

Abstract
SIRT3 is a key mitochondrial protein deacetylase proposed to play key roles in regulating mitochondrial metabolism but there has been considerable debate about its actual size, the sequences required for activity, and its subcellular localization. A previously cloned mouse SIRT3 has high sequence similarity with the C-terminus of human SIRT3 but lacks an N-terminal mitochondrial targeting sequence and has no detectable deacetylation activity in vitro. Using 5′ rapid amplification of cDNA ends, we cloned the entire sequence of mouse SIRT3, as well as rat and rabbit SIRT3. Importantly, we find that full-length SIRT3 protein localizes exclusively to the mitochondria, in contrast to reports of SIRT3 localization to the nucleus. We demonstrate that SIRT3 has no deacetylation activity in vitro unless the protein is truncated, consistent with human SIRT3. In addition, we determined the inhibition constants and mechanism of action for nicotinamide and a small molecule SIRT3 inhibitor against active mouse SIRT3 and show that the mechanisms are different for the two compounds with respect to peptide substrate and NAD+. Thus, identification and characterization of the actual SIRT3 sequence should help resolve the debate about the nature of mouse SIRT3 and identify new mechanisms to modulate enzymatic activity.
Keywords: sirtuin, NAD+-dependent deacetylation, inhibitor, mitochondrial function
Introduction
The silent information regulator 2 (Sir2 or sirtuin) proteins comprise a family of NAD+-dependent protein deacetylases and ADP-ribosyltransferases that are highly conserved in both prokaryotes and eukaryotes.1 Sirtuins are involved in many cellular processes including gene silencing,2 cell cycle regulation,3,4 metabolism,5–8 apoptosis9–11 and the lifespan-extension effects of calorie restriction.12,13 Mammals express seven sirtuins, SIRT1-7.14 SIRT1, 2, 3, and 5 possess deacetylase activity, whereas SIRT4 and 6 are ADP-ribosyltransferases. These proteins display a range of subcellular localization, including nuclear (SIRT1, 6, 7), cytosolic (SIRT2),15 and mitochondrial stores (SIRT3, 4, 5)16. These diverse locations in the cell reflect varying functions. SIRT1 has been studied most extensively and has been linked to glucose homeostasis,17–19 inflammation,20 cancer,21–23 and neuropathology.24,25 We have previously described small molecule activators of SIRT1 that improve insulin sensitivity, lower plasma glucose and increase mitochondria capacity in diet-induced obese and genetically obese mice.19
SIRT3 is a major mitochondrial deacetylase. Mitochondrial proteins show hyperacetylation in SIRT3 knockout mice, but not in SIRT4 or SIRT5 knockout mice.26 Acetyl-CoA synthetase 2 (AceCS2), a mitochondrial enzyme that converts acetate into acetyl-CoA, was the first mitochondrial substrate of SIRT3 identified.6,27 Deacetylation of AceCS2 at lysine 642 by SIRT3 activates acetyl-CoA synthetase activity, providing increased acetyl-CoA to feed into the tricarboxylic acid cycle. Glutamate dehydrogenase (GDH), another mitochondrial protein involved in energy production, is deacetylated by SIRT3.26 GDH can also be ADP-ribosylated by SIRT4 in turn to decrease its enzyme activity.28 This indicates that SIRT3 could play an important role in cell metabolism. SIRT3 has also been shown to be involved in selective apoptosis pathways and cell growth control.29 SIRT3 and SIRT4, but not SIRT5, have been implicated in the NAD+ salvage pathway that regulates the NAD+ level relating to cell survival.30 In addition, variability in the hSIRT3 gene has been linked to human longevity.31,32
Human SIRT3 (hSIRT3) has been shown to localize to mitochondria33–35 in a majority of the reported studies. One controversial report suggests that full length hSIRT3 is a nuclear protein under basal conditions but translocates into mitochondria during cellular stress including over-expression of hSIRT3.36 A more recent report disagrees and shows full length hSIRT3 is localized only in mitochondria with either transient or stable over-expression in various human cell lines, and the hSIRT3 lacking the N-terminal 142 residues is present in the cytoplasm and nucleus at high expression levels.35
The N-terminus of hSIRT3 contains a mitochondrial targeting sequence, an amphipathic α-helix rich in basic residues.34 The full length hSIRT3 is enzymatically inactive. When the first 101 residues are cleaved by mitochondrial matrix processing peptidase (MPP) in vitro, the protein becomes activated. Both a full-length 44 kDa hSIRT3 and a smaller 28 kDa product were observed in whole-cell lysates of HEK293T cells transfected with FLAG tagged hSIRT3, whereas a 28-kDa product was the only form detected in the mitochondrial lysates. Only SIRT3 purified from fractionated mitochondrial lysates showed NAD+-dependent protein deacetylase activity.33 However, Scher et al.36 reported that both the full length and 142-residue truncated hSIRT3 have similar deacetylation activity and substrate specificity.
