tools to develop new therapies aimed at delaying or preventing tumor formation in Nf1 patients.
Keywords: neurofibromin, tumor microenvironment, mast cell, neurofibroma, NF1, GAP
von Recklinghausen's Neurofibromatosis Type I
Neurofibromatosis type I (Nf1) was first recorded by Tilesius in 1793 and described in organized detail by the German pathologist Friedrich Daniel von Recklinghausen in 1882. Fifty years later, the Viennese ophthalmologist Lisch described the presence of iris nodules, which are now an important criterion for the clinical diagnosis of Nf1 (Riccardi, 1992). It is one of the most common human genetic diseases. It has a de novo incidence of one in 3000–4000 individuals and affects male and female subjects equally in all races (Szudek et al., 2000; Trovo-Marqui and Tajara, 2006). Although it is inherited as an autosomal-dominant trait with essentially 100% penetrance, spontaneous mutations occur in 50% of cases (Yohay, 2006). This means the disease will likely remain in the human population and will have increasing incidence over time. Nf1 patients have defects in neural crest-derived tissues, leading to a wide spectrum of clinical presentations, including developmental, pigment and neoplastic aberrations (Cichowski and Jacks, 2001; Zhu et al., 2001). The cardinal features of Nf1 are café au lait macules, axillary and groin freckling, combined with multiple peripheral and central nerve tumors. They also exhibit a less penetrant variety of additional pathologies of the skin, nervous system, bones, endocrine organs, blood vessels and the eyes (Ward and Gutmann, 2005) (Table 1).
Table 1.
Key clinical features of Neurofibromatosis type I
| Skin | Neurologic |
| Café au lait macules | UBO on MRI |
| Axillary and inguinal freckling | Learning disabilities |
| Dermal neurofibroma | Seizure |
| Plexiform neurofibroma | Mental retardation |
| Juvenile xanthogranuloma | Aqueduct sternosis |
| Neoplasia | Skeletal |
| Optic glioma | Macrocephaly |
| MPNST | Sphenoid wing dysplasia |
| Pheochromocytoma | Scoliosis |
| Juvenile chronic myelogenous leukemia | Spinal bifida |
| Other CNS tumors (astrocytoma) | Pseudoarthrosis |
| Rhabdomyosarcoma | Thinning of long bone cortex |
| Duodenal carcinoid | Absence of patella |
| Somatostatinoma | Vertebral disc dysplasia |
| Parathyroid adenoma | Short stature |
| Cardiovascular | Eyes |
| Hypertension | Lisch nodules |
| Pulmonic sternosis | Hyperterrorism |
| Renal artery sternosis | Glaucoma |
| Gastrointestinal | Psychiatric |
| Constipation | Heavy psychosocial burden |
Abbreviations: CNS, central nervous system; MPNST; malignant peripheral nerve sheath tumor; UBO: unidentified bright object.
Biology of Neurofibromin
The NF1 gene is located on human chromosome 17q11.2 and was identified by positional cloning in 1990 (Ballester et al., 1990; Xu et al., 1990). It spans over 350 kb of genomic DNA, and has at least 60 exons (Jentarra et al., 2006). It encodes a tumor suppressor, known as Neurofibromin (NF1), which is ubiquitously expressed but most abundant in neurons, Schwann cells, astrocytes, oligodendrocytes and leukocytes (Gutmann et al., 1991; Daston et al., 1992). Despite its large size, very little is known about its function. Previous studies have shown a variety of human mutations associated with Nf1 across the entire gene (Castle et al., 2003; Jentarra et al., 2006). The majority of NF1 mutations predict truncations in the protein, and there is no clear correlation between specific mutation and clinical presentations (Castle et al., 2003). To date, it is known to have two functional domains, Sec14 and RasGAP (Trovo-Marqui and Tajara, 2006) (Figure 1). Sec14-interactive domain is located between amino acids 1545–1816 and is homologous to the yeast Sec14p, which is known to regulate intracellular proteins and lipid trafficking in yeast (Mousley et al., 2006). However, the biological role of Sec14 domain in NF1 is currently unknown.
Figure 1.
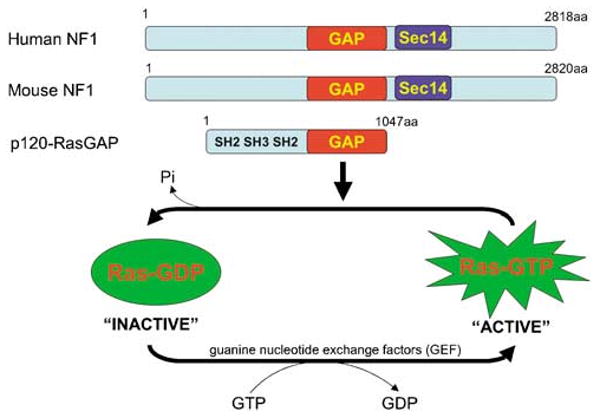
Functional domains of NF1. Mouse NF1 is more than 98.5% identical to its human homologue. Both contain the p120Ras-GRD, which accelerates the conversion of the active, GTP-bound Ras into its inactive GDP-bound form.
