

Abstract
The tail needle, gp26, is a highly stable homo-trimeric fiber found in the tail apparatus of bacteriophage P22. In the mature virion, gp26 is responsible for plugging the DNA exit channel, and likely plays an important role in penetrating the host cell envelope. In this article, we have determined the 1.98 Å resolution crystal structure of gp26 bound to xenon gas. The structure led us to identify a calcium and a chloride ion intimately bound at the interior of α-helical core, as well as seven small cavities occupied by xenon atoms. The two ions engage in buried polar interactions with gp26 side chains that provide specificity and register to gp26 helical core, thus enhancing its stability. Conversely, the distribution of xenon accessible cavities correlates well with the flexibility of the fiber observed in solution and in the crystal structure. We suggest that small internal cavities in gp26 between the helical core and the C-terminal tip allow for flexible swinging of the latter, without affecting the overall stability of the protein. The C-terminal tip may be important in scanning the bacterial surface in search of a cell-envelope penetration site, or for recognition of a yet unidentified receptor on the surface of the host.
Keywords: tail needle gp26, bacteriophage P22, xenon, ion-trap, trimeric coiled-coil
Introduction
P22 is a Salmonella infecting phage of the Podoviridae family, which is characterized by a short noncontractile tail.1 The proteins in the tail apparatus, which is roughly 2.8 MDa in size,2 participate in packaging the viral genome into the virion during replication, and in injecting the viral DNA into the host during infection. Five P22-encoded proteins are present in various stoichiometries in the P22 tail, which is far simpler than the tail from other bacteriophages, such as long-tailed phage like T43,4 or even most other short-tailed phages like T7.5 This simplicity has made it considerably easier to assign distinct functions to each of the components of the tail. At the “top” of the tail, most proximal to the capsid head, is the portal protein, which forms the DNA conduit through which DNA is translocated during packaging and injection.6 Surrounding the inner hub of the tail are six tailspike homotrimers that enable the phage to adsorb to the lipopolysaccharide (LPS) protruding from the host cell surface.7 The portal protein channel extends as a hollow tube formed by the tail accessory factors gp4 and gp10.8–11 Gp4 binds to portal protein and is reported to have murein hydrolase activity, which may be important in digestion of the peptidoglycan layer of the Salmonella cell wall during infection.12 Directly below the dodecamer of gp4, a hexamer of gp10 extends channel to the tail needle gp26,11 which forms a strikingly elongated protein fiber.8,9,13,14 Gp26 serves as the plug for the portal protein channel and thus stabilizes DNA within the capsid by preventing its leakage out of the capsid.15 After infection, gp26 is found inside the host, suggesting the protein is ejected to allow delivery of viral genome into the Salmonella cell.16 It remains unknown if the ejection of gp26 into the host is mediated by a direct penetration of gp26 through the host cell envelope or by interaction with a yet unidentified receptor.
The crystal structure of the P22 tail needle was recently determined to 2.1 Å resolution.14 The protein folds into a homo-trimeric triple stranded coiled-coil fiber ∼240 Å in length. Gp26 is composed of four domains: an extended N-terminal domain presenting a 310 helix between residues Pro4 and Asn7 responsible for binding to gp10,17 a ∼100 residue long triple stranded coiled-coil (Domain II) followed by a triple stranded beta-helix (Domain III), and a C-terminal helical domain termed the lazo-domain (Domain IV). The latter represents the outermost tip of the virion and presents a conserved tyrosine residue, hypothesized to be important for lipid puncturing.14 Biophysical investigations of the tail needle stability have revealed some of the extraordinary properties of gp26: the fiber is stable up to 88°C, ∼6.5M guanidine hydrochloride, or 10% SDS and remains trimeric in an anhydrous state.17 The main determinant for gp26 stability resides in helical Domain II, which consists of a continuous trimeric helical coiled-coil. Domain II is stabilized by a continuous spine of hydrophobic residues stacked at the internal helical interface, together with a network of salt bridges latching across the outside of the gp26 helices.14 The C-terminal Domains III and IV are not trimeric when expressed alone but dramatically enhance the structural stability of the gp26 helical core in the context of the full fiber. Although the molecular basis for this stabilization is unknown, replacing the gp26 C-terminal domain with the short (27 residues) foldon domain of the phage T4 fibritin results in a fully stable and self-refolding protein fiber,18 suggesting the foldon rescues the role played by Domains III and IV in the full length fiber.
Almost counterintuitive to its remarkable stability, gp26 appears to have intrinsic structural flexibility. The asymmetric unit of the gp26 crystal contains two trimeric gp26 fibers, which show dramatic structural differences in the C-terminal tip (Domains III and IV).14 This region of the fiber swings up to 18° with respect to the helical core, suggesting the existence of a hinge between the C-terminal tip and the stiff fibrous Domain II.14 This flexibility in a protein that melts at 88°C is likely to have profound biological implications, and raises the question of how the tail needle forms a simultaneously ultra-stable and flexible fiber. In this report, we investigated the structural basis for gp26 flexibility using crystallographic analysis of gp26 crystals pressurized under xenon gas. We show that xenon gas identifies a network of internal cavities at the internal trimerization interface, whose distribution and location fit well with the C-terminal flexibility previously observed both in solution17 and in the crystal structure.14.
Methods
Protein expression, purification, and crystallization
The expression, purification, and crystallization of gp26 were carried out as previously described.14,19 To obtain larger, single crystals of gp26, powdered borosilicate glass (1.5–2.5 μm particle size) was added to the protein to a final concentration of 0.0067% (w/v), just before setting up the crystal drops. The presence of the glass shards aided in crystal nucleation, resulting in fewer, larger crystals in the drop. Crystals appeared within 1 week, and continued to grow for ∼1 month. Crystals were transferred to cryo-solution (mother liquor plus 27% ethylene glycol) before flash freezing in liquid nitrogen.
Xenon derivatization, data collection, and structure determination
Gp26 crystals were harvested in cryo-protectant with a 0.5 mm cryo-loop and derivatized with xenon gas using a Hampton Research Xenon derivatization chamber. Crystals were pressurized with xenon gas between 250–550 psi for various times and immediately flash frozen in liquid nitrogen. The crystals used for data collection (Xe-HR, Xe-High, and Xe-Low) were derivatized for 2 h at 450 psi in cryo-protectant. The NaBr dataset was measured using a gp26 crystal soaked for ∼30 min in cryo-protectant supplemented with 500 mM sodium bromide (NaBr) prior to freezing in liquid nitrogen. Datasets Xe-HR and NaBr were collected at the Cornell High Energy Synchrotron Source (CHESS) beamlines F1 and F2, respectively, while datasets Xe-High and Xe-Low were collected at the National Synchrotron Light Source (NSLS) beamline X6a. Xe-HR was collected on an ADSC Quantum-270 CCD detector, and Xe-High, Xe-Low, and NaBr were collected on an ADSC Quantum-210. All datasets were integrated and scaled using the HKL-2000 suite20 and analyzed further using CCP4 programs.21 The crystals belonged to space group P21 and contained two gp26 homotrimers per asymmetric unit (also referred to as Bundle A and Bundle B). The structure of gp26 bound to xenon gas was determined by molecular replacement with PHASER22 using the underivatized structure of gp26 (PDB ID: 2POH) as a search model. Initial refinement by rigid body and positional refinement using REFMAC23 lowered the Rfree to ∼35%. After manual rebuilding of misplaced residues, the solvent was built in Coot24 and the structure further refined using SHELX, which resulted in a final Rfactor of 23.4% and an Rfree of 26.8%. The final deposited model has good geometry, with RMSD of bonds = 0.006 Å and RMSD of angles = 1.2°, with no disallowed Ramachandran residues. Data collection and refinement statistics are summarized in Table I.