Mouse SIRT3 (mSIRT3) was first cloned by Yang et al.37 It was reported to be a 257 amino acid protein that aligned well with the C-terminal portion of hSIRT3 (residues 143–399) and localized in certain areas of the cytoplasm in discrete stacks. Mouse SIRT3 is expressed in many tissues, with high expression in brain, kidney, testes, heart and liver and is also expressed in brown adipose tissue.38 The expression is up-regulated during caloric restriction and cold exposure. Although the reported mSIRT3 does not contain the N-terminal mitochondrial targeting sequence, Shi et al. showed that over-expressed mSIRT3 is localized to the inner mitochondrial membrane of NIH3T3 cells.38 In a more recent report, when subcellular fractions prepared from mouse liver were probed with SIRT3-specific antibody, mSIRT3 is found to be a soluble mitochondrial protein, as with hSIRT3, under both basal and stress conditions.26
Here we report the cloning of a longer form of mSIRT3 (mSIRT3L), its tissue distribution, subcellular localization, the fragments that retain deacetylation activity, as well as its inhibition by nicotinamide and SRT1720, a small molecule SIRT3 inhibitor. Our rationale for attempting to clone a longer mSIRT3 was the lack of deacetylase activity for the recombinant short mSIRT3 with 257 amino acids and the equivalent hSIRT3-143-399. We also questioned the mitochondrial localization of mSIRT3 given the lack of an obvious mitochondrial targeting sequence. The long mSIRT3 that we have cloned matches the predicted mSIRT3 reported by Cooper and Spelbrink.35
Results and Discussion
Cloning of the long mouse SIRT3
To identify cDNA that encodes the longer mouse SIRT3, we performed 5′-RACE PCR with mRNA from mouse liver. Two sequences were identified with an 8 base pair (bp) difference (TGTTACAG) at the 5′ end of exon 2 of the mouse SIRT3 gene on chromosome 7 [Fig. 1(A)]. The sequence containing the 8 bp insertion encodes an open reading frame (ORF) for a protein of 257 residues (gene BC025878, protein AAH258219). This is the reported short form of mSIRT3 without the mitochondrial targeting sequence. Interestingly, the sequence lacking the 8 bp insertion encodes a much longer ORF for a protein of 334 residues (matching GenBank EST database AW258219). This longer ORF [Fig. 1(B)] is the same as predicted by Cooper and Spelbrink.35 Both sequences had been reported by Yang et al.37 previously. In their study, the longer ORF was not detected by reverse transcription-PCR (RT-PCR) in mouse tissues. The additional sequence was considered to have weak identity to human SIRT3 and its existence was dismissed. The two variants of mSIRT3 gene could be caused by alternative splicing. Alternative splicing at donor or acceptor sites located just a few nucleotides apart has been reported to be widespread in many species and results in subtle changes in the transcripts that could cause changes in the encoded proteins.39 We have not yet determined the frequencies of the two variants in mouse and what controls the splicing.
Figure 1.

The cDNA and amino acid sequence of the longer mouse SIRT3. (A) Schematic representation of the mouse SIRT3 gene. The exons are presented as rectangular boxes with the exon number labeled in each box and the base pair numbers of the coding sequences are labeled above each box. The 5′ and 3′ untranslated regions (UTR) are colored in dark blue. Coding sequences are colored in light blue. The location of the eight base pair insertion is colored in red. The three potential start codons and the stop codon are indicated. (B) The sequence of long mSIRT3. The three methionine residues that have been considered as the starting residue are in bold character and underlined. The other residues in bold are the starting residues for different N-terminal truncations that we generated and listed in Figure 1(C). (C) Schematic representation of all the mSIRT3 proteins that were generated for enzyme activity testing. The starting and ending residue numbers are labeled. The deacetylation activity for each protein is indicated as “−” no activity, “++” high activity, and “+” low activity.
The longer mSIRT3 has three methionine residues at positions 1, 15, and 78 near the N-terminus. Residue Met78 is the first residue for the reported short mSIRT3 containing 257 amino acids. When we aligned the mSIRT3L with human SIRT3 by TCOFFEE40 (Fig. 3), it was Met15 (not Met1) of mSIRT3L that aligned with Met1 of hSIRT3. Residues 15–30 of mSIRT3L have high homology with the N-terminus of hSIRT3, whereas there is a 77-residue insertion in hSIRT3 that is absent in mSIRT3L. The rest of the additional sequence in the mSIRT3L (residues 31–77) is highly similar to hSIRT3. It is unclear which methionine, Met1 or Met15, is the starting residue for the mSIRT3L (discussed in more detail below).
Figure 3.

Sequence alignment of SIRT3 from human, mouse, rat, and rabbit. Conserved residues are labeled by “*”, functionally highly conserved residues are labeled by “:”, and less conserved residues are labeled by “.”. The arrow points to the putative MPP cleavage site. The three potential starting residues, Met1, Met15, and Met 78 of mSIRT3L, are in bold.
Cloning of SIRT3 from rat and rabbit
The SIRT3 of rat and rabbit have not been previously cloned. We performed RACE reactions as we did with cloning mSIRT3L. The predicted rat SIRT3 (XM_215124) has 257 amino acids like the short mouse SIRT3. Based on the rat genome, rat has the sequence equivalent to mSIRT3L-1-334. We performed 5′-RACE PCR multiple times and obtained two sequences with or without the 8 bps insertion (AGTATTAC) like the 5′-RACE PCR result for mSIRT3. The sequence without the 8 bps codes for 320 amino acid protein [Fig. 2(A)] and has high identity with mSIRT3-15-334 (Fig. 3).
Figure 2.

The cDNA and amino acid sequences of rat and rabbit SIRT3. (A) The sequence of rat SIRT3. (B) The sequence of rabbit SIRT3. The italic DNA sequence before the coding sequence was obtained by a BLAST search of the rabbit genome. The underlined portion was obtained from our 5′-RACE PCR. The potential start codon (ATG) before the coding codon is in bold.
The sequence we obtained for rabbit SIRT3 contains 319 amino acids [Fig. 2(B)] and aligned well with mouse SIRT3-15-334 (Fig. 3). A matching sequence was found by a BLAST search of the rabbit genome showing additional 5′ sequence upstream of the coding sequence. We cloned 17 bps before the coding start codon ATG in our 5′-RACE product. There is another ATG in the upstream 5′ sequence [Fig. 2(B)] that is not in the current ORF (44 bps upstream). Considering the high similarity of rabbit SIRT3 with SIRT3 from human, mouse and rat, we believe that we have obtained the full-length rabbit SIRT3.