The RasGAP-related domain (Ras-GRD) in NF1 spans between amino acids 1125–1537 and is corresponding to exons 20–27a (Ballester et al., 1990; Trovo-Marqui and Tajara, 2006). It accelerates the conversion of the active, GTP-bound Ras into its inactive GDP-bound form (Figure 1). Ras is activated at the plasma membrane upon binding of growth factor receptors to specific ligands, triggering the recruitment of a complex containing the adapter protein growth factor receptor bound protein 2 (Grb2) and the Ras guanine nucleotide exchange factor Sos to the site of receptor tyrosine kinase activation. Here, Ras is catalysed to switch to its GTP-bound state. This active form of Ras then binds and activates the kinase Raf and phosphatidylinositol 3′-kinase (PI3K), which then sets off a kinase cascade, culminating the activation of mitogen-activated protein kinases (MAPK)/mitogen-activated protein kinases kinase and PI3K pathways. Some of these signals are then transmitted to the nucleus, regulating the expression of genes controlling cell proliferation, cell death, differentiation and migration (Figure 2). In fact, it is established that constitutively active mutations of Ras are frequent and associated with multiple human cancers as a result of permanent stimulation of the Raf–MAPK and/or PI3K signaling cascades that lead to uncontrolled cell proliferation and escape of apoptosis (Weiss et al., 1999).
Figure 2.
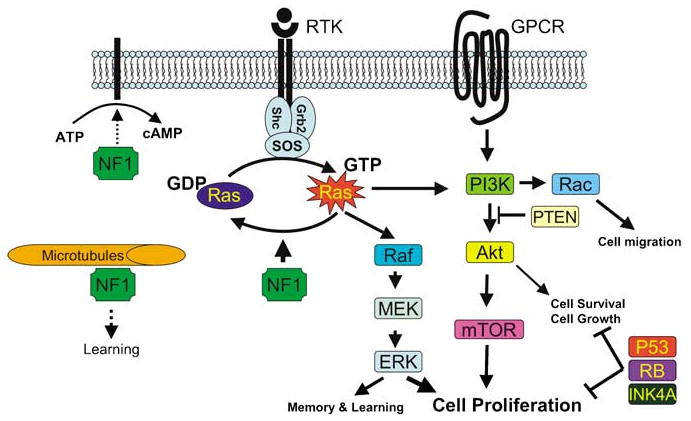
Representation of NF1 interactions with the Ras and PI3K pathways. NF1 constrains Ras activity in the normal cell. Therefore, loss of NF1 expression leads to elevated Ras activity, dysregulated cell growth and tumorigenesis. NF1 may also associate with microtubules and modulate the cAMP-PKA signaling pathway.
NF1, via its Ras-GRD, exerts a reverse effect on Ras by increasing the GTP hydrolysis rate. Therefore, its function as a tumor suppressor is believed to occur by constraining Ras activity in the normal cell. It is this biological property of NF1 that is believed to be the major key to pathophysiologic mechanisms underlying the clinical presentations of NF1 mutations in both mice and humans, ranging from learning disability to malignant tumors (Costa et al., 2002). In fact, beside the tuberous sclerosis complex, NF1 is the only other mammalian RasGAP that is known to cause tumor predisposition disorder in humans.
Li et al. (2005) reported that the learning deficits in NF1+/− mice can be rescued by genetic and pharmacologic manipulations that decrease Ras function. Previous studies by this group and others have shown that Ras activity and its downstream effectors are elevated in the cortex and hippocampus of NF1+/− mice, leading to impairments in long-term potentiation, which is a key cellular apparatus of learning and memory (Costa et al., 2002; Li et al., 2005). When NF1+/− mice were crossed with the K-ras+/− heterozygote to reduce the level of K-ras expression by half, the NF1+/−/K-ras+/− mice perform as well as the wild-type mice in cognitive functions. In addition, the learning deficits in the NF1+/− mice were also rescued when they were treated with either farnesyl-transferase inhibitor, which blocks a key post-translational modification essential for Ras function, or lovastatin, a known Ras isoprenylation inhibitor (Costa et al., 2002; Li et al., 2005). These results indicate that increased Ras activity, as a consequence of NF1 mutations, is at least partially responsible for the learning and memory disabilities seen in NF1 mutant mice and by analogy, suggest a link to the intellectual deficits described in patients. These learning deficits may also relate to the ability of NF1 to associate with microtubules, which are expressed at high level in axonal and dendritic processes of neurons. Gregory et al. (1993) and Xu and Gutmann (1997) have shown that the region of NF1 that is critical for this interaction resides within the Ras-GRD. Although microtubules may be important for neuronal connection and neurite outgrowth, the exact function of NF1 relative to microtubules is currently unknown.
