

Abstract
UV damage endonuclease is a DNA repair enzyme that can both recognize damage such as UV lesions and introduce a nick directly 5′ to them. Recently, the crystal structure of the enzyme from Thermus thermophilus was solved. In the electron density map of this structure, unexplained density near the active site was observed at the tip of Lys229. Based on this finding, it was proposed that Lys229 is post-translationally modified. In this article, we give evidence that this modification is a carboxyl group. By combining activity assays and X-ray crystallography on several point mutants, we show that the carboxyl group assists in metal binding required for catalysis by donating negative charge to the metal-coordinating residue His231. Moreover, functional and structural analysis of the K229R mutant reveals that if His231 shifts away, an increased activity results on both damaged and undamaged DNA. Taken together, the results show that T. thermophilus ultraviolet damage endonuclease is carboxylated and the modified lysine is required for proper catalysis and preventing increased incision of undamaged DNA.
Keywords: carboxylated lysine, UV DNA repair, endonuclease, X-ray crystallography
Introduction
Repairing damage in DNA is essential for maintaining genomic integrity. Therefore, several protein systems have evolved to remove DNA lesions. One of them is the ultraviolet damage endonuclease (UVDE) repair system, initially found in the yeast Schizosaccharomyces pombe, and described as an alternative repair system for UV-induced lesions.1 The UVDE enzyme was shown to introduce a nick 5′ to both of the main UV-induced lesions: cyclobutane pyrimidine dimers (CPD) and 6-4 photoproducts (6-4PP).2 Later studies showed, however, that UVDE from S. pombe has a much broader substrate specificity than originally thought—recognizing and nicking DNA lesions significantly different from UV induced damage such as abasic sites and small loops.3 UVDE enzymes have been found in both eukaryotes (e.g., S. pombe, Neurospora crassa) and in prokaryotes (e.g., Thermus thermophilus, Bacillus subtilis).
The crystal structure of the T. thermophilus UVDE has recently been solved to 1.55 Å resolution.4 This structure shows UVDE to be a single domain TIM-barrel with extensive positive charges positioned on both sides of a 29 Å groove, which was proposed to be the DNA-binding site. At the bottom of the groove, a cluster of three metal ions was found. Furthermore, a protein pocket was identified in UVDE near Tyr6 and Asn10, into which UVDE might flip damaged base(s). Unexplained electron density at the tip of Lys229 was observed and it was suggested that Lys229 is post-translationally modified. Here, we investigate the identity of this modification by X-ray crystallography and conclude that it is a carboxyl group. Carboxylated lysines have been observed in several proteins.5–11 Most often, the carboxyl group has a structural role (e.g., bridging two active site zinc atoms), but a more direct role in reaction mechanisms has also been reported.6 In addition to identifying this modification, we study, by means of X-ray crystallography and activity assays on several point mutants, why the protein uses a carboxylated lysine instead of a standard amino acid.
Results
Identity of the modification
The unexplained electron density observed at the tip of Lys229 is situated between Arg58, Ser99, and His231. His231 was shown to coordinate one of the metal ions (Mn1). By comparing the metal ion cluster of TthUVDE to that of endonuclease IV,12 Mn1 is probably one of the two metal ions that together activate the water molecule (bridging Mn1 and Mn2) that serves as a nucleophile to attack the DNA phosphodiester backbone.
Several options are possible for the unexplained electron density at the tip of Lys229 among which are an acetyl group or a carboxyl group. Considering the potential hydrogen bonding to the neighboring residues (Arg58, Ser99, and His231), a carboxyl group is the more likely option (see Fig. 1). Moreover, if an acetyl group is modeled into the density, its methyl group's B-factor is 13.94 Å2 whereas the Nζ and Cɛ of the lysine and the carbonyl of the acetyl group all have B-factors between 20 and 22 Å2. If a carboxyl group is modeled into the density, all atoms of the carboxyl group and the Nζ and Cɛ of the lysine have B-factors between 18 and 21 Å2.
Figure 1.