Mouse SIRT3 tissue distribution
We performed real-time RT-PCR against RNA from different mouse tissues with primers and a probe which is derived from the N-terminal region of mSIRT3L that is not present in the short mSIRT3. The mRNA expression of mSIRT3L is highest in kidney, brain and heart, followed by liver and testes, and is relatively low in lung, ovary and thymus (Fig. 4). We also performed RT-PCR using primers and a probe designed against the short mSIRT3 sequence and obtained a similar result (data not shown). Our results are consistent with previous studies of mSIRT3 mRNA expression.26,33,37,38
Figure 4.
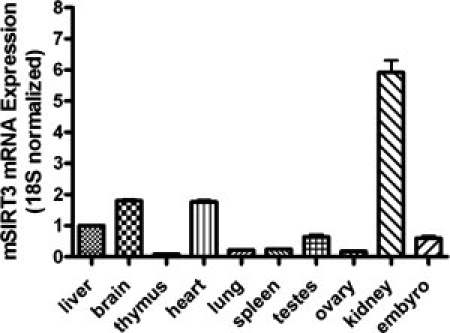
Tissue distribution of mSIRT3L. The mRNA levels for mSIRT3L were analyzed by real-time RT-PCR in several mouse tissues. The mSIRT3L mRNA level in each tissue was normalized to 18S mRNA. The mRNA for mSIRT3L in the liver was set to 1 and those in the other tissues were normalized to that in the liver.
Long mSIRT3 is a mitochondrial protein
The predicted longer form of mSIRT3 may contain N-terminal mitochondrial targeting sequences that are also present in human SIRT3. As noted by Cooper et al., this longer mSIRT3 could begin at the most N-terminal methionine (Met1) or a second downstream methionine (Met15).35 Therefore, we compared the localization of both of these proteins (mSIRT3L-1-334 and mSIRT3L-15-334) to that of the originally predicted mouse SIRT3 (mSIRT3L-78-334). To determine the subcellular distribution of these proteins we transfected mouse embryonic fibroblasts with mSIRT3L-1-334, 15-334, and 78-334 expressed with a C-terminal FLAG tag [Fig. 5(A)]. In immunoflourescence assays, over-expressed mSIRT3 proteins were detected by anti-FLAG antibody (green) and mitochondrial compartments were labeled by staining for endogenous cytochrome c (red) [Fig. 5(B)]. Both mSIRT3L-1-334 and 15-334 colocalized with cytochrome c in the mitochondria in all transfected cells, with no apparent alteration of mitochondrial morphology. In contrast, mSIRT3L-78-334 was localized uniformly in the cytosol in 82% of cells and aggregated within the cytosol with partial cytochrome c overlap in 14% of transfected cells. mSIRT3L-78-334 was also found in the nucleus when highly over-expressed.
Figure 5.
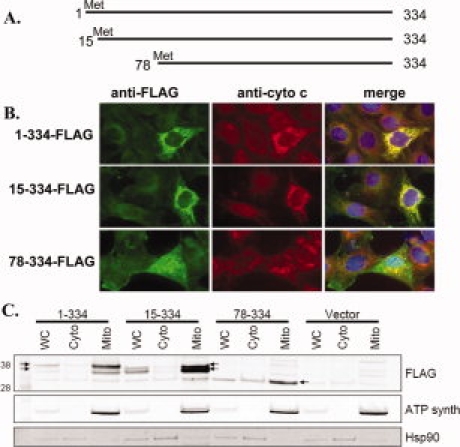
Long mSIRT3 localizes to mitochondria. (A) Schematic drawing of SIRT3 constructs. mSIRT3L-1-334 begins at the most N-terminal Met, mSIRT3L-15-334 starts at the second Met, and mSIRT3L-78-334 represents the short mSIRT3 previously believed to be the full-length protein. (B) Mouse embryonic fibroblasts were transfected with C-terminal FLAG-tagged SIRT3 constructs and stained with both anti-FLAG (green) antibody to detect over-expressed mSIRT3 and anti-cytochrome c (red) to label mitochondria. Data are representative of three independent experiments. (C) Cell fractionation of MEFs transfected with either empty vector or plasmids shown in A. mSIRT3L-1-334 and mSIRT3L-15-334 are found predominantly in the mitochondrial fraction, whereas mSIRT3L-78-334 is distributed throughout both cytosolic and mitochondrial fractions. Arrows point out the mSIRT3 protein bands that are discussed in the text.
Cell fractionation demonstrated that mSIRT3L-1-334 and mSIRT3L-15-334 are almost exclusively mitochondrial, whereas mSIRT3L-78-334 is distributed amongst all fractions [Fig. 5(C)]. The mSIRT3L-78-334 ran overlapping with another protein band that was present in all the fractions of the cell transfected with control vector. Our result is consistent with data obtained by Cooper et al., where the authors demonstrate that the full-length hSIRT3 is exclusively mitochondrial but an N-terminal truncation mutant that corresponds to the original mouse start codon (hSIRT3 −143-399) remains dispersed throughout the cell.35 Taken together, our data suggest mitochondrial localization of the longer mSIRT3 isoforms, whether they begin at the first (Met1) or second (Met15) methionine. In contrast, the originally predicted mouse protein (78-334) remains largely cytosolic and nuclear. The mitochondrial staining observed with mSIRT3L-78-334 appears to alter mitochondrial morphology, similar to staining observed by Shi et al.38 and noted as atypical by Cooper and Spelbrink.35
Our data demonstrate that over-expressed mSIRT3L, starting at either position 1 or 15, is a mitochondrial protein. To predict which methionine is the possible starting residue for mSIRT3L, we analyzed the sequences with three programs that apply different approaches for predicting mitochondrial targeting sequences, MitoProt,41 Predotar,42 and TargetP.43 The scores for mitochondrial targeting are highest for mSIRT3L-15-334 in all predictions (Table I). The score for rat SIRT3-1-320 is higher than the predicted rat SIRT3-1-334 as well. In addition, we calculated the scores for human and rabbit SIRT3. Based on the results from sequence alignment [Fig. 3(A)], the prediction of mitochondrial protein (Table I) and the homology among SIRT3 of mouse, human, rat and rabbit, mSIRT3L-15-334 is most likely to be the full-length mouse SIRT3 protein that contains 320 amino acids.