Table I.
X-ray Data Collection and Refinement Statistics
| Data set name | Xe-HR | Xe-High | Xe-Low | NaBr |
|---|---|---|---|---|
| Data collection statistics | ||||
| Beam line | CHESS F1 | BNL-X6a | BNL-X6a | CHESS-F2 |
| Wavelength (Å) | 0.9179 | 0.9541 | 1.746 | 0.9202 |
| Space group | P21 | P21 | P21 | P21 |
| Ref (total/unique) | 233,295/103,949 | 463,519/72,781 | 280,225/50,583 | 205,964/57,513 |
| Unit cell (Å) | a = 40.539 | a = 40.135 | a = 40.254 | a = 40.19 |
| b = 114.53 | b = 114.30 | b = 114.94 | b = 114.71 | |
| c = 172.045 | c = 171.38 | c = 171.71 | c = 171.39 | |
| β-angle (°) | 91.014 | 90.477 | 90.628 | 90.65 |
| Resolution (Å) | 20.0–1.95 | 40.0–2.2 | 30.0–2.5 | 30.0–2.4 |
| Completeness (%) | 91.9 (74.1) | 92.8 (66.9) | 94.1 (68.1) | 93.9 (81.2) |
| Redundancy | 2.4 (1.9) | 6.4 (4.0) | 5.5 (4.1) | 3.6 (3.1) |
| Rsyma | 8.3 (37.6) | 6.5 (41.3) | 8.6 (25.7) | 7.6 (35.6) |
| 〈I〉 / 〈σ(I) 〉 | 11.1 (1.3) | 17.6 (2.3) | 14.2 (3.7) | 16.2 (3.4) |
| Refinement statistics (Xe-HR) | ||||
| Resolution reflection used (Å) | 20–1.98 | |||
| Protein residues in the model | ||||
| Bundle A | 708 | |||
| Bundle B | 668 | |||
| Water molecules | 1590 | |||
| Ligand atoms | 18 | |||
| Rfactor/Rfreeb (%) | 23.4/26.8 | |||
| Rmsd bond lengths (Å) | 0.007 | |||
| Rmsd bond angles (deg) | 1.26 | |||
| ϕψ most favored (%) | 90.1 | |||
| ϕψ additionally favored (%) | 7.2 | |||
| ϕψ generously favored (%) | 2.6 | |||
| ϕψ disallowed (%) | 0 | |||
| Average B-factor (Å2) | ||||
| Wilson B-factor | 22.4 | |||
| Bundle A | 37.9 | |||
| Bundle B | 41.8 | |||
| Water | 44.2 | |||
| Calcium (Ca1) | 21.4 | |||
| Chloride (Cl1) | 28.2 | |||
| Xenon (Xe1, Xe2, Xe3, Xe4, Xe5, X5, Xe6, Xe7) | 59.9, 34.0, 39.4, 73.9, 33.8, 31.6, 96.2 | |||
The numbers in parenthesis refer to the statistics for the outer resolution shell.
Rsym = Σi,h|I(i,h) − <I(h)> |/ Σi,h|I(i,h) | where I(i,h) and <I(h)> are the ith and mean measurement of intensity of reflection h.
The Rfree value was calculated using 5% of the data.
Analysis of cavities, fitting in the cryo-EM structure, and illustrations
Analysis of cavities, and calculation of cavity maps was performed with the program VOIDOO,25 using a water molecule as the probe sphere. The structure of gp26 derivatized with xenon gas was manually fit into the asymmetric cryo-EM reconstructions of the P22 virion (accession codes EMD-1220 and EMD-1222)8,9 and the fit refined using the program Chimera.26 Sequence alignments were generated using ClustalW27 with default parameters and displayed using JalView.28 All other figures were created with Pymol, using FFT (1994) to create maps with one half the normal grid spacing; with the exception of figures of cryo-EM reconstructions, which were displayed using Chimera.26
Results
Investigating the structural plasticity of gp26 with xenon gas
We have studied the interaction of the noble gas xenon with gp26. Large crystals of the tail needle were obtained by adding 0.0067% (w/v) powdered borosilicate glass (1.5–2.5 μm particle size) to the protein prior to crystallization. This procedure was found to aid in crystal nucleation, resulting in more reproducible and larger crystals of gp26, which survived prolonged pressurization with xenon gas. Several datasets were collected from gp26 crystals derivatized between 5 min and 18 h. Although short derivatization with xenon reproducibly improved gp26 diffraction quality, prolonged pressurization (>8 h) had a negative effect on diffraction quality, likely due to partial dehydration in the pressurization chamber. A complete dataset to 1.95 Å resolution was collected at the CHESS beamline F1, at a wavelength of 0.92 Å (named Xe-HR in Table I) using a large gp26 crystal derivatizated with xenon for 2 h at 450 psi. The structure was solved by molecular replacement using as the search model the structure of underivatized gp26 (PDB ID: 2POH).14 Although pressurization in the presence of xenon did not change the unit cell parameter and lattice geometry, xenon-pressurized gp26 crystals were not pseudo-merohedrally twinned29 as observed for all wild type crystals.14 This remarkable improvement in the crystal order is likely attributed to the pressurization process itself, which has been shown to improve the diffraction quality and lattice order of biological crystals.30 Using phase angles from the refined model, nine potential xenon sites were identified in a FObs − FCal electron density difference map contoured at 4σ above background [Fig. 1(a)]. All putative sites were located in the interior of gp26, at the trimerization interface, which spans the entire length of the fiber (∼240 Å). To confirm that the putative sites were indeed xenon atoms, anomalous difference maps were also computed. At the wavelength used for data collection, λ = 0.92 Å (Table I), the imaginary component of the anomalous scattering (f″) for xenon is ∼5 electrons, which is comparable to that of selenium at the anomalous edge. For a well-occupied site, this usually results in an anomalous peak of density 5–15σ above background. However, two of the nine potential sites in the helical core [named Ca1 and Cl1 in Fig. 1(a)] had significant lower anomalous signal (∼1–2σ) and were only marginally visible above noise. To conclusively determine if these sites belonged to poorly occupied xenon sites or ions possibly bound to gp26, we carried out a two-wavelength anomalous diffraction (MAD) experiment using a single xenon-pressurized gp26 crystal (Xe-High and Xe-Low datasets in Table I). The first dataset, Xe-Low, was measured in the vicinity of xenon anomalous edge, at 7.0 keV (λ = 1.75 Å), where xenon exhibits a remarkably high anomalous signal (f″ ∼10 electrons). The second dataset, Xe-High, was a high energy remote measured at 12.7 keV (λ = 0.95 Å), where xenon theoretical f″ is roughly half than at low energy (∼5 vs. ∼10 electrons). Anomalous difference maps computed using diffraction data from the Xe-High and Xe-Low datasets, revealed that for seven of the nine potential sites [named Xe1–Xe7 in Fig. 1(a)], the intensity of the anomalous peaks at lower energy was ∼1/3 to 1/2 greater than that at higher energy [blue density and red density, respectively in Fig. 1(b–d)], as expected for genuine xenon atoms. Likewise, an underivatized control dataset collected at low and high energy showed no density for any of these seven sites (data not shown). Thus, seven peaks of density buried at the interior of tail needle gp26 core were assigned to xenon atoms [Xe1–Xe7 in Fig. 1(a)].