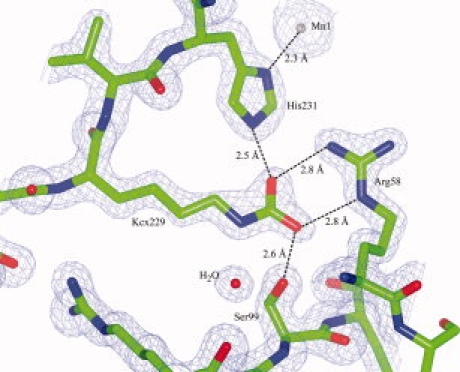
Detail of the original structure of TthUVDE wild type with electron density map (contoured at 1.5 σ) showing a carboxyl group modeled into the additional density at the tip of Lys229 (Kcx229). Distances between the oxygen atoms of the carboxyl group and neighboring residues, and the distance of His231 to Mn1 are indicated. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
To begin the investigation on the modification's identity we performed mass spectrometry on trypsinated UVDE. In the resulting mass spectrum, however, only a peptide fragment with an unmodified Lys229 was found. Considering the preparation of the sample (trichloroacetic acid (TCA) precipitation and trypsin digestion), an acetyl group probably would have been observed if it had been present. In contrast, a carboxyl group on a lysine residue can be expected to fall off during the sample preparation, since at low pH the ζ-nitrogen of the lysine is protonated, which results in a loss of the carboxyl group as carbon dioxide.
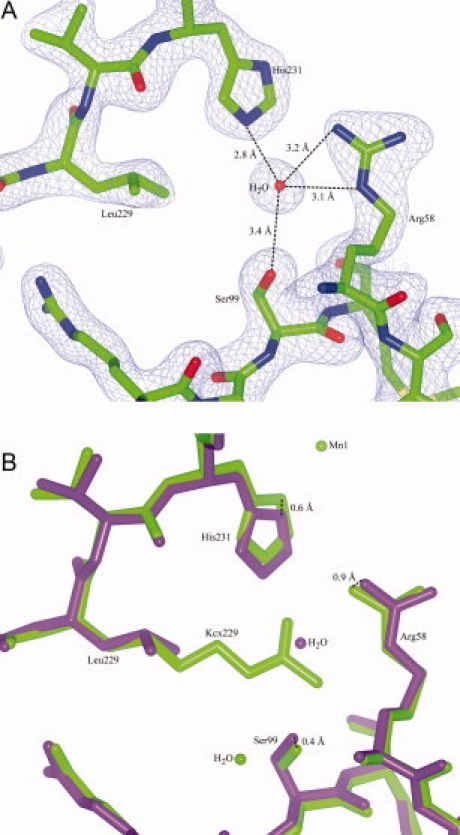
To confirm the above result, we obtained UVDE crystals at low pH (expecting that the carboxylation will not be present, due to the acid liability of the carboxylation). For this, TthUVDE was crystallized in 0.1M acetate buffer pH 4.4, 2M sodium formate, and 1 mM MnCl2 (UVDE pH 4.4) and crystallized in a different space group (P6122) with different packing than the wild type structure (P1) (see Table I for the crystallographic statistics). In this structure Lys229 is indeed not modified (see Fig. 2). Two waters are at the place where the modified lysine originally was. His231 seems to have moved inwards (over 0.96 Å) to form hydrogen bonds with one of these waters. This shift seems to have cause a 1.36 Å shift to Glu175. In the structure of UVDE pH 4.4, no metal ions were found, probably because of protonation of the metal-coordinating histidines due to the low pH. A water molecule was found near the site (0.81 Å distance) where originally Mn1 was located.
Table I.