Table I.
Mitochondrial Targeting Scores for Long Mouse SIRT3 with Three Different Starting Methionine Residues (Met1, Met15, and Met78), as well as for SIRT3 of Human, Rat, and Rabbit
| Mouse SIRT3 |
Rat SIRT3 |
Human SIRT3 |
Rabbit SIRT3 |
||||
|---|---|---|---|---|---|---|---|
| 1–334 aa | 15–334 aa | 78–334 aa | 1–334a aa | 1–320 aa | 1–399 aa | 1–319 aa | |
| MitoProt II | 0.6043 | 0.7847 | 0.0086 | 0.5418 | 0.7376 | 0.7455 | 0.941 |
| Predotar | 0 | 0.54 | 0 | 0.01 | 0.4 | 0.65 | 0.78 |
| TargetP | 0.12 | 0.814 | 0.18 | 0.109 | 0.819 | 0.555 | 0.938 |
Three different mitochondria targeting prediction programs were used.
Scores range from 0 to 1, with a higher score indicating higher probability for the protein to contain a mitochondria targeting sequence.
Indicates the rat SIRT3 that was predicted from the rat genome, but was not cloned.
Mouse SIRT3 deacetylase activity
The full length and several fragments of mSIRT3L have been expressed and purified [Fig. 1(C)]. The presence of chaperone was required to obtain soluble protein. The proteins containing residues 1–334, 15–334, and 78–334 of mSIRT3L do not have detectable deacetylation activity, whereas the other N-terminal truncated proteins except 64-334 displayed robust enzyme activity and have similar activity to hSIRT3-118-334, a very active hSIRT3 fragment. The 64-334 fragment of mSIRT3L had lower deacetylation activity, indicating that truncating mSIRT3L beyond residue 59 might adversely affect the proper folding of the protein. The proteins which include the mitochondrial targeting sequence (1-334 and 15-334) did not have detectable deacetylation activity similar to the full length hSIRT3.34
MPP (matrix processing peptidase) is the putative mitochondrial enzyme that cleaves hSIRT3 between residue 101 and 102 to activate the enzymatic activity.34 Based on the sequence alignment between mSIRT3L and hSIRT3, mSIRT3L-38-334 is equivalent to hSIRT3-102-399 (Fig. 3). The two arginine residues at −2 and −3 relative to the cleavage site of MPP and the hydrophobic residue at +1 position in hSIRT3 (MPP recognition site requirements) are conserved in mSIRT3L, as well as in rat and rabbit SIRT3. Like hSIRT3, mSIRT3L became active when the N-terminal residues were removed [Fig. 1(C)].
We observed [Fig. 5(C)] that there is another protein band under each of the protein bands of mSIRT3L-1-334 and mSIRT3L-15-334 that migrates to a similar location relative to each other but slightly higher than mSIRT3L-78-334, the originally predicted mSIRT3 protein. This size difference suggests that the 78-334 protein is not the fragment produced through endogenous peptide cleavage. As mentioned above, the 78-334 fragment is not active in our in vitro deacetylation assay. This leaves open the possibility that mSIRT3L-38-334, the fragment which corresponds to the human MPP processed SIRT3 (hSIRT3 102-399) and active in our in vitro assay, is more likely to be the endogenous active form of mouse SIRT3L.
In our experiments, the human SIRT3-143-399 does not have deacetylation activity in vitro, although the predicted MPP cleaved form 102-399 is active (data not shown). These truncated proteins are similar in size, with 143–399 28.7 kD and 102–399 31.7 kD. Upon more careful inspection of the gel pictures in the report of Schwer et al.,34 the mitochondrial hSIRT3 and MPP cleaved hSIRT3 were migrating slightly higher than the 28.4 molecular weight marker. It could be that the 28-kDa hSIRT3 that was referred to be the active hSIRT3 is actually the 31.7-kDa hSIRT3-102-399.
Shi et al. have reported that mSIRT3 78-334 is a mitochondrial protein that can increase basal oxygen consumption of HIB1B cells by 1.8-fold when stably over-expressed.38 Although we do see some amount of this protein in the mitochondria, over-expression of mSIRT3L-78-334 appeared to distort mitochondrial structure and was localized to additional cellular compartments. Therefore over-expression of this form of mSIRT3 may not be reflective on the true activity of endogenous protein. We are in the process of immunoprecipitating over-expressed mSIRT3L-1-334 and 15-334 to isolate sufficient amount of the processed form of mSIRT3 for N-terminal sequence identification.
SIRT3 inhibition studies
As part of the characterization of the purified mSIRT3L-54-334 protein, we have tested two deacetylase inhibitors, nicotinamide (NAM) and SRT1720 as described in Material and Methods. NAM is a known inhibitor of the deacetylation activity of sirtuins,26,44–47 but the inhibition mechanism of NAM toward its substrates for SIRT3 has not been determined before. We also identified a small molecule SIRT3 inhibitor, SRT1720. Figure 6 shows the curves and the Km, Vmax, Ki and αKi values calculated by the global fitting for each compound against NAD+ or AceCS2 peptide. The Km and Vmax values attained for NAD+ and AceCS2 peptide are within experimental error to previously obtained values without inhibitor used in the inhibition mechanism study.
Figure 6.