Figure 1.
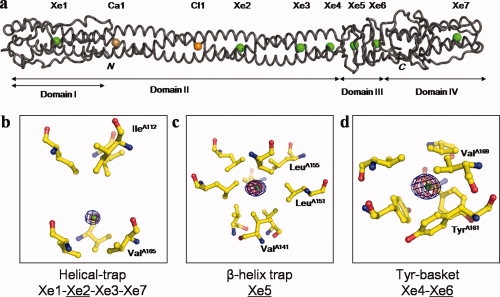
Three-dimensional structure of phage P22 tail needle bound to xenon gas. (a) Worm representation of tail needle gp26 bound to seven xenon atoms, one calcium, and one chloride atom (represented as green, gold, orange spheres, respectively). The structure was determined to 1.98 Å resolution and refined to an Rfree of 26.8% (Table I). The tail needle is ∼240 Å homotrimer, consisting of four structural domains (indicated by horizontal arrows). All xenon and chloride atoms were found in cavities at the gp26 internal trimerization interface. (b–d) Close-up view of the three classes of xenon sites observed inside gp26 core: the orientation of gp26 shown is perpendicular to that in (a). Gp26 residues lining the binding site are depicted as ball-and-sticks with nitrogen and oxygen atoms colored in blue and red, respectively. Only residues of protomer A of gp26 are labeled. The blue and red densities around xenon atoms represent anomalous difference maps computed using X-ray data measured at low energy (λ = 1.75 Å) and high energy (λ = 0.95 Å), respectively (datasets Xe-Low and Xe-High in Table I). All anomalous maps were computed to 2.5 Å resolution and contoured at 15σ above background. The refined B-factor to 1.98 Å resolution for xenon atoms Xe2, Xe3, Xe5, and Xe6 ranges between 32 and 39 Å2, whereas Xe1, Xe4, and Xe7 have significantly higher B-factor, between 60 and 96 Å2 (Table I).
Chemistry of xenon binding to tail needle gp26
The seven xenon sites bound to gp26 can be classified into three categories [Fig. 1(b–d)]. The first set of xenon sites is composed of xenon sites Xe1, Xe2, Xe3 found in the helical Domain II of gp26, and Xe7, which is located in the inverted helices of Domain IV. Each of these xenon atoms resides in a “helical trap” [Fig. 1(b)], formed by two planes of hydrophobic residues, typically a triplet of isoleucines and a triplet of valines (or isoleucines, as in the case of Xe7). Each plane is formed by residues protruding inward from the helices of three identical gp26 protomers (Val42 and Ile45 for Xe1; Val105 and Ile112 for Xe2; Val126 and Ile133 for Xe3; Ile205 and Ile208 for Xe7). The xenon atom is held in the hydrophobic cavity, centered between the two hydrophobic planes, with xenon:carbon distances ranging from 3.38 to 5.70 Å. This is fairly representative of the average distances observed in most protein crystals derivatized with xenon gas and is consistent with xenon engaging in weak hydrophobic and van der Waals contacts with the amino acid side chains.31–34 Two of these helical xenon traps, the most N- and C-terminal, show the lowest anomalous difference peak heights (Xe1: 15σ, Xe7: 12σ), whereas the other two xenon helical traps, Xe2 and Xe3, have much larger peaks of 31σ and 21σ for Xe2 and Xe3, respectively. Consistently, the refined B-factors of the two terminal Xe sites (59.9 Å2 for Xe1 and 96.2 Å2 for Xe7) are considerably higher than the B-factors of the internal helical traps (34.0 Å2 for Xe2 and 39.4 Å2 for Xe3) (Table I).
The second type of xenon binding site has only one example in gp26, Xe5, which is found in a “β-helix trap” [Fig. 1(c)]. This site is located toward the base of the β-helix of gp26, centered in the first layer of the β-helix. Nine hydrophobic residues line the xenon trap, three donated by each of the gp26 protomers. Val141 forms the base of the pocket, Leu151 lines the sides of the cavity, and Leu155 caps the top of the pocket. This site is a 31σ peak in an anomalous Fourier difference map computed using X-ray data measured at low energy (Xe-Low dataset, Table I). The strong anomalous signal is likely due to a well-occupied xenon atom that engages in hydrophobic and van der Waals contacts with nine, instead of six residues as in the “helical trap,” surrounding a cavity ∼13.4 Å3 in volume. The highly occupied Xe5 slightly perturbs the structure of the cavity where it is found, causing Leu155 to swing upward to enlarge the diameter of the cavity from 7.8 to 8.4 Å.
The last set of xenon sites observed in the gp26 structure is tapped in a novel “tyrosine basket motif” [Fig. 1(d)]. These motifs are built by a plane of three leucine or valine residues protruding from each gp26 strand, and a basket of three tyrosines (Leu136 and Tyr140 for Xe4; Val169 and Tyr161 for Xe6). The two sites, Xe4 and Xe6, are very similar in the architecture, with the hydrophobic side chains of leucines/valines on one face, and the bulky aromatic rings of the tyrosines resembling a basket on the other face. Additionally, the two xenon-occupied cavities have similar size, with a diameter of 9.3 Å for the Xe6 site and 8.4 Å for the Xe4 site. Despite the similarities in the two-tyrosine basket motifs, the xenon sites have vastly different anomalous peak sizes. The Xe4 site, which is located near the end of the helical core, is the second smallest site in gp26 with a peak height of 13σ, whereas the largest site in the tail needle was Xe6, with a peak height of 37σ. The B-factors for the two sites, 73.9 Å2 for Xe4 and 31.6 Å2 for Xe6 (Table I), support the idea that the height of a xenon peak is not linearly related to the volume of the cavity occupied by the noble gas. The inequality in the refined B-factor of Xe6 when compared with Xe4 can be explained by the position of the site in the protein. The Xe4 site is located in the stable and rigid helical core Domain II, which is less likely to allow the xenon gas to enter and therefore results in a less well-defined site. On the other hand, the Xe6 site, located in the flexible β-helix domain, can allow the gas to readily enter the structure between the strands of the β-helix. Consistently, the two strongest xenon sites in the protein (Xe5 and Xe6) are located in the β-helix domain.
Ions trapped at gp26 trimeric coiled coil interface
In addition to the seven xenon sites, two additional weak anomalous peaks were detected inside the gp26 helical core. These sites, named Ca1 and Cl1 in Figure 2(a), appeared as 3.5σ and 2σ anomalous peaks, respectively, at low energy [Fig. 2(c,d), blue density] but had negligible signal at high energy. However, in a difference electron density map computed using Fourier coefficient |FObs − FCal|, both sites appeared as ∼5σ peaks [Fig. 2(c,d), green density], which suggested the weak anomalous signal was due to the presence of atoms with low anomalous signal, as opposed to poorly occupied xenon atoms. In the previously reported lower resolution structure of gp26,14 both sites were erroneously interpreted as water molecules, which gave unusually low B-factor values (∼1 Å2) in the refined deposited model. Both sites are located in the center of gp26 helical core Domain II, between residues 60–100. To assign chemical identities to the two peaks of density, we carefully analyzed the size of the binding cavity where the two peaks of density are localized as well as the chemistry of the residues surrounding the density.