There is increasing evidence indicating that NF1 maybe involved in other cellular functions besides Ras regulation. Indeed, there exist multiple mutation sites outside of the NF1 Ras-GRD identified in Nf1 patients (Castle et al., 2003; Jentarra et al., 2006). In addition, Ismat et al. (2006) showed that mice expressing the NF1 GRD only partially rescue the phenotypes seen in NF1−/− mice, suggesting that other regions of NF1, besides the Ras-GRD, are also critical for NF1 function. This group has engineered a mouse line where they inserted the HA-tagged NF1 GRD (HA-GRD) coding sequence under floxed PGK-Neo into the Rosa26 locus. They then generated compound mice that have CMV-Cre; HA-GRD; NF1−/−, in which they showed that Ras activity in these mice has returned to wild-type level by the expression of the HA-GRD. Although NF1−/− mice are lethal at E13.5 because of cardiac defect, the CMV-Cre; HA-GRD; NF1−/− mice survive to birth with normal cardiac anatomy. However, these mice succumb to death at perinatal period because of overgrowth of neural crest-derived tissues (Ismat et al., 2006). These findings could be interpreted to indicate that not all of the phenotypes seen in NF1 mutations are solely secondary to Ras dysregulation. However, the precise regulation of NF1 activity is not mimicked in the knock-in mice and, therefore, a ras dysregulation could persist in these mice.
In another instance, studies in Drosophila, where a highly conserved homologue of NF1 is present, have implicated the involvement of NF1 in the cAMP pathway. Guo et al. (1997) observed that Drosophila NF1 is crucial for G protein-mediated activation of adenylyl cyclase, a key enzyme in the cAMP signaling pathway. In addition, The et al. (1997) showed that Drosophila homozygous for null mutation of NF1 have apparently normal Ras1-mediated signaling. However, these flies have reduced body size. This phenotype was rescued by increasing the expression of activated protein kinase A (PKA), an intermediate signaling molecule between cyclic adenosine monophosphate (cAMP) and BRAF-MAPK pathway. Subsequently, Tong et al. (2002) and Dasgupta et al. (2003) have reported that NF1 inactivation in mouse primary neuronal or astrocyte culture, respectively, results in decreased level of cAMP production in response to the neuropeptide pituitary adenylyl cyclase-activating polypeptide. Therefore, in addition to the Ras pathway, NF1 may also function upstream of PKA to modulate cell proliferation via the cAMP pathway. However, whether the PKA-pathway modulation is a ras-dependent or -independent event has not been fully resolved. Another remaining caveat is that the biological consequences of this interaction between NF1 and adenylyl cyclase may not conserved across species from fly to mouse to human.
NF1 and tumorigenesis
NF1 is a tumor suppressor gene that is inherited as an autosomal-dominant trait, suggesting a possible gene dosage effect. In fact, haploinsufficiency is apparently enough to bring about many of the clinical manifestations seen in Nf1 patients. However, consistent with the Knudson ‘two-hit’ model of tumorigenesis, loss of heterozygosity (LOH) is responsible for the formation of neurofibromas (Cichowski et al., 1999). On the other hand, the development of malignant cancers in Nf1 individuals requires further acquisition of additional genetic aberrations, whether it is inactivation of PTEN, TP53, CDKN2A or amplification of platelet-derived growth factor receptor or epidermal growth factor receptor (Zhu and Parada, 2002; Castle et al., 2003; Levy et al., 2004). Reminiscent with the malignancies seen in Nf1 patients, compound heterozygous mice for both NF1 and p53 develop malignant peripheral nerve sheath tumors with full penetrance (Cichowski et al., 1999; Vogel et al., 1999). In addition, as Nf1 has a spectrum of specific tumors, modifying genes and epigenetic phenomena have been shown to play a role in modulating Nf1-associated tumor susceptibility (Easton et al., 1993; Reilly et al., 2004).