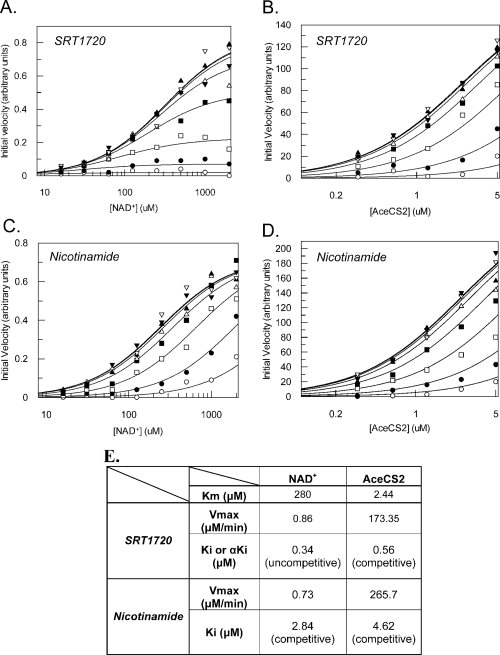
Inhibition of mSIRT3L-54-334 by SRT1720 and nicotinamide. The global fitting curves of the observed rate of reaction with respect to varying substrate and inhibitor concentrations are displayed. (A) SRT1720 inhibition against NAD+. Eight concentrations of nicotinamide were used: ○ 15 μM, • 3.75 μM, □ 0.94 μM, ▪ 0.23 μM, Δ 0.06 μM, ▴ 0.015 μM, ▿ 0.004 μM and ▾ 0.001 μM. (B) SRT1720 inhibition against AceCS2 peptide. The same eight concentrations of SRT1720 were used as in A. (C) Nicotinamide inhibition against NAD+. Eight concentrations of nicotinamide were used as follows: ○ 80 μM, • 20 μM, □ 5 μM, ▪ 1.25 μM, Δ 0.31 μM, ▴ 0.08 μM, ▿ 0.02 μM and ▾ 0.005 μM. (D) Nicotinamide inhibition against AceCS2 peptide. Eight concentrations of nicotinamide were used: ○ 85 μM, • 28.3 μM, □ 9.4 μM, ▪ 3.2 μM, Δ 1.05 μM, ▴ 0.35 μM, ▿ 0.12 μM and ▾ 0.04 μM. (E) The Km and Vmax values of the enzyme, Vmax value in μM/min, Ki or αKi values in μM, and the mechanism of inhibition for SRT1720 and nicotinamide are listed.
SRT1720 inhibits mSIRT3L-54-334 with αKi of 0.34 μM against NAD+ [Fig. 6(A,E)]. The α value is significantly smaller than 1 indicating uncompetitive inhibition. However SRT1720 inhibits mSIRT3L-54-334 in a competitive manner (α value ≫ 1) against AceCS2 peptide with a Ki value of 0.56 μM [Fig. 6(B,E)]. This suggests that SRT1720 binding requires NAD+ to bind first to the enzyme and that SRT1720 competes for the same binding site of AceCS2 peptide or works as an allosteric competitive inhibitor to mSIRT3.
NAM has an α value much greater than one against NAD+ and AceCS2 peptide suggesting a competitive inhibition mechanism toward both substrates with Ki values of 2.84 μM [Fig. 6(C,E)] against NAD+ and 4.62 μM [Fig. 6(D,E)] against AceCS2 peptide. NAM has been reported as a non-competitive inhibitor toward both NAD+ and the peptide substrate and is part of the base-exchange reaction in SIRT1, SIRT2, yeast and bacterial sirtuins.44–48 However, it is likely that mSIRT3 has a different mechanism of inhibition for NAM. A more complete kinetic analysis of SIRT3 is ongoing to propose a reaction mechanism.
Under the current experimental conditions, the mechanism of inhibition is different for SRT1720 and NAM against NAD+ and AceCS2 peptide. These data suggest that SRT1720 and NAM bind to different sites on mSIRT3 and that mSIRT3 could be inhibited by different inhibitors via different sites and mechanisms.
It is still unclear what the biological and physiological roles of SIRT3 are and how inhibition of SIRT3 would affect glucose metabolism and insulin sensitivity. SRT1720 has previously been shown to activate SIRT1.19 However we have developed many other small molecule SIRT1 activators that are as effective as SRT1720 in controlling glucose homeostasis in ob/ob and diet-induced obesity mouse models but activate or have no effect on SIRT3. These data suggest that SRT1720, along with other SIRT1 activators, are potentially useful in treating type 2 diabetes. Developing SIRT3 selective modulators will help to address the functional roles of SIRT3 in vivo.
In summary, we have identified the correct form of mSIRT3. This longer form is more similar in length, localization, and activity to human SIRT3 than the shorter, originally cloned mouse ortholog. The identification of this protein increases our confidence of using mouse models to design small molecule modulators of human SIRT3 that could one day treat human disease.
Materials and Methods
Cloning of long mouse, rat, and rabbit SIRT3
To identify the 5′ end of mSIRT3, 5′-RACE (rapid amplification of cDNA ends) polymerase chain reaction (PCR) was performed on the cDNA prepared using the SMART RACE cDNA Amplification kit (Clontech) from RNA isolated from the liver of a female mouse (Institute of Cancer Research, CD-1) using TRI-reagent (Sigma). The mSIRT3 specific reverse primer was designed based on the gene with Genbank number of BC025878. The gene specific primer TTATCTGTCCTGTCCATCCA and universal primer mix were used for the PCR reaction. Nested PCR was performed on the 5′-RACE PCR product with nested universal primers and mSIRT3 specific reverse primer: CCTGTCCGCCATCACATCAGCCCAT. Specific DNA bands of mSIRT3 from the 5′-RACE products were generated and ligated into T-vector (Takara). Positive clones were obtained with two coding sequences. The novel isoform of mSIRT3 has 334 amino acids which contains 77 additional amino acids at the N-terminus of previously reported mSIRT3 containing 257 amino acids.
The longer mSIRT3 was cloned into a modified pET21a vector with an N-terminal His6 tag. Several N-terminal truncations of mouse SIRT3 were also generated in the same vector. The truncated proteins start at residue 15, 38, 44, 49, 54, 59, 64, or 78 and end at residue 334.