Crystallographic Data and Refinement Statistics
| UVDE pH 4.4 | UVDE K229L | UVDE K229R | UVDE E175A | |
|---|---|---|---|---|
| Data collection | ||||
| Beam line | ESRF ID14-2 | ESRF ID14-2 | ESRF ID14-2 | ESRF ID14-2 |
| Detector | MAR225 CCD | MAR225 CCD | MAR225 CCD | MAR225 CCD |
| Resolution range (Å) | 28.78–1.91 Å | 37.11–2.30 Å | 46.37–3.15 Å | 40.46–2.74 Å |
| (2.01–1.91 Å)a | (2.42–2.30 Å) | (3.32–3.15 Å) | (2.89–2.74 Å) | |
| Multiplicity | 6.4 (6.5) | 11.3 (11.5) | 10.7 (11.0) | 4.4 (4.4) |
| Completeness (%) | 99.2 (100) | 99.2 (99.8) | 95.3 (96.2) | 99.3 (99.8) |
| Rmergeb (%) | 0.048 (0.282) | 0.078 (0.367) | 0.134 (0.366) | 0.087 (0.364) |
| Rpimc(%) | 0.020 (0.113) | 0.025 (0.122) | 0.043 (0.117) | 0.048 (0.203) |
| I/sigma | 10.2 (2.0) | 8.3 (2.0) | 5.4 (2.0) | 8.0 (2.0) |
| Space group | P6122 | P6122 | P6122 | P6122 |
| No. of molecules in asymmetric unit | 1 | 1 | 1 | 1 |
| Unit cell parameters a/b/c (Å) | 107.14 × 107.14 × 90.83 | 113.39 × 113.39 × 89.39 | 107.11 × 107.11 × 91.12 | 113.38 × 113.38 × 89.08 |
| Crystallization condition | 0.1M Ac pH 4.4 | 0.5M (NH4)2SO4 | 0.1M Ac pH 5.6 | 0.5M (NH4)2SO4 |
| 2M Naformate | 0.1M Ac pH 6 | 1M Naformate | 0.1M Ac pH 6 | |
| 1 mM MnCl2 | 1M Li2SO4 | 1M Li2SO4 | ||
| Molecular replacement | ||||
| Correlation coefficent | 0.613 | 0.658 | NA | 0.639 |
| R factor of correct solution/second peak | 0.515/0.643 | 0.442/0.625 | NA | 0.416/0.610 |
| Refinement | ||||
| No. of reflections used in refinement | 22,835 | 14,615 | 5122 | 8773 |
| Cutoff | None | None | None | None |
| Resolution range (Å) | 28.78–1.91 Å | 34.28–2.30 Å | 46.18–3.15 Å | 35.03–2.74 Å |
| (1.96–1.91 Å)a | (2.36–2.30Å) | (3.23–3.15 Å) | (2.81–2.74 Å) | |
| R factord | 0.196 (0.286) | 0.188 (0.198) | 0.195 (0.232) | 0.198 (0.295) |
| Rfreee | 0.244 (0.363) | 0.244 (0.278) | 0.249 (0.360) | 0.245 (0.363) |
| Ramanchandran statisticsf (%) | 91.1/7.6/0.8/0.4 | 89.8/8.9/0.4/0.8 | 91.5/7.7/0.0/0.9 | 91.9/6.8/0.9/0.4 |
| R.m.s. deviations (bonds, Å/angle, °)g | 0.019/1.689 | 0.014/1.545 | 0.006/0.945 | 0.009/1.320 |
| Average atomic B-factor for protein/Mn/solvent atoms | 25.4/NA/33.0 | 30.6/65.2/30.6 | 32.6/NA/32.4 | 22.8/74.3/11.0 |
| Wilson plot B-factor | 25.0 | 35.9 | 54.4 | 47.3 |
Values in parentheses are for the highest resolution bin, where applicable.
Rmerge = Σ|I − 〈I〉|/Σ| I|.
R = Σ||Fobs(hkl) | − |Fcalc(hkl) || /Σ|Fobs(hkl) |.
About 5% of the reflections were used for the crossvalidation set. These reflections were randomly chosen.
According to the program PROCHECK15. The percentages are indicated of residues in the most favored, additionally allowed, generously allowed, and disallowed regions of the Ramachandran plot, respectively.
Estimates provided by the program REFMAC16.
Figure 2.
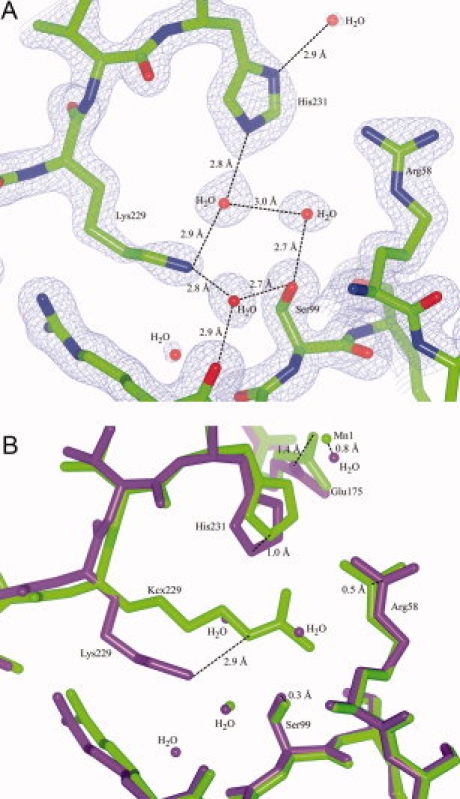
Structure of UVDE pH 4.4. Panel A: Model and map (contoured at 1.7 σ) of UVDE pH 4.4, showing an unmodified Lys229. Hydrogen network with Ser99, Lys229, and His231 is indicated. Panel B: Detail of the superposition of the original structure of UVDE (light green) and UVDE pH 4.4 (dark purple). Shift of residues Arg58 and Ser99. Glu175, Lys229, and His231 and of Mn1 compared with the water in UVDE pH 4.4 are indicated at the atoms where the shift was measured. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
To check that the above result is caused by the low pH of the crystallization condition and not due to the different space group (P6122) of the previously published structure (P1), we also solved a structure of TthUVDE in P6122 that was crystallized at a higher pH [0.5M (NH4)2SO4, 0.1M sodium acetate buffer pH 5.6, 1M Li2SO4, 1 mM MnCl2; results not shown]. In this crystal structure, Lys229 was seen to be modified, showing that the modification can be accommodated in P6122.