Figure 2.
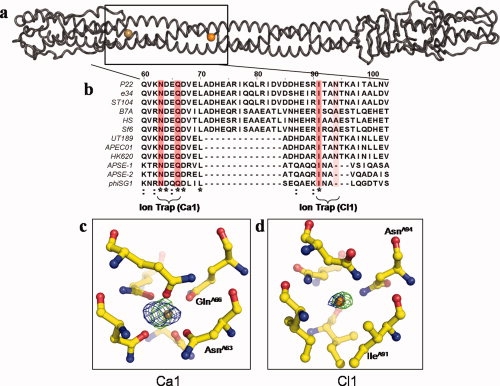
Ions trapped at the interior of gp26 trimerization interface. (a) Worm representation of tail needle gp26, with the calcium and chlorine ions displayed as a gold sphere and orange sphere, respectively. (b) Sequence alignment of the putative tail needle sequence from 12 P22-like phages. For simplicity, only the amino sequence spanning residues 60–103 is displayed. Invariant residues interacting with the two ions are highlighted in dark pink, and partially conserved interacting residues in light pink. Other conserved residues are indicated below the alignment, where an asterisk (*) indicates an invariant residue, and a colon (:) a highly conserved residue. (c, d) Close-up view of the calcium and chloride ions observed at the gp26 internal trimerization interface. In both panels, the green density around the ions represents a FObs − FCal difference map computed using structure factor amplitudes from the final refined model missing the Ca1 and Cl1 ions. The density was computed to 2.5 Å and contoured at 5σ. The calcium site has a refined B-factor of ∼21 Å2 and the chlorine site ∼28 Å2, respectively. The blue density around the ions represents an anomalous map computed using anomalous differences measured at low energy (λ = 1.75 Å, dataset Xe-Low in Table I) and contoured at 2σ.
The first site, Ca1 [Fig. 2(c)] is held in an octahedral cage formed by the side chains of Asn63 and Gln66 protruding from each of the three-gp26 protomers [Fig. 2(b)]. The putative density occupies the center of the octahedron, whose center of mass is only 2.3–2.5 Å from the side chains of Asn63 and Gln66. Although chlorine is commonly found within the inner core of trimeric coiled-coil structures trapped by the ɛ- and/or δ-nitrogen of Asn/Gln side chains,35,36 stereochemical considerations suggested that chloride was too large to fit inside this region of gp26 helical core. In fact, as chloride has a van der Waals radius of 1.81 Å, and nitrogen ∼1.5 Å,37 the minimal distance possible between a chloride ion and the side chains of Asn/Gln is ∼3.25 Å.35,38 This was roughly 1 Å longer that the distance between the observed peak of density and the refined atomic positions of Asn63 and Gln66 nitrogens. In contrast to halogens, metals are known to form ionic complexes of much shorter distance. Eight metals (M) have been observed bound to the oxygen (O) of Asn/Gln side chains with M-O distances ranging from 2.07 to 2.41 Å,39 namely Na, Mg, Ca, Mn, Fe, Co, Cu, Zn. Although the standard deviation for the M-O distance varies considerably (∼0.05 to 0.20 Å), and thus all of the afore mentioned metals could potentially occupy the experimentally observed density, among them, calcium is the only element to have significantly higher anomalous scattering at λ = 1.75 Å (f″ ∼1.5 electrons) compared with λ = 0.95 Å (f″ ∼0.5 electrons). The small anomalous signal observed at low energy (3.5σ) compared with the negligible signal at high energy was fully consistent with the presence of an octahedrally coordinated calcium ion. Additionally, when a calcium ion was placed in the density, the refined B-factor was ∼20 Å2, which is comparable to the B-factors of the surrounding atoms (∼15–25 Å2). Thus we interpret the first peak of density trapped between Asn63 and Gln66 as calcium (Ca1) [Fig. 2(c)].
The second peak of density buried at the gp26 core is held between the side chains of Ile91 and Asn94 protruding from each of the three protomers of gp26 [Fig. 2(d)]. We interpret this second peak of density as a chloride atom (Cl1) based on four considerations. First, the center of mass of the electron density is ∼3.2 Å from the δ-nitrogen of Asn94 and ∼4.3 Å from the methylpropyl group of Ile91, which is in good agreement with the distance expected for a chloride atom interacting with a protein side chain. Second, the greater anomalous signal observed at low energy [Fig. 2(d)] is consistent with the anomalous signal of chloride that is negligible at λ = 0.95 Å (f″ ∼0.4 electron) and slightly above background at λ = 1.75 Å (f″ ∼0.8 electron). Third, the refined B-factor value for this atom, ∼28 Å2, is also comparable to the B-factors of the surrounding atoms (∼15–25 Å2). Fourth, the chloride site found inside the gp26 helical core is very similar to the chloride seen in the five-stranded coiled-coil protein, COMP,38 which is trapped by an analogous set of glutamines and leucines.
To determine if this chloride ion can freely exchange with the outside, we soaked gp26 crystals in 500 mM sodium bromide for 30 min (Table I, NaBr dataset) followed by a data collection at the bromide anomalous edge (λ = 0.92 Å). As the anomalous signal for bromine at λ = 0.92 Å is about 10 times greater than that of chloride (f″ ∼4 vs. ∼0.4 electrons), exchanging the native Cl− with Br− would result in a strong peak of density in an anomalous difference map. Indeed several strong Br− sites were found bound to the outside of the gp26 helical core but no apparent exchange of bromide was seen for the Cl− ion (data not shown). This suggests that the chloride is specifically and tightly bound to the interior of the coiled-coil core of gp26, where it fails to exchange with the outside. Although the exact biological function of the two ions identified inside gp26 helical core (Ca1 and Cl1) remains to be elucidated, the two ions may play a structural role and stabilize the trimeric coiled-coil helical interface by providing a register to insure the proper alignment of the protomer helices during assembly.35,36,40 In agreement with the idea of a conserved biological function, gp26 residues forming the two ion traps, Asn63/Gln66 and Ile91/Asn94 are well conserved. Sequence alignment of 12 putative gp26 homologues, identified based on the conservation of the N-terminal gp10-binding domain (residues 1–50)17 [Fig. 2(b)], revealed the calcium trap residues Asn63/Gln66 are invariant, whereas the chloride trap pair residues Ile91/Asn94 are conserved in at least half of the putative gp26 homologues.