5′-RACE PCR was performed as above on cDNA derived from the liver and kidney mRNA purified from a Wistar male rat. Gene specific primers were designed according to the predicted rat SIRT3 sequence (XM_215124). The reverse primer used for 5′-RACE PCR was TCAGCTGTCTAGCGATGGGCATG and for nested PCR was TACGGGATGTCATACTGCTGAAGGTTGCT. The cloned rat SIRT3 contains 320 amino acids.
For cloning rabbit SIRT3, mRNA was purified from the liver of a male New Zealand white rabbit. The cDNA for 5′-RACE and 3′-RACE was prepared using the Clontech SMART RACE kit, and PCR was performed using a universal primer mix and gene specific primer. The gene specific reverse primer was CAACCAGCTTTGAGGCAGGGAT for 5′-RACE and the forward primer was TCACAACCCCAAGCCCTTTTTCA for 3′-RACE. Both RACE reactions yielded product of interest. The cloned rabbit SIRT3 contains 319 amino acids.
For immunoflourescence and cell fractionation studies, the mSIRT3L-1-334, 15-334, and 78-334 were cloned into the p3XFLAG-CMV-14 vector (Sigma) between KpnI and XbaI sites. Plasmid DNA was isolated using Endofree Plasmid Maxi kit (QIAgen).
Reverse transcription-PCR amplification
Total RNA was purchased from Ambion and reverse transcribed in duplicate using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real Time PCR reactions were performed following conditions recommended by the manufacturer and analyzed using the 7300 Fast Real-Time PCR System (Applied Biosystems). Genes of interest were normalized to 18S ribosomal RNA (Integrated DNA Technologies). RT-PCR primers and probes were synthesized by Integrated DNA Technologies. The mouse SIRT3 probe was labeled at the 5′ end with 6-FAM™ dye (6-carboxyfluorescein) and at the 3′ end with BHQ-1 (Black Hole Quencher-1™). The mouse 18S RNA probe was labeled at the 5′ end with JOE (6-carboxy-4′, 5′-dichloro-2′, 7′- dimethoxyfluorescein) and at the 3′ end with BHQ-1. The probe sequence was CTGCTTCGGACCAGAGCCTGCAGT from the N-terminal sequence unique to mSIRT3L. The forward primer was GGCTTTGGAGGTGGAGGAA and reverse primer was GCCACTGGGTGTGCTGATG for mSIRT3 PCR. The probe sequence for 18S was TGCTGGCACCAGACTTGCCCTC. The forward primer for 18S amplification was CGGCTACCACATCCAAGGAA and reverse primer was GAGCTGGAATTACCGCGGCT.
Immunoflourescence
Mouse Embryonic Fibroblasts (MEFs) (2 × 106) were tranfected with 5 μg plasmid DNA per reaction using Amaxa nucleofector kit V (Amaxa Biosystems). Cells were then plated in 2-well chamber slides (LabTekII) at a density of 2 × 105 cells per well. Twenty-four hours following transfection, cells were washed three times with PBS and fixed with 4% paraformaldehye/PBS for 15 min at room temperature. Cells were washed as above, then permeabilized with 0.2% Triton X-100/PBS for 10 min on ice and incubated with 5%BSA/PBS blocking buffer for 1 h at room temperature. Cells were incubated for 1 h in 5% BSA/PBS with mouse anti-FLAG (1:100, Sigma) and rabbit anti-cytochrome c (1:100, Santa Cruz) primary antibodies. Cells were washed three times with 1% BSA/PBS and then incubated for 1 h in 5%BSA/PBS with FITC-conjugated anti-mouse (1:200, Jackson ImmunoRes) and TRITC-conjugated anti-rabbit (1:100, Jackson ImmunoRes) secondary antibodies. All antibody incubations were performed at room temperature. After three washes, mounting media with DAPI was added (VectaShield H-1200; Vector Bioloabs) and a coverslip was placed on each slide. Staining was visualized using a Zeiss Axiovert 200M fluorescence microscope and imaged with AxioVision 4.5 software. Quantification of subcellular localization of mSIRT3 constructs was based on scoring of 50 random transfected cells per slide for mitochondrial or cytosolic localization. Data are representative of three independent experiments.
Cell fractionation and western blotting
Mouse Embryonic Fibroblasts (MEFs) (4 × 106) were tranfected with 5 μg plasmid DNA per reaction using Amaxa nucleofector kit V (Amaxa Biosystems) and were plated in 10 cm dishes. Eighteen hours following transfection, mitochondria were isolated from whole cell lysate and cytosol using the Dounce homogenization protocol of the Pierce Mitochondria Isolation Kit (Pierce). Protein concentration of whole cell, cytosol, and mitochondrial fractions was measured by A280 and by the BCA Protein Assay Kit (Pierce). Each fraction (20 μg) was run on a NuPAGE 4–12% SDS-PAGE gel (Invitrogen). Gels were transferred to nitrocellulose membranes (iBlot, Invitrogen) and membranes were blocked in Odyssey Blocking buffer (Li-cor) for 30 min. Membranes were incubated overnight with anti-FLAG (1:1000, Sigma), anti-ATP synthase subunit alpha (1:1000, Invitrogen), or anti-Hsp90 (1:500, Abcam) primary antibody in 0.1% Tween/Odyssey buffer. Blots were washed three times for 10 min with 0.1% Tween/PBS and incubated with IR Dye800 donkey anti-mouse secondary antibody (1:10,000, Rockland) for 1 h in 0.1% Tween/Odyssey buffer. Following three washes as above, blots were imaged using the Odyssey Infrared Imager (Li-cor).