Neurofibromas and malignant peripheral nerve sheath tumors
Neurofibromas are the most common tumor in Nf1 and classified into three subtypes. Dermal and subcutaneous forms appear at puberty and increase in number with age and during pregnancy. They occur as a result of proliferation of all supporting elements of the nerve fibers, including Schwann cells, perineurial cells, fibroblasts, blood vessels, as well as infiltration of mast cells. Plexiform neurofibromas occur in about 30% of Nf1 individuals and are virtually pathognomonic of the disease. They are congenital and progressively enlarge throughout life. Although dermal neurofibromas are most frequently benign, patients with plexiform neurofibromas have a 10% lifetime risk of developing malignant peripheral nerve sheath tumors (MPNST), which can metastases widely and are often signaling a fatal outcome (Lakkis and Tennekoon, 2000; Ferner, 2006). In addition, owing to their unusual capacity for growth, plexiform neurofibromas can be life threatening by their physical impairment of organ or neural function.
Research over the past decade using mouse models has greatly enhanced our knowledge of neurofibroma development and malignant progression in Nf1. Mice homozygous for NF1 mutation are embryonic lethal at E13.5 secondary to defective heart and malformation of the major cardiac outflow tracts. They also exhibit hyperplasia of neural crest-derived sympathetic ganglia (Brannan et al., 1994; Jacks et al., 1994). To the contrary, heterozygous NF1 mutant mice are viable and only have increased incidence of pheochromocytomas and myeloid leukemias beyond 10- to 12-month old (Parada, 2000). Puzzlingly, in contrast to the human condition, NF1+/− mice do not develop neurofibromas (Brannan et al., 1994; Jacks et al., 1994) (Figure 3). One possible explanation for this observation is that LOH necessary for neurofibroma development is impaired in mice. Perhaps, given the short time of gestation and lifespan and a smaller neural crest compartment compared with human, the NF1+/− mice do not have the necessary window of opportunity to undergo effective LOH in target cells to initiate neurofibroma formation. Cichowski et al. (1999) addressed this issue elegantly when they created chimeric mice by injecting LacZ-positive NF1−/− ES cells into wild-type C57BL/6 blastocysts. These mice developed microscopic neurofibromas derived from the injected ES cells, demonstrating the requirement of NF1 homozygosity for tumor formation. However, the degree of chimerism in these mice occurs randomly and cannot be controlled genetically. As a result, it is difficult to establish the target cell or whether other cell types contribute to the tumorigenesis.
Figure 3.
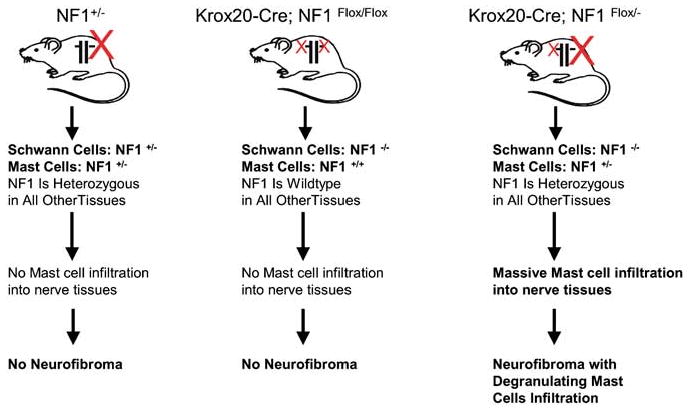
Schwann cell origin and the role of tumor microenvironment in neurofibroma formation. Both nullizygosity at NF1 locus in Schwann cells and haploinsufficiency of NF1 in the somatic tissue are required for neurofibroma tumorigenesis. In addition, the NF1-heterozygous mast cells infiltration into nerve tissues precedes development of NF1flox/−;Krox20-Cre plexiform neurofibromas, implying a unique affinity between NF1−/− Schwann cells and NF1+/− mast cells and a causal role for mast cells in tumor initiation.
To circumvent this problem and to develop a more precise mouse model for evaluating the evolution of Nf1-associated tumors, Cre/loxP technology that allows tissue-specific ablation of NF1 function has been adopted. Schwann cell-specific NF1-deficient mice were derived by crossing NF1flox/flox mice with the Krox20-cre transgenic mice, an embryonic Schwann cell-specific promoter (Zhu et al., 2002). Mice with a conditional knockout (KO) of NF1 only in embryonic Schwann cells, but wild-type in all other cell lineages (NF1flox/flox; Krox20-Cre), exhibit microscopic hyperplasia in sensory ganglia but do not develop neurofibromas. However, when mice homozygous for NF1 mutation (NF1−/−) in Schwann cells but heterozygous for NF1 (NF1+/−) in all other somatic cells (NF1flox/−; Krox20-Cre) were generated, these mice developed multiple classic plexiform neurofibromas with a massive degranulating mast cell infiltration, modeling human neurofibroma (Zhu et al., 2002) (Figure 3). These genetic studies implied the Schwann cell origin for neurofibroma. Nevertheless, in addition to nullizygosity at NF1 locus in Schwann cells, haploinsufficiency of NF1 in the tumor microenvironment is also required for the tumorigenesis.