Cavities and flexibility in tail needle gp26
For xenon gas to enter a protein structure, there must be some structural plasticity so the gas can diffuse into the structure and occupy hydrophobic cavities, without significantly perturbating the overall conformation of main and side chains. In the case of proteins which have an internal lumen or channel, such as the COMP protein,38 the xenon gas can funnel into the structure and is a useful indicator of possible tunnels to transfer reaction substrates and products.41 In gp26, however, there is no lumen to provide a channel for xenon to travel through the needle [Fig. 3(a)]. The dense packing of hydrophobic residues at the gp26 inner core, as well as the polar buried surfaces occupied by Ca1 and Cl1 prevent the movement of xenon along the trimeric axis of the protein. Rather, the xenon gas must enter the tail needle during transient movement of the protein side chains, which occurs with higher propensity in regions of local flexibility. The only chance for xenon to become trapped within the needle is for it to occupy an existing cavity and engage in weak hydrophobic and van der Waals contacts with the protein side chains of its inner surface.31–34 This idea became readily apparent when the gross distribution of internal cavities in gp26 was analyzed using the software VOIDOO,25 which detects cavities in protein structures and measures their volume. In VOIDOO, cavities are defined based on the protein accessible surface, rather than the van der Waals radius.42 After stripping all xenon atoms and using a probe radius of 1.4 Å,25 VOIDOO identified seven cavities inside gp26, which correlated perfectly with the distribution of xenon sites observed crystallographically [Fig 3(a)]. Among them, cavities occupied in our structure by xenon atoms Xe2–Xe6 are completely surrounded by protein atoms and thus isolated from the outside (defined as “voids”). These cavities are completely self-contained, and show no interconnectivity. This strengthens the idea that the xenon gas must enter the tail needle during transient movement of the residues lining the cavities, which then relax into place, trapping the xenon inside. In contrast, the cavities occupied by Xe1 and Xe7 were “invaginations,” which show some degree of connectivity with the outside but would be closed off if the atomic radius of the probe were to be increased.25 The most C-terminal cavity of gp26, 17.4 Å3 in volume and occupied by Xe7, shows a clear path to the exterior of the protein, which likely explains why Xe7 has the highest B-factor in the structure (∼96 Å2). Likewise, all cavities in gp26 existed as an empty void in the deposited, underivatized structure, with the exception of the Xe1 site, which is at the far N-terminal tip of gp26 [Fig. 3(a)] and is occupied by a water molecule in the native structure.14 The fact that xenon gas is able to displace the water further confirms this cavity is an invagination that is not closed off from the outside but allows for water molecules to diffuse out of the structure. Overall, both invaginations and voids predicted in gp26 and filled by xenon gas in our study, were considerably smaller than in other deposited structures. Gp26 cavities range from a volume of 5.8 Å3 for the extreme N-terminal Xe1, to 13.4 Å3 for the β-helix site, Xe5. This is about one tenth to one thirtieth of the volume occupied by well-characterized cavities engineered in the hydrophobic core of GCN4 coiled coil homotetramers, which measure ∼80–370 Å3.43
Figure 3.
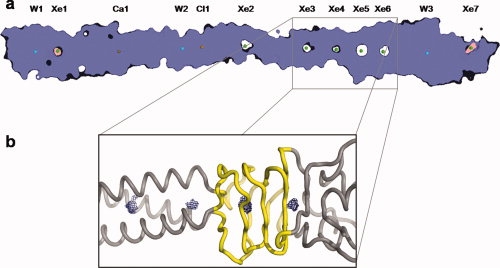
Organization of internal cavities in tail needle gp26. (a) Slice-view of the gp26 space-filling model. Xenon atoms are represented by green spheres, calcium by a gold sphere, chlorine by an orange sphere, and waters as blue spheres. Seven major cavities are detected at the trimeric binding interface using the program, VOIDOO25 all filled by xenon atoms in our crystal structure. The two terminal cavities filled by Xe1 and Xe7 and shaded in pink are “invaginations,” which are defined as cavities in partial exchange with the exterior of protein. Cavities occupied by Xe2, Xe3, Xe4, Xe5, and Xe6 and shaded in white are “voids” completely closed off from the exterior of the protein. (b) Representation of the four cavities clustered at the boundary between gp26 helical Domain II and the triple β-helix, Domain III. The gp26 backbone is represented as a grey tube, with the flexible β-helix highlighted in yellow, and the volume of cavities computed by VOIDOO25 shown as a blue mesh.
In addition to internal cavities occupied by xenon gas in our structure, gp26 interior also hosts three water molecules [indicated as W1–W3 in Fig. 3(a)] which hydrogen bond polar residues. Although W1 contacts a triplet of Tyr34 and Gln38, both W2 and W3 contact the δ-nitrogen of two sets of three histidines (His73 and His87, respectively), and form a plane with the water molecule trapped in the center. Although this type of contact is also observed for small ions, the density coordinated by the three histidines appears as a 3σ peak in an FObs − FCal difference map but shows no anomalous signal at any of the data collection energy. Thus these two sites were assigned to water molecules. The B-factors for the refined W2 and W3 sites were 19.2 and 10.3 Å2, respectively, which is consistent with well-ordered water molecules.
Discussion
It is an established concept in biology that a critical step in the folding pathway of globular proteins is the formation of a hydrophobic core.44 The amino acid residues within the interior of a folded protein are packed at very high density, analogous to the packing density seen in a crystal of small organic molecule.42 Thus, the interior hydrophobic core of proteins should not be thought like a liquid but rather as a crystal, and, consequentially, it does not retain the flexibility of liquid systems. Although the high packing density inside the hydrophobic core of a protein enhances the overall thermodynamic stability of the folded protein, cavities inside proteins exert an opposite function and typically destabilize the folded state.45 In contrast to globular proteins, the tail needle gp26 is a homotrimeric fiber, whose internal hydrophobic core is intermolecular as opposed to intramolecular. The cooperative nature of the gp26 unfolding17 and the presence of an intermolecular hydrophobic spine of 22 hydrophobic residues17 suggest that similar thermodynamic considerations made for globular proteins can be used to describe the folding stability of gp26. Most of the stabilization energy for the gp26 Domain II is generated by the hydrophobic trimerization interface that binds together the three gp26 protomers. In addition to this hydrophobic effect, the gp26 strands are latched together by a network of nineteen intermolecular salt bridges running along the surface of Domain II. These electrostatic interactions shield the external charge on gp26 surface and are responsible for the extreme stability of gp26 observed in anhydrous environment, under which the hydrophobic effect is greatly reduced.17
Buried polar residues in the hydrophobic core of tail needle gp26
In this article, we have expanded the structural characterization of the tail needle trimerization interface and identified a Ca2+ and a Cl− ion tightly bound inside the otherwise largely hydrophobic core of Domain II. These two atoms, previously erroneously interpreted as water molecules, engage in extensive buried polar interactions with gp26 side chains, and thus contribute significantly to the stabilization of the helical core. The calcium interacts in an octahedral cage formed by Asn63 and Gln66, whereas an Ile91/Asn94 triplet traps the chloride ion. What is function of polar buried interactions at the inner core of a protein fiber? The burial of polar residues in the hydrophobic interior of a protein is known to usually play a destabilizing role,46 albeit buried polar residues are commonly found in proteins.47,48 At least in the case of oligomeric coiled-coil proteins, one explanation for the occurrence of buried polar residues is that they play a role in the specificity of protein folding. In this respect, Oakley and Kim49 showed that a single polar interaction by an asparagine residue buried in the interface between an heterodimeric α-helical coiled-coil is sufficient to determine the relative orientation of the coiled-coil. In the case of phage P22 tail needle, which is a homotrimeric coiled coil fiber, the side chains of residues Asn63, Gln66 and Asn94 engage in polar contacts with Ca2+ and Cl− ions, which are tightly bound to the gp26 core and fail to exchange with bromide. This represents an extension of the classical Asn-Asn polar buried interface extensively characterized in coiled-coil peptides.49,50 In the case of the calcium binding site, the metal ligand geometry Asn63-Ca2+-Gln66 positions the δ-oxygen of Asn63 only ∼5 Å away from the ɛ-oxygen of Gln66, at a distance comparable to that of a weak hydrogen bond. But in contrast to H-bonds, which can form between folded, misfolded and/or unfolded gp26 protomers, the octahedrally coordinated Ca2+ ion and the selective trapping of Cl− are very sensitive to the correct folded state of the fiber, as a homotrimer is the only conformation of gp26 that yields the correct binding geometry. We propose that chelating ions at the interior of α-helical core provides specificity and register to the otherwise purely repetitive “Protein Velcro” generated by the hydrophobic spine of 22 residues that include the four trimerization octads.14 These ion traps could function to prevent any sliding of the protomer helices relative to each other, which the array of hydrophobic interactions may not be able to abolish. Alternatively, the ion traps may “seed” the trimerization of gp26 protomers by kinetically bringing the helices together before formation of octad-mediated hydrophobic interactions. In either instance, the nonspecific assembly of fiber aggregates is an important problem to overcome in the folding pathway of fibrous proteins like gp26. Interestingly, neither the previously characterized salt bridges latching gp26 protomers from the outside, nor the core of hydrophobic residues inside gp26 are biologically useful to discriminate right versus wrong gp26 oligomers.