Expression of mSIRT3 proteins
The full length and fragments of mSIRT3L were expressed in BL21(DE3) Star cells (Novagen) with co-expression of chaperone groES-groEL-tig (pG-Tf2, Takara). A single colony was inoculated in LB media containing 20 μg/mL chloramphenicol and 100 μg/mL ampicillin for plasmid selection and 5 ng/mL tetracycline for chaperone induction at 37°C, 250 rpm until the OD600 reached 0.3. The culture was then transferred to 18°C, 250 rpm until the OD600 reached 0.6. IPTG was added to a final concentration of 0.3 mM and expression was continued at 18°C, 180 rpm overnight. Cells were collected by centrifugation and the pellet was resuspended in Lysis buffer (50 mM Tris-HCl, 300 mM NaCl, 10% glycerol, 10 mM MgCl2, pH 8.8, 5 mL/gm of cell pellet) and sonicated to open the cells. Supernatant was separated from cell debris by centrifugation at 10,000 g for 40 min at 4°C and loaded onto a Ni-NTA column (Qiagen). The column was washed with 10 CV (column volume) of the Binding buffer (50 mM Tris-HCl, 10% glycerol, 300 mM NaCl, 10 mM MgCl2, pH 8.0), 10 CV of the Wash buffer (50 mM Tris-HCl, 10% glycerol, 300 mM NaCl, 20 mM imidazole, 10 mM MgCl2, pH 8.0) and eluted with the Elution buffer (50 mM Tris-HCl, 10% glycerol, 300 mM NaCl, 250 mM imidazole, 10 mM MgCl2, pH 8.0). The eluted protein was dialyzed against the Binding buffer and then digested with TEV protease (Invitrogen) to remove the N-terminal His tag. The tag-free enzyme was isolated by a 2nd Ni–NTA column. The mSIRT3L-54-334 was further purified to >95% purity by an S200 column (GE) judged by SDS-PAGE.
Enzyme assay
The partially purified full length and different fragments of mSIRT3L were tested for deacetylation activity with the mass spectrometry based assay reported previously.19 Saturating amounts of peptide substrate (20 μM) and βNAD+ (3 mM) were added to the reaction with enzyme concentration starting at 4 μM and serial diluted in a 1:2 ratio. Reactions were incubated at 25°C and stopped at 0, 15, 30, 60, 90, 120, 150, 180 minute time points with 10% formic acid with 50 mM nicotinamide. The conversion of substrate to product was determined by mass spectrometry in conjunction with a RapidFire system (BioTrove).
Inhibition analyses
To identify the mechanism of inhibition of SRT1720, a small molecule SIRT3 inhibitor, and nicotinamide against mSIRT3L-54-334, we used a single acetylated peptide derived from the SIRT3 substrate AceCS2 (EILVVKRLPKTRSG-KAc-VMRRLLRKIITSEAQ, KAc is acetylated lysine). For compound inhibition against acetylated AceCS2 peptide, eight concentrations of nicatinamide (15, 3.75, 0.94, 0.23, 0.06, 0.015, 0.004, and 0.001 μM) or nicotinamide (85, 28.3, 9.4, 3.2, 1.05, 0.35, 0.12, and 0.04 μM) were used in the reaction. For each of the respective compound concentrations, the deacetylation rate was measured at five concentrations of acetylated peptide (5, 2.5, 1.25, 0.62, and 0.31 μM) with mSIRT3L-54-334 (1.47 nM) and NAD+ (220 μM) kept constant. Substrate inhibition was detected at high concentrations forcing the use of lower amounts of acetylated peptide. The reactions were carried out at 25°C. The time course was determined from the linear portions of the AceCS2 Km curve (ran at NAD+ Km) and the reaction was stopped with 10% formic acid with 50 mM nicotinamide and the conversion of substrate to product was determined by mass spectrometry.
For compound inhibition against NAD+, eight concentrations of SRT1720 (15, 3.75, 0.94, 0.23, 0.06, 0.015, 0.004, 0.001 μM) or nicotinamide (80, 20, 5, 1.25, 0.31, 0.08, 0.02, and 0.005 μM) were used. For each of the respective compound concentrations, the deacetylation rate was measured at eight fixed concentrations of NAD+ (2000, 1000, 500, 250, 125, 63, 31, and 16 μM) with mSIRT3L-54-334 (1.47 nM) and acetylated AceCS2 peptide (2 μM) kept constant at 25°C. The time course was determined from the linear portions of the NAD Km curve (ran at AceCS2 Km) and the reaction was stopped with 10% formic acid with 50 mM nicotinamide and the conversion of substrate to product was determined by mass spectrometry.
To determine the inhibition constants (Ki) and mechanism of action toward NAM and SRT1720, the initial velocities of several series of reactions were calculated by global non-linear regression. Briefly, varying concentrations of inhibitor were titrated against fixed concentrations of substrate (NAD+ or AceCS2). The observed rates of reaction with respect to substrates and inhibitors were then globally fit to the mixed noncompetitive inhibition equation [eq. (1)] using the software package GraFit 6.0.5 (Erithacus Software).
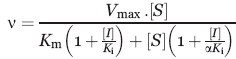 |
(1) |
where, Ki and αKi are the competitive and uncompetitive inhibition constants, respectively. The mechanisms of inhibition are determined from the alpha constant value. α = 1, indicates noncompetitive inhibition, α ≫ 1, competitive inhibition and α ≪ 1 uncompetitive inhibition.49
GenBank deposition
The sequences for mouse, rat, and rabbit SIRT3 have been deposited at GenBank with accession numbers of EU886466, EU886468, and EU886467, respectively.
Acknowledgments
The authors thank Drs. Peter J. Elliott, Olivier Boss and Eneida Pardo for helpful comments on the manuscript.
Glossary
Abbreviations:
- AceCS2
acetyl-CoA synthetase 2
- GDH
glutamate dehydrogenase
- MPP
mitochondrial matrix processing peptidase
- mSIRT3L
a longer form of mouse SIRT3
- NAM
nicotinamide.