The p53 and NF1 genes are linked on chromosome 17 in human and on chromosome 11 in mouse. To model malignant progression in human neurofibromatosis, two groups have generated mice that are compound heterozygous for both p53 and NF1 in cis configuration. These mice develop malignant peripheral nerve sheath tumor (MPNST) with full penetrant, recapitulated malignancies seen in Nf1 patients. DNA analysis of tumors reveals loss of both wild-type alleles, indicating that the loss of both genes orchestrate to cause MPNST development (Cichowski et al., 1999; Vogel et al., 1999).
Optic pathway gliomas
The second most common tumor in Nf1 is optic pathway gliomas, originally reportes in about 15% of Nf1 individuals although current imaging technology suggests that the incidence of non-pathologic tumors is considerably higher. These are low-grade but potentially debilitating pilocytic astrocytomas as they often arise along the optic nerves and chiasm (Arun and Gutmann, 2004). To elucidate the role of NF1 in astrocytoma tumorigensis, mice lacking NF1 in astrocytes and neural precursors have been generated using two different hGFAP-Cre transgenic lines (Bajenaru et al., 2002, 2003; Zhu et al., 2005). Glial fibrillary acidic protein (GFAP) is a marker for astrocyte and neural stem/progenitor cells. The hGFAP-Cre transgene drives Cre in both astrocytes and glial precursors and neurons adjacent to the retina. However, the hGFAP*-Cre transgene only drives Cre in astrocytes, not in glial precursor and neuron in the retina. Intercrossing the hGFAP-Cre transgene into NF1flox/flox or NF1flox/− backgrounds gives phenotypically identical offspring, termed NF1hGFAP KO. The NF1hGFAP KO mice develop neoplasia at the optic nerves reminiscent of human optic glioma, especially at areas adjacent to the retina. On the other hand, the NF1flox/flox; hGFAP*-Cre mutant mice did not develop glioma (Zhu et al., 2005). These results show that loss of NF1 in microglias and neurons in the surrounding adjacent environment may contribute to optic glioma tumorigenesis. In other studies, Bajenaru et al. (2002, 2003) did not observe astrocytoma formation in the NF1flox/flox; hGFAP*-Cre mice but did detect optic glioma in the NF1flox/−; hGFAP*-Cre mice, further stressing the importance of haploinsufficiency of NF1 in the tumor microenvironment for Nf1-associated glioma formation.
Other tumors
In addition to MPNST, Nf1 patients are predisposed to develop other malignancies. They include pheochromocytomas, juvenile chronic myelogenous leukemia, malignant astrocytoma, rhadomyosarcoma of the geni-to-urinary tract, duodenal carcinoid, somatostatinoma and parathyroid adenoma. The fact that not all Nf1 patients succumb to malignancy implies that other genetic and non-genetic factors greatly influence Nf1-associated tumor susceptibility (Castle et al., 2003). As such, Tyler Jacks and co-workers have shown that mice mutant for both p53 and NF1 in cis configuration develop malignant astrocytoma, progression to glioblastoma, in C57BL/6J background but not in 129S4/SvJae background (Reilly et al., 2004), indicating roles for modifying genes and epigenetic phenomena. In addition, identical twins that have Neurofibromatosis type I have been shown to have similar pattern of café au lait macules and other cutaneous presentations but differ greatly on malignant susceptibility (Easton et al., 1993).
Neurofibromas and tumor microenvironment
In the conventional KO model, the NF1+/− mice (containing NF1+/− Schwann cells and NF1+/− mast cell) did not have mast cell infiltration into nerves and did not develop neurofibroma (Parada et al., 2005). Subsequently, in the tissue-specific KO model using Cre/loxP technology, the NF1flox/flox;Krox20-Cre mice (containing NF1−/− Schwann cells and NF1+/+ mast cell) also did not exhibit mast cell infiltration into peripheral nerves and did not develop neurofibromas. On the other hand, the NF1flox/−;Krox20-Cre mice (containing NF1−/− Schwann cells and NF1+/− mast cell) have massive mast cell infiltration and develop plexiform neurofibromas (Zhu et al., 2002) (Figure 3). This fact indicates the essential role for the heterozygous environment and specifically implicates heterozygote mast cells as critical accomplices in the development of neurofibroma.
However, exactly how mast cells promote tumor development remains unknown. In regard to neurofibroma tumorigenesis, the interaction between Schwann cells and their environment could be bi-directional, where the NF1−/− Schwann cells can alter their microenvironment to promote neurofibroma formation. In this regard, we observed that there are massive NF1-heterozygous mast cells infiltration into nerve tissues long before the NF1flox/−;Krox20-Cre mice develop frank plexiform neurofibromas (Zhu et al., 2002).