Conformational flexibility
Despite its great stability, the gp26 helical core is flexibly connected to the C-terminal tip (Domains III and IV). This tip is built by a short helical coiled-coil and a triple-β helix and its flexibility with respect to the helical core has been observed in solution,17 in crystal,14 and possibly in ice-embedded particles of mature P22 virion.8,9 As the latter, the asymmetric cryo-electron microscopic reconstructions of mature phage P22 overlaid to the crystal structure of the tail needle show weak density for the C-terminus of the tail needle [Fig. 4(a,b)], likely due to small misalignments in the tip of the tail needle in each of the particles used in the reconstructions. A comparison of the position of the xenon sites in the structure and the density for gp26 in the cryo-EM reconstructions of the mature phage show that six of the seven xenon sites map to regions that have weak or no density in the cryo-EM maps [Fig. 4(a,b)]. This data supports the strong correlation between gp26 flexibility and xenon binding to internal cavities. The only xenon atom that apparently contradicts this is Xe1, which occupies an invagination at the N-terminal tip of the fiber, and is in equilibrium with the exterior. Thus, Xe1 does not directly sense the flexibility of the fiber, but fills a cavity otherwise occupied by a water molecule in the underivatized structure.14 We propose that the flexibility of the gp26 C-terminal tip with respect of Domain II is generated by a constellation of adjacent, yet noncommunicating cavities at the inner core of gp26. These cavities, occupied in our structure by Xe3, Xe4, Xe5, Xe6, are clustered at the boundary between the helical core Domain II and the triple β-helix Domain III. Small cavities in the interior of gp26 allow for flexibility and multiple conformations that would be otherwise impossible to attain given the highly rigid native conformation of the gp26 helical core. Superimposition of the two gp26 bundles in the crystallographic asymmetric unit reveals that the C-terminal tip (Domains III and IV) can swing up to ∼18° in one direction with respect to the helical core. If we consider that the conformations seen in the crystals are stabilized by crystal contacts, it is plausible that in an in vivo setting, the C-terminal tip has even greater degree of freedom and swings in all directions like a shoulder-joint [Fig. 4(c)]. The rigid body movement seen in the lazo-domain of two gp26s in the asymmetric unit is made possible by the flexibility of the main chain combined with the lateral compression of side chains in the hydrophobic core, as opposed to a global “repacking” of buried residues.
Figure 4.
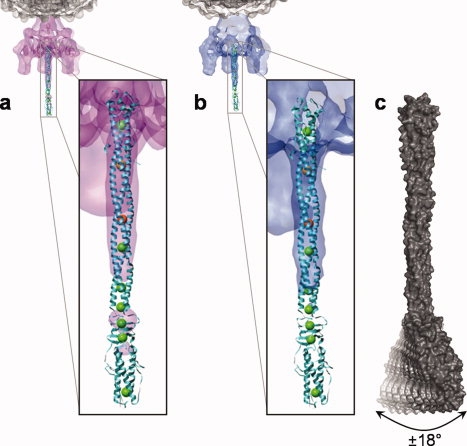
Xenon gas probes the flexibility of tail needle gp26. (a, b) The structure of gp26 bound to xenon gas was fit into the cryo-EM reconstructions of P22 mature viron determined by Lander et al.9 (a) and Chang et al.8 (b). In both panels (a, b) the seven xenon atoms, the calcium ion, and the chloride ion bound to gp26 are shown as green, gold, and orange spheres, respectively. The C-terminal tip of gp26 has poor density in the cryo-EM reconstructions, likely indicating high flexibility or multiple conformations. In contrast, the two ion traps (gold/orange spheres) are found in the more rigid areas of the protein, characterized by strong density in both cryo-EM reconstructions. (c) Space filling model of gp26, indicating the flexibility observed in the crystal structure and supported by the EM reconstructions. Domains III and IV of the tail needle are flexibly connected the helical core Domain II, which can bend ∼18° relative to the rigid helical core, thus generating a putative scanning device that interacts with the host cell envelope.
How can cavities in gp26 contribute to the flexibility of the tail needle? We support a model where the partial loss of the closely packed core promotes extensive dynamic motion of the protein backbone atoms, which increase the overall breathing of the fiber. In a crystal lattice this forces one of the two fibers in the asymmetric unit to bend in a specific orientation, with the lazo-domain significantly tilted with respect to the helical core. In solution, the breathing of the fiber is likely to be unbiased and simply result in an overall flexibility. Similar to the cavities seen in gp26, it is believed that cavities in enzymes increase the local dynamic movement of the protein backbone to define local areas of flexibility, which may be important in establishing the path for ligand entry. In direct support of this idea is the observation that removal of ligands or cofactors from flavodoxin51 or intestinal fatty acid binding protein52 that generates empty cavities inside the protein also results in a dramatic increase in the μs-ms time scale dynamics of the protein backbone. In gp26, which is not an enzyme and serves a structural role in the virion, the flexible connection between the C-terminal Domains III and IV and the helical core likely enables the fiber to function as a scanning tip that probes the Salmonella cell surface for either a point of injection or a proteinaceous receptor.
Flexibility in viral fibers used to interact with the host has been observed for several other systems and likely plays a critical role in the virus:host interaction. For instance in phage SPP1, the distal tip of the tail that binds the extracellular ectodomain of the Bacillus subtilis receptor YueB is flexibly connected to the tail tube by a protein, gp19.1, which allows bending of the tip with respect to the tube at angles as high as 50°, which enhances the ability of the virus tail to scan for receptors on the host surface,53 and T4 tail fibers are flexibly attached to its tail baseplate.4,54 Flexibility in the adenovirus fiber was reported to be critical for efficient interaction with the coxsackievirus and adenovirus receptor. When the flexibility in the Adenovirus 5 (Ad5) shaft is reduced, both a decrease in virus infection and cell attachment is observed.55 Interestingly, the adenovirus shaft folds as a triple β-helix similar to the gp26 Domain III, where the largest Xenon sites are observed. Therefore, we speculate the short triple β-helix downstream of the gp26 Domain II interrupts the rigidity and stiffness of the helical core and represents a useful structural adaptor to connect the surface-probing moiety (Domain IV) to the helical core (Domain II). The real challenge remaining is to understand if gp26 flexibility is used by the phage to penetrate the lipid bilayer or to interact with a yet uncharacterized surface receptor.