References
- 1.Dutnall RN, Pillus L. Deciphering NAD-dependent deacetylases. Cell. 2001;105:161–164. doi: 10.1016/s0092-8674(01)00305-1. [DOI] [PubMed] [Google Scholar]
- 2.Rusche LN, Kirchmaier AL, Rine J. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem. 2003;72:481–516. doi: 10.1146/annurev.biochem.72.121801.161547. [DOI] [PubMed] [Google Scholar]
- 3.Straight AF, Shou W, Dowd GJ, Turck CW, Deshaies RJ, Johnson AD, Moazed D. Net1, a Sir2-associated nucleolar protein required for rDNA silencing and nucleolar integrity. Cell. 1999;97:245–256. doi: 10.1016/s0092-8674(00)80734-5. [DOI] [PubMed] [Google Scholar]
- 4.Dryden SC, Nahhas FA, Nowak JE, Goustin AS, Tainsky MA. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol Cell Biol. 2003;23:3173–3185. doi: 10.1128/MCB.23.9.3173-3185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 6.Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci USA. 2006;103:10230–10235. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell. 2007;28:91–106. doi: 10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 8.Schwer B, Verdin E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008;7:104–112. doi: 10.1016/j.cmet.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 9.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 10.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 11.Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG, Kouzarides T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002;21:2383–2396. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 13.Haigis MC, Guarente LP. Mammalian sirtuins–emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 14.North BJ, Verdin E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 2004;5:224. doi: 10.1186/gb-2004-5-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 16.Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C, Permutt MA, Imai S. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2:105–117. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 18.Bordone L, Motta MC, Picard F, Robinson A, Jhala US, Apfeld J, McDonagh T, Lemieux M, McBurney M, Szilvasi A, et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006;4:e31. doi: 10.1371/journal.pbio.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:861–870. doi: 10.1164/rccm.200708-1269OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saunders LR, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007;26:5489–5504. doi: 10.1038/sj.onc.1210616. [DOI] [PubMed] [Google Scholar]
- 22.Vaitiekunaite R, Butkiewicz D, Krzesniak M, Przybylek M, Gryc A, Snietura M, Benedyk M, Harris CC, Rusin M. Expression and localization of Werner syndrome protein is modulated by SIRT1 and PML. Mech Ageing Dev. 2007;128:650–661. doi: 10.1016/j.mad.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 23.Firestein R, Blander G, Michan S, Oberdoerffer P, Ogino S, Campbell J, Bhimavarapu A, Luikenhuis S, de Cabo R, Fuchs C, et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE. 2008;3:e2020. doi: 10.1371/journal.pone.0002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Zhao W, Thiyagarajan M, MacGrogan D, Rodgers JT, et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006;281:21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- 25.Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci USA. 2006;103:10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006;126:941–954. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 29.Allison SJ, Milner J. SIRT3 is pro-apoptotic and participates in distinct basal apoptotic pathways. Cell Cycle. 2007;6:2669–2677. doi: 10.4161/cc.6.21.4866. [DOI] [PubMed] [Google Scholar]
- 30.Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rose G, Dato S, Altomare K, Bellizzi D, Garasto S, Greco V, Passarino G, Feraco E, Mari V, Barbi C, et al. Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp Gerontol. 2003;38:1065–1070. doi: 10.1016/s0531-5565(03)00209-2. [DOI] [PubMed] [Google Scholar]
- 32.Bellizzi D, Rose G, Cavalcante P, Covello G, Dato S, De Rango F, Greco V, Maggiolini M, Feraco E, Mari V, et al. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics. 2005;85:258–263. doi: 10.1016/j.ygeno.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 33.Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci USA. 2002;99:13653–13658. doi: 10.1073/pnas.222538099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol. 2002;158:647–657. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cooper HM, Spelbrink JN. The human Sirt3 protein deacetylase is exclusively mitochondrial. Biochem J. 2008;411:279–285. doi: 10.1042/BJ20071624. [DOI] [PubMed] [Google Scholar]
- 36.Scher MB, Vaquero A, Reinberg D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007;21:920–928. doi: 10.1101/gad.1527307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang YH, Chen YH, Zhang CY, Nimmakayalu MA, Ward DC, Weissman S. Cloning and characterization of two mouse genes with homology to the yeast Sir2 gene. Genomics. 2000;69:355–369. doi: 10.1006/geno.2000.6360. [DOI] [PubMed] [Google Scholar]
- 38.Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem. 2005;280:13560–13567. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- 39.Hiller M, Platzer M. Widespread and subtle: alternative splicing at short-distance tandem sites. Trends Genet. 2008;24:246–255. doi: 10.1016/j.tig.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 40.Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 41.Claros MG, Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- 42.Small I, Peeters N, Legeai F, Lurin C. Predotar: a tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics. 2004;4:1581–1590. doi: 10.1002/pmic.200300776. [DOI] [PubMed] [Google Scholar]
- 43.Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- 44.Landry J, Slama JT, Sternglanz R. Role of NAD(+) in the deacetylase activity of the SIR2-like proteins. Biochem Biophys Res Commun. 2000;278:685–690. doi: 10.1006/bbrc.2000.3854. [DOI] [PubMed] [Google Scholar]
- 45.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 46.Sauve AA, Schramm VL. Sir2 regulation by nicotinamide results from switching between base exchange and deacetylation chemistry. Biochemistry. 2003;42:9249–9256. doi: 10.1021/bi034959l. [DOI] [PubMed] [Google Scholar]
- 47.Jackson MD, Schmidt MT, Oppenheimer NJ, Denu JM. Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J Biol Chem. 2003;278:50985–50998. doi: 10.1074/jbc.M306552200. [DOI] [PubMed] [Google Scholar]
- 48.Borra MT, Langer MR, Slama JT, Denu JM. Substrate specificity and kinetic mechanism of the Sir2 family of NAD+-dependent histone/protein deacetylases. Biochemistry. 2004;43:9877–9887. doi: 10.1021/bi049592e. [DOI] [PubMed] [Google Scholar]
- 49.Copeland RA. Enzymes. 2nd ed. New York: Wiley-VCH, Inc; 2000. [Google Scholar]