Through elegant tissue culture studies, Clapp and coworkers have shown convincing evidence that NF1+/− mast cells are hyperproliferative and hypermotile toward stem cell factors (SCF), a natural ligand for the tyrosine receptor kinase C-kit. In addition, they demonstrated that the NF1−/− Schwann cells produce elevated levels of SCF, which can stimulate this mast cell migration and proliferation (Yang et al., 2003). Recently, Clapp and co-workers also established that NF1+/− mast cells secreted elevated level of tumor growth factor (TGF)-β, a profibrotic factor that stimulate NF1+/− fibroblast to proliferate and synthesize excessive collagen (Yang et al., 2006), a histopathologic feature of neurofibroma. These results are in striking alignment with the mouse modeling data. A compelling scenario is that NF1−/− Schwann cells recruit NF1+/− mast cells via SCF. Once in place, the NF1-heterozygous mast cells produce mitogens and facilitate a permissive microenvironment for tumor formation. Mast cells are also known to make vascular endothelial growth factor and other angiogenic factors leading to neovascularization, which could sustain neurofibroma tumorigenesis (Theoharides and Conti, 2004) (Figure 4).
Figure 4.
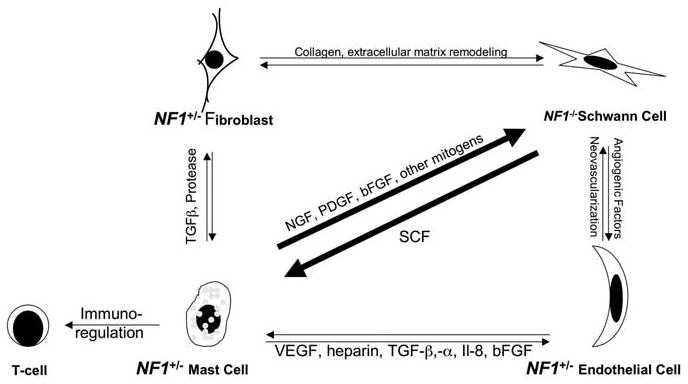
The possible interactions between NF1 heterozygous (abnormal) local cells within the neurofibroma tumor microenvironment. NF1+/− mast cells could be recruited to the site by the NF1−/− Schwann cells via chemoattractant such as SCF. Once in place, the mast cells may selectively secrete factors such as mitogens to initiate tumorigenesis, angiogenic molecules to promote neovascularization or TGF-β to stimulate collagen production for extracellular matrix remodeling.
Future therapies for Neurofibromatosis type I
Clinical management for the Nf1 patients requires a multidisciplinary approach and the use of a multi-speciality Neurofibromatosis clinic is desirable. However, current treatment options for Nf1 are primarily limited to surgery and longitudinal surveillance. The advances provided by transgenic mouse models as the cloning of NF1 may afford new opportunity for specific target therapies, which are beginning to emerge. Therapeutic approaches aimed at the reduction of Ras-GTP levels in affected tissues of the neural crest-derived cells can be expected to relieve some of the Nf1 symptoms, from learning disability to tumor susceptibility. In fact, several clinical trials are under way, from lovastatin for Nf1-associated learning disability to Ras inhibitor farnesyl-transferase for plexiform neurofibroma (http://clinicaltrials.gov/). It is reasonable to speculate that lessons learned from the molecular interactions between Schwann cells and their microenvironment will provide us novel approaches to develop new therapies to delay and to prevent neurofibroma formation in Nf1 patients.
Conclusions
The clinical presentations of Nf1 in an individual, in addition to the emotional burden of carrying the disease and social stigma, have a significant impact on the patient's quality of life (Page et al., 2006). Although the mortality is not high in Neurofibromatosis type I, the morbidity is tremendous. The hope for Nf1 patients, therefore, is that new understanding into molecular pathogenesis will lead to novel treatment. Surprising advances have been made in the research of neurofibromatosis type I in the past decade. Mouse modeling in the Nf1 arena may represent a breakthrough in the use of mouse models of human cancer, not through recapitulation of the disease, which has been achieved for many human cancers, but through the derivation of important insights into the biology of the disease that may guide toward novel therapeutic windows. If expounded upon, this recent knowledge could be used to develop effective treatment for one of the most common human genetic diseases in the world today.
Acknowledgments
LFP is funded by NINDS and DOD (Grant # DAMD 17-02-1-0638 & DAMD 17-03-1-0216) and ACS RP-04-084-01. We thank members of the Parada lab for helpful discussions and Stephanie Bates for assistance in the preparation of this manuscript.