Accession number
The structure factor and atomic coordinates for gp26 bound to xenon gas determined to 1.98 Å resolution have been deposited in RCSB Protein Data Bank with the accession code 3C9I.
Acknowledgments
The authors thank Vivian Stojanoff and the rest of the NSLS X6A staff, as well as the macCHESS staff for beam time and assistance in data collection.
Glossary
Abbreviations:
- cryo-EM
cryo-electromicroscopy
- gp
gene product
- MAD
multiwavelength anomalous diffraction
- SDS
sodiumdodecyl sulfate.
References
- 1.Ackermann HW. Bacteriophage observations and evolution. Res Microbiol. 2003;154:245–251. doi: 10.1016/S0923-2508(03)00067-6. [DOI] [PubMed] [Google Scholar]
- 2.Tang L, Marion WR, Cingolani G, Prevelige PE, Johnson JE. Three-dimensional structure of the bacteriophage P22 tail machine. EMBO J. 2005;24:2087. doi: 10.1038/sj.emboj.7600695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fokine A, Chipman PR, Leiman PG, Mesyanzhinov VV, Rao VB, Rossmann MG. Molecular architecture of the prolate head of bacteriophage T4. Proc Natl Acad Sci USA. 2004;101:6003–6008. doi: 10.1073/pnas.0400444101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leiman PG, Chipman PR, Kostyuchenko VA, Mesyanzhinov VV, Rossmann MG. Three-dimensional rearrangement of proteins in the tail of bacteriophage T4 on infection of its host. Cell. 2004;118:419–429. doi: 10.1016/j.cell.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 5.Molineux IJ. No syringes please, ejection of phage T7 DNA from the virion is enzyme driven. Mol Microbiol. 2001;40:1–8. doi: 10.1046/j.1365-2958.2001.02357.x. [DOI] [PubMed] [Google Scholar]
- 6.Bazinet C, Benbasat J, King J, Carazo JM, Carrascosa JL. Purification and organization of the gene 1 portal protein required for phage P22 DNA packaging. Biochemistry. 1988;27:1849–1856. doi: 10.1021/bi00406a009. [DOI] [PubMed] [Google Scholar]
- 7.Goldenberg D, King J. Trimeric intermediate in the in vivo folding and subunit assembly of the tail spike endorhamnosidase of bacteriophage P22. Proc Natl Acad Sci USA. 1982;79:3403–3407. doi: 10.1073/pnas.79.11.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang J, Weigele P, King J, Chiu W, Jiang W. Cryo-EM Asymmetric reconstruction of bacteriophage P22 reveals organization of its DNA packaging and infecting machinery. Structure. 2006;14:1073–1082. doi: 10.1016/j.str.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 9.Lander GC, Tang L, Casjens SR, Gilcrease EB, Prevelige P, Poliakov A, Potter CS, Carragher B, Johnson JE. The structure of an infectious p22 virion shows the signal for headful DNA packaging. Science. 2006;312:1791–1795. doi: 10.1126/science.1127981. [DOI] [PubMed] [Google Scholar]
- 10.Olia AS, Al-Bassam J, Winn-Stapley DA, Joss L, Casjens SR, Cingolani G. Binding-induced stabilization and assembly of the phage P22 tail accessory factor gp4. J Mol Biol. 2006;363:558–576. doi: 10.1016/j.jmb.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 11.Olia AS, Bhardwaj A, Joss L, Casjens S, Cingolani G. Role of gene 10 protein in the hierarchical assembly of the bacteriophage P22 portal vertex structure. Biochemistry. 2007;46:8776–8784. doi: 10.1021/bi700186e. [DOI] [PubMed] [Google Scholar]
- 12.Moak M, Molineux IJ. Peptidoglycan hydrolytic activities associated with bacteriophage virions. Mol Microbiol. 2004;51:1169. doi: 10.1046/j.1365-2958.2003.03894.x. [DOI] [PubMed] [Google Scholar]
- 13.Andrews D, Butler JS, Al-Bassam J, Joss L, Winn-Stapley DA, Casjens S, Cingolani G. Bacteriophage P22 tail accessory factor GP26 is a long triple-stranded coiled-coil. J Biol Chem. 2005;280:5929–5933. doi: 10.1074/jbc.C400513200. [DOI] [PubMed] [Google Scholar]
- 14.Olia AS, Casjens S, Cingolani G. Structure of phage P22 cell envelope-penetrating needle. Nat Struct Mol Biol. doi: 10.1038/nsmb1317. (in press) [DOI] [PubMed] [Google Scholar]
- 15.Strauss H, King J. Steps in the stabilization of newly packaged DNA during phage P22 morphogenesis. J Mol Biol. 1984;172:523–543. doi: 10.1016/s0022-2836(84)80021-2. [DOI] [PubMed] [Google Scholar]
- 16.Israel V. E proteins of bacteriophage P22. I. Identification and ejection from wild-type and defective particles. J Virol. 1977;23:91–97. doi: 10.1128/jvi.23.1.91-97.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhardwaj A, Olia AS, Walker-Kopp N, Cingolani G. Domain organization and polarity of tail needle GP26 in the portal vertex structure of bacteriophage P22. J Mol Biol. 2007;371:374–387. doi: 10.1016/j.jmb.2007.05.051. [DOI] [PubMed] [Google Scholar]
- 18.Bhardwaj A, Walker-Kopp N, Wilkens S, Cingolani G. Foldon-guided self-assembly of ultra-stable protein fibers. Protein Sci. 2008;17:1475–1485. doi: 10.1110/ps.036111.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cingolani G, Andrews D, Casjens S. Crystallogenesis of bacteriophage P22 tail accessory factor gp26 at acidic and neutral pH. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:477–482. doi: 10.1107/S1744309106013856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otwinowski Z, Minor W. Processing of X-ray diffraction Data Collected in Oscillation Mode. In: Carter JCW, Sweet RM, editors. macromolecular crystallography, part A. New York: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 21.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 22.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 24.Jahn TR, Radford SE. The Yin and Yang of protein folding. FEBS J. 2005;272:5962–5970. doi: 10.1111/j.1742-4658.2005.05021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kleywegt GJ, Jones TA. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr D Biol Crystallogr. 1994;50:178–185. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
- 26.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 27.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 28.Clamp M, Cuff J, Searle SM, Barton GJ. The Jalview Java alignment editor. Bioinformatics. 2004;20:426–427. doi: 10.1093/bioinformatics/btg430. [DOI] [PubMed] [Google Scholar]
- 29.Parsons S. Introduction to twinning. Acta Crystallogr D Biol Crystallogr. 2003;59:1995–2003. doi: 10.1107/s0907444903017657. [DOI] [PubMed] [Google Scholar]