Functional studies of Lys229 mutants
To investigate the role of the carboxylated lysine in the activity of UVDE, we mutated this residue into an alanine (UVDE K229A) and a leucine (UVDE K229L). UVDE K229A showed an extremely reduced catalytic activity on all tested DNA substrates (see Fig. 3). The K229L mutant showed severely reduced catalytic activity on the CPD (10%) and the abasic site lesion (less than 1%). On the 6-4PP the incision efficiency of this mutant is slightly reduced compared with that of the wild type (see Fig. 3). These results indicate that Lys229 has an important role in the function of TthUVDE.
Figure 3.
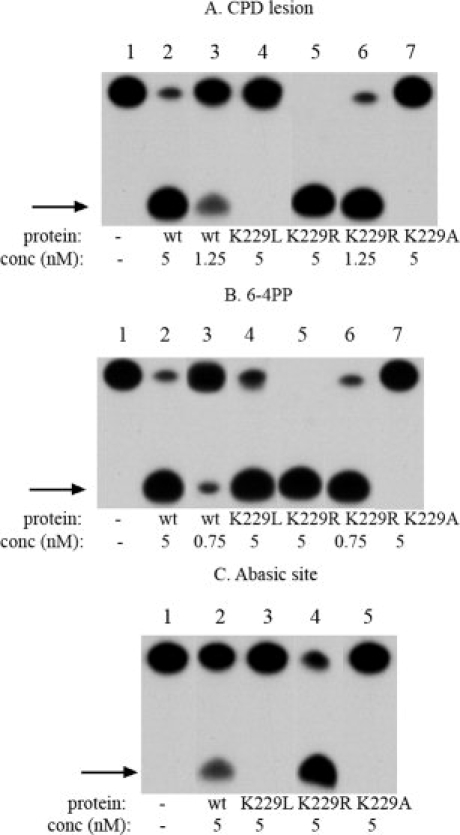
Activity of wild type and mutant UVDE proteins. Activity assay with terminally labeled 30 bp DNA substrates containing a CPD (A), 6-4PP lesion (B), and abasic site (C). The incision product is indicated with an arrow. Below the lanes is indicated which protein is used and the concentration of protein used (in nM).
To investigate if the observed reduction in the incision efficiency is due to an effect on the formation of protein—DNA complexes, we tested the binding properties of UVDE K229A and UVDE K229L in a filter binding assay. As can be seen in Table II the binding capacity of both mutants is similar to the wild type. This indicates that the reduced incision activity is not due to changes in the enzymes ability to bind DNA, but an impairment in catalysis. Moreover, DNA-binding is similar either in absence and presence of Mn2+ (for wild type and mutants) showing that Mn2+ does not influence DNA binding.
Table II.
DNA binding Properties of TthUVDE wt and Mutants Tested in a Filter Binding Assay
| % Binding |
% Binding |
||
|---|---|---|---|
| DNA lesion | UVDE | no Mn2+ | 1 mM Mn2+ |
| CPD | Wild type | 34 ± 2 | 33 ± 1.7 |
| CPD | K229L | 31 ± 0.7 | 31 ± 0.7 |
| CPD | K229R | 31 ± 0.5 | 32 ± 0.2 |
| 6-4PP | Wild type | 19 ± 3.7 | 20 ± 1 |
| 6-4PP | K229L | 20 ± 0.5 | 21 ± 1.8 |
| 6-4PP | K229R | 17 ± 2.8 | 19 ± 0.7 |
| Abasic site | Wild type | 20 ± 2 | 23 ± 0.7 |
| Abasic site | K229L | 21 ± 0.7 | 21 ± 1.4 |
| Abasic site | K229R | 21 ± 2 | 21 ± 0.7 |
| No damage | Wild type | 2.5 ± 0.1 | 2.8 ± 0.4 |
| No damage | K229L | 3.7 ± 0.2 | 3.8 ± 0.4 |
| No damage | K229R | 7.6 ± 0.7 | 5.8 ± 1 |
Binding is expressed as the percentage of the input DNA retained on the filter. Binding efficiencies were tested in presence and absence of 1 mM MnCl2.