References
- Arun D, Gutmann DH. Recent advances in neurofibromatosis type 1. Curr Opin Neurol. 2004;17:101–105. doi: 10.1097/00019052-200404000-00004. [DOI] [PubMed] [Google Scholar]
- Bajenaru ML, Hernandez MR, Perry A, Zhu Y, Parada LF, Garbow JR, et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003;63:8573–8577. [PubMed] [Google Scholar]
- Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22:5100–5113. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, et al. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell. 1990;63:851–859. doi: 10.1016/0092-8674(90)90151-4. [DOI] [PubMed] [Google Scholar]
- Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW, et al. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994;8:1019–1029. doi: 10.1101/gad.8.9.1019. [DOI] [PubMed] [Google Scholar]
- Castle B, Baser ME, Huson SM, Cooper DN, Upadhyaya M. Evaluation of genotype-phenotype correlations in neurofibromatosis type 1. J Med Genet. 2003;40:e109. doi: 10.1136/jmg.40.10.e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104:593–604. doi: 10.1016/s0092-8674(01)00245-8. [DOI] [PubMed] [Google Scholar]
- Cichowski K, Shih TS, Schmitt E, Santiago S, Reilly K, McLaughlin ME, et al. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999;286:2172–2176. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, et al. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002;415:526–530. doi: 10.1038/nature711. [DOI] [PubMed] [Google Scholar]
- Dasgupta B, Dugan LL, Gutmann DH. The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. J Neurosci. 2003;23:8949–8954. doi: 10.1523/JNEUROSCI.23-26-08949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daston MM, Scrable H, Nordlund M, Sturbaum AK, Nissen LM, Ratner N. The protein product of the neurofibromatosis type 1 gene is expressed at highest abundance in neurons, Schwann cells, and oligodendrocytes. Neuron. 1992;8:415–428. doi: 10.1016/0896-6273(92)90270-n. [DOI] [PubMed] [Google Scholar]
- Easton DF, Ponder MA, Huson SM, Ponder BA. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): evidence for modifying genes. Am J Hum Genet. 1993;53:305–313. [PMC free article] [PubMed] [Google Scholar]
- Ferner RE. Neurofibromatosis 1. Eur J Hum Genet. 2006 doi: 10.1038/sj.ejhg.5201676. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Gregory PE, Gutmann DH, Mitchell A, Park S, Boguski M, Jacks T, et al. Neurofibromatosis type 1 gene product (neurofibromin) associates with microtubules. Somat Cell Mol Genet. 1993;19:265–274. doi: 10.1007/BF01233074. [DOI] [PubMed] [Google Scholar]
- Guo HF, The I, Hannan F, Bernards A, Zhong Y. Requirement of Drosophila NF1 for activation of adenylyl cyclase by PACAP38-like neuropeptides. Science. 1997;276:795–798. doi: 10.1126/science.276.5313.795. [DOI] [PubMed] [Google Scholar]
- Gutmann DH, Wood DL, Collins FS. Identification of the neurofibromatosis type 1 gene product. Proc Natl Acad Sci USA. 1991;88:9658–9662. doi: 10.1073/pnas.88.21.9658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismat FA, Xu J, Lu MM, Epstein JA. The neurofibromin GAP-related domain rescues endothelial but not neural crest development in Nf1 mice. J Clin Invest. 2006;116:2378–2384. doi: 10.1172/JCI28341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet. 1994;7:353–361. doi: 10.1038/ng0794-353. [DOI] [PubMed] [Google Scholar]
- Jentarra G, Snyder SL, Narayanan V. Genetic aspects of neurocutaneous disorders. Semin Pediatr Neurol. 2006;13:43–47. doi: 10.1016/j.spen.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Lakkis MM, Tennekoon GI. Neurofibromatosis type 1. I. General overview. J Neurosci Res. 2000;62:755–763. doi: 10.1002/1097-4547(20001215)62:6<755::AID-JNR1>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Levy P, Vidaud D, Leroy K, Laurendeau I, Wechsler J, Bolasco G, et al. Molecular profiling of malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1, based on large-scale real-time RT-PCR. Mol Cancer. 2004;3:20. doi: 10.1186/1476-4598-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Cui Y, Kushner SA, Brown RA, Jentsch JD, Frankland PW, et al. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol. 2005;15:1961–1967. doi: 10.1016/j.cub.2005.09.043. [DOI] [PubMed] [Google Scholar]
- Mousley CJ, Tyeryar KR, Ryan MM, Bankaitis VA. Sec14p-like proteins regulate phosphoinositide homoeostasis and intracellular protein and lipid trafficking in yeast. Biochem Soc Trans. 2006;34:346–350. doi: 10.1042/BST0340346. [DOI] [PubMed] [Google Scholar]