- 30.Girard E, Kahn R, Mezouar M, Dhaussy AC, Lin T, Johnson JE, Fourme R. The first crystal structure of a macromolecular assembly under high pressure: CpMV at 330 MPa. Biophys J. 2005;88:3562–3571. doi: 10.1529/biophysj.104.058636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubin SM, Lee SY, Ruiz EJ, Pines A, Wemmer DE. Detection and characterization of xenon-binding sites in proteins by 129Xe NMR spectroscopy. J Mol Biol. 2002;322:425–440. doi: 10.1016/s0022-2836(02)00739-8. [DOI] [PubMed] [Google Scholar]
- 32.Duff AP, Trambaiolo DM, Cohen AE, Ellis PJ, Juda GA, Shepard EM, Langley DB, Dooley DM, Freeman HC, Guss JM. Using xenon as a probe for dioxygen-binding sites in copper amine oxidases. J Mol Biol. 2004;344:599–607. doi: 10.1016/j.jmb.2004.09.075. [DOI] [PubMed] [Google Scholar]
- 33.Nienhaus K, Maes EM, Weichsel A, Montfort WR, Nienhaus GU. Structural dynamics controls nitric oxide affinity in nitrophorin 4. J Biol Chem. 2004;279:39401–39407. doi: 10.1074/jbc.M406178200. [DOI] [PubMed] [Google Scholar]
- 34.Colloc'h N, Sopkova-de Oliveira Santos J, Retailleau P, Vivares D, Bonnete F, Langlois d'Estainto B, Gallois B, Brisson A, Risso JJ, Lemaire M, Prange T, Abraini JH. Protein crystallography under xenon and nitrous oxide pressure: comparison with in vivo pharmacology studies and implications for the mechanism of inhaled anesthetic action. Biophys J. 2007;92:217–224. doi: 10.1529/biophysj.106.093807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fass D, Harrison SC, Kim PS. Retrovirus envelope domain at 1.7 angstrom resolution. Nat Struct Biol. 1996;3:465–469. doi: 10.1038/nsb0596-465. [DOI] [PubMed] [Google Scholar]
- 36.Duquerroy S, Vigouroux A, Rottier PJ, Rey FA, Bosch BJ. Central ions and lateral asparagine/glutamine zippers stabilize the post-fusion hairpin conformation of the SARS coronavirus spike glycoprotein. Virology. 2005;335:276–285. doi: 10.1016/j.virol.2005.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reviewer JV. CRC handbook of chemistry and physics. In: Weast RC, editor. 1st Student Edition. Vol. 16. Boca Raton, FL: CRC Press Inc.; 1989. pp. 145–145. Medical physics. [Google Scholar]
- 38.Malashkevich VN, Kammerer RA, Efimov VP, Schulthess T, Engel J. The crystal structure of a five-stranded coiled coil in COMP: a prototype ion channel? Science. 1996;274:761–765. doi: 10.1126/science.274.5288.761. [DOI] [PubMed] [Google Scholar]
- 39.Harding MM. Metal-ligand geometry relevant to proteins and in proteins: sodium and potassium. Acta Crystallogr D Biol Crystallogr. 2002;58:872–874. doi: 10.1107/s0907444902003712. [DOI] [PubMed] [Google Scholar]
- 40.Gut H, Pennacchietti E, John RA, Bossa F, Capitani G, De Biase D, Grutter MG. Escherichia coli acid resistance: pH-sensing, activation by chloride and autoinhibition in GadB. EMBO J. 2006;25:2643–2651. doi: 10.1038/sj.emboj.7601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doukov TI, Blasiak LC, Seravalli J, Ragsdale SW, Drennan CL. Xenon in and at the end of the tunnel of bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase. Biochemistry. 2008;47:3474–3483. doi: 10.1021/bi702386t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richards FM. The interpretation of protein structures: total volume, group volume distributions and packing density. J Mol Biol. 1974;82:1–14. doi: 10.1016/0022-2836(74)90570-1. [DOI] [PubMed] [Google Scholar]
- 43.Yadav MK, Redman JE, Leman LJ, Alvarez-Gutierrez JM, Zhang Y, Stout CD, Ghadiri MR. Structure-based engineering of internal cavities in coiled-coil peptides. Biochemistry. 2005;44:9723–9732. doi: 10.1021/bi050742a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ventura S, Serrano L. Designing proteins from the inside out. Proteins. 2004;56:1–10. doi: 10.1002/prot.20142. [DOI] [PubMed] [Google Scholar]
- 45.Eriksson AE, Baase WA, Zhang XJ, Heinz DW, Blaber M, Baldwin EP, Matthews BW. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science. 1992;255:178–183. doi: 10.1126/science.1553543. [DOI] [PubMed] [Google Scholar]
- 46.Sauer RT, Lim WA. Mutational analysis of protein stability. Curr Opin Struct Biol. 1992;2:46–51. [Google Scholar]
- 47.Barlow DJ, Thornton JM. Ion-pairs in proteins. J Mol Biol. 1983;168:867–885. doi: 10.1016/s0022-2836(83)80079-5. [DOI] [PubMed] [Google Scholar]
- 48.Rashin AA, Honig B. On the environment of ionizable groups in globular proteins. J Mol Biol. 1984;173:515–521. doi: 10.1016/0022-2836(84)90394-2. [DOI] [PubMed] [Google Scholar]
- 49.Oakley MG, Kim PS. A buried polar interaction can direct the relative orientation of helices in a coiled coil. Biochemistry. 1998;37:12603–12610. doi: 10.1021/bi981269m. [DOI] [PubMed] [Google Scholar]
- 50.Lumb KJ, Kim PS. A buried polar interaction imparts structural uniqueness in a designed heterodimeric coiled coil. Biochemistry. 1995;34:8642–8648. doi: 10.1021/bi00027a013. [DOI] [PubMed] [Google Scholar]
- 51.Steensma E, van Mierlo CP. Structural characterisation of apoflavodoxin shows that the location of the stable nucleus differs among proteins with a flavodoxin-like topology. J Mol Biol. 1998;282:653–666. doi: 10.1006/jmbi.1998.2045. [DOI] [PubMed] [Google Scholar]
- 52.Hodsdon ME, Cistola DP. Ligand binding alters the backbone mobility of intestinal fatty acid-binding protein as monitored by 15N NMR relaxation and 1H exchange. Biochemistry. 1997;36:2278–2290. doi: 10.1021/bi962018l. [DOI] [PubMed] [Google Scholar]
- 53.Plisson C, White HE, Auzat I, Zafarani A, Sao-Jose C, Lhuillier S, Tavares P, Orlova EV. Structure of bacteriophage SPP1 tail reveals trigger for DNA ejection. EMBO J. 2007;26:3720–3728. doi: 10.1038/sj.emboj.7601786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wood WB. Bacteriophage T4 assembly and the morphogenesis of subcellular structure. Harvey Lect. 1979;73:203–223. [PubMed] [Google Scholar]
- 55.Wu E, Pache L, Von Seggern DJ, Mullen TM, Mikyas Y, Stewart PL, Nemerow GR. Flexibility of the adenovirus fiber is required for efficient receptor interaction. J Virol. 2003;77:7225–7235. doi: 10.1128/JVI.77.13.7225-7235.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]