To gain a more detailed insight into the function of the carboxylated lysine, a mutant was constructed in which Lys229 was changed to an arginine (UVDE K229R). An arginine has a positive charge like a lysine, but an arginine cannot be carboxylated. The positive side-chain of an arginine can therefore not be turned into a negative group by carboxylation.
Surprisingly, the K229R mutant was found to be active and shows very high activity on CPD [Fig. 3(A)] and 6-4PP [Fig. 3(B)] lesions, even higher than that of wild type. Also abasic site, which is not an optimal substrate for the TthUVDE wild type (20% incision), is cleaved by the K229R mutant with high efficiency [80%, Fig. 3(C)].
We also tested the activity of K229R on UV-damaged DNA in a supercoiled incision assay [Fig. 4(A); plasmid I]. In the lanes with UVDE K229R (Lanes 6–8), much more relaxed and even linear plasmid DNA as a result of damage-specific incision can be seen compared with the lanes with UVDE wild type (Lanes 2–4). This confirms that UVDE K229R is much more active on UV damage sites than the wild type protein. UVDE K229R also has incision activity on the undamaged supercoiled plasmid DNA present in the assay [Fig. 4(A), plasmid II, Lane 8], though this activity is very small compared with the activity on UV-damaged supercoiled plasmid DNA. To investigate this activity on undamaged DNA more closely, we performed an assay with only undamaged supercoiled plasmid DNA [Fig. 4(B)]. In this assay we could see that, surprisingly, UVDE wild type has some activity on undamaged plasmid DNA (Lane 2). The activity of K229R on undamaged DNA, however, was again significantly higher (Lane 4).
Figure 4.
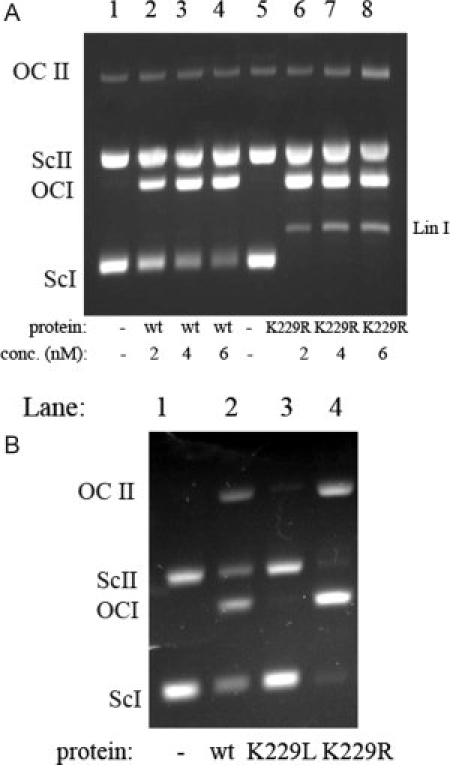
Activity of UVDE wild type and K229R on UV-irradiated and nonirradiated plasmid DNA. Panel A: A mixture of UV-irradiated supercoiled DNA of pUC18 (I, 2686 bp) and nonirradiated pNP228 (II, 4686 bp) was incubated with different concentrations (as indicated) UVDE wild type and K229R mutant for 15 min. The positions of the supercoiled (sc), open circle (oc), and linear (lin) forms of the plasmids are indicated. Panel B: A mixture of undamaged supercoiled DNA of pUC18 (I, 2686 bp) and pNP228 (II, 4686 bp) was incubated with 10 nM UVDE wild type and mutant proteins as indicated. The positions of the supercoiled (sc) and open circle (oc) forms of the plasmids are indicated.
Filter-binding assays (Table II) showed that K229R has similar DNA binding properties to wild type UVDE. Thus, the higher activity of this mutant on both damaged and undamaged DNA is not caused by a change to DNA binding, but by an increased efficiency of the incision reaction.