- Page PZ, Page GP, Ecosse E, Korf BR, Leplege A, Wolkenstein P. Impact of neurofibromatosis 1 on quality of life: a cross-sectional study of 176 American cases. Am J Med Genet A. 2006;140:1893–1898. doi: 10.1002/ajmg.a.31422. [DOI] [PubMed] [Google Scholar]
- Parada LF. Neurofibromatosis type 1. Biochim Biophys Acta. 2000;1471:M13–9. doi: 10.1016/s0304-419x(00)00014-7. [DOI] [PubMed] [Google Scholar]
- Parada LF, Kwon CH, Zhu Y. Modeling neurofibromatosis type 1 tumors in the mouse for therapeutic intervention. Cold Spring Harb Symp Quant Biol. 2005;70:173–176. doi: 10.1101/sqb.2005.70.025. [DOI] [PubMed] [Google Scholar]
- Reilly KM, Tuskan RG, Christy E, Loisel DA, Ledger J, Bronson RT, et al. Susceptibility to astrocytoma in mice mutant for Nf1 and Trp53 is linked to chromosome 11 and subject to epigenetic effects. Proc Natl Acad Sci USA. 2004;101:13008–13013. doi: 10.1073/pnas.0401236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi VM. Neurofibromatosis phenotype, natural history and pathogenesis. The Johns Hopkins University Press; Baltimore: 1992. [Google Scholar]
- Szudek J, Birch P, Riccardi VM, Evans DG, Friedman JM. Associations of clinical features in neurofibromatosis 1 (NF1) Genet Epidemiol. 2000;19:429–439. doi: 10.1002/1098-2272(200012)19:4<429::AID-GEPI13>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- The I, Hannigan GE, Cowley GS, Reginald S, Zhong Y, Gusella JF, et al. Rescue of a Drosophila NF1 mutant phenotype by protein kinase A. Science. 1997;276:791–794. doi: 10.1126/science.276.5313.791. [DOI] [PubMed] [Google Scholar]
- Theoharides TC, Conti P. Mast cells: the Jekyll and Hyde of tumor growth. Trends Immunol. 2004;25:235–241. doi: 10.1016/j.it.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Tong J, Hannan F, Zhu Y, Bernards A, Zhong Y. Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nat Neurosci. 2002;5:95–96. doi: 10.1038/nn792. [DOI] [PubMed] [Google Scholar]
- Trovo-Marqui AB, Tajara EH. Neurofibromin: a general outlook. Clin Genet. 2006;70:1–13. doi: 10.1111/j.1399-0004.2006.00639.x. [DOI] [PubMed] [Google Scholar]
- Vogel KS, Klesse LJ, Velasco-Miguel S, Meyers K, Rushing EJ, Parada LF. Mouse tumor model for neurofibromatosis type 1. Science. 1999;286:2176–2179. doi: 10.1126/science.286.5447.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BA, Gutmann DH. Neurofibromatosis 1: from lab bench to clinic. Pediatr Neurol. 2005;32:221–228. doi: 10.1016/j.pediatrneurol.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Weiss B, Bollag G, Shannon K. Hyperactive Ras as a therapeutic target in neurofibromatosis type 1. Am J Med Genet. 1999;89:14–22. [PubMed] [Google Scholar]
- Xu GF, O'Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell. 1990;62:599–608. doi: 10.1016/0092-8674(90)90024-9. [DOI] [PubMed] [Google Scholar]
- Xu H, Gutmann DH. Mutations in the GAP-related domain impair the ability of neurofibromin to associate with microtubules. Brain Res. 1997;759:149–152. doi: 10.1016/s0006-8993(97)00328-4. [DOI] [PubMed] [Google Scholar]
- Yang FC, Chen S, Clegg T, Li X, Morgan T, Estwick SA, et al. Nf1+/− mast cells induce neurofibroma like phenotypes through secreted TGF-beta signaling. Hum Mol Genet. 2006;15:2421–2437. doi: 10.1093/hmg/ddl165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang FC, Ingram DA, Chen S, Hingtgen CM, Ratner N, Monk KR, et al. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/− mast cells. J Clin Invest. 2003;112:1851–1861. doi: 10.1172/JCI19195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohay KH. The genetic and molecular pathogenesis of NF1 and NF2. Semin Pediatr Neurol. 2006;13:21–26. doi: 10.1016/j.spen.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920–922. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Harada T, Liu L, Lush ME, Guignard F, Harada C, et al. Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development. 2005;132:5577–5588. doi: 10.1242/dev.02162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Parada LF. The molecular and genetic basis of neurological tumours. Nat Rev Cancer. 2002;2:616–626. doi: 10.1038/nrc866. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, et al. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15:859–876. doi: 10.1101/gad.862101. [DOI] [PMC free article] [PubMed] [Google Scholar]