Structural studies of the Lys229 mutants
To obtain a structural basis for the above results, the mutants K229L, K229R, and E175A were crystallized and their structures were solved by molecular replacement (see Table I for statistics). In all three mutants, the overall structure is the same as wild type.
In the structure of K229L, a water molecule is present at the place of the carboxylated lysine (see Fig. 5). Only one of the three metal ions is present: Mn3. This is the metal ion near residues His244 and His203 and it corresponds to Zn3 in endonuclease IV.12 The two metal ions (Mn1 and Mn2) that are proposed to activate the catalytic water molecule (based on comparison with EndoIV) are absent. These results suggest a correlation between the presence of the metal ions and the carboxyl group.
Figure 5.
Structure of UVDE K229L. Panel A: Detail of the model and the map of UVDE K229L showing the environment of Leu229. Distances of the water molecule to neighboring residues are indicated. Panel B: Detail of a superposition of UVDE K229L (in dark purple) with the original structure of UVDE (in light green). The slight shifts of residues Arg58, Ser99, and His231 are indicated. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
To study whether the carboxyl group can still be present if there are no metal ions, the previously constructed mutant4 E175A was studied in detail. Glu175 bridges Mn1 and Mn2. The mutant E175A was previously found to have no observable activity on DNA containing CPD and a severely reduced activity on DNA containing 6-4PP (5% activity left). In the structure of this mutant, Mn1 and Mn2 were not observed, as expected. Instead of these ions, there is one water molecule (see Fig. 6). The metal ion near His244 and His203, Mn3, is present. Most importantly, the modification on Lys229 was still observed. This result shows that the modification can be present in the absence of the Mn1 and Mn2.
Figure 6.
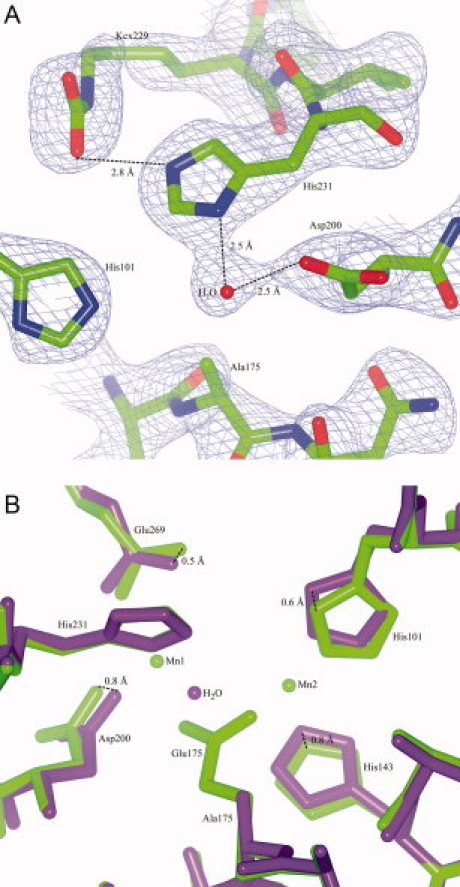
Structure of UVDE E175A. Panel A: Detail of the map (contoured at 1.25 σ) and the model of UVDE E175A showing the environment of Ala175 and the presence of a carboxylated lysine. Distance between an oxygen atom of Kcx229 and His231 is indicated as well as the distances to the water taking the place of Mn1 and Mn2 to neighboring residues. Panel B: Detail of the superposition of the original structure of UVDE (in light green) and UVDE E175A (in dark purple) showing the surroundings of residue Glu175/Ala175. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Together, the above results suggest that the carboxyl group can be present on Lys229 in absence of Mn1 (as in E175A), but that perhaps this metal ion cannot bind stably in the absence of the carboxyl group (as in structure K229L).
In K229R, Arg229 has moved compared with the carboxylated lysine [Fig. 7(A,B)]. A water molecule was found at the place where the tip of the carboxylated lysine originally was. Significant shifts are seen for the side-chains of His231 (2.5 Å) and Glu269 (3.2 Å), which have moved outwards away from Arg229. Indeed, His231 moves away from the positive charge of Arg229 and Glu269 moves accordingly. It should be noted that UVDE K229R is actually a double mutant (K229R L133I) due to a misincorporation during the PCR reaction. No influence of the second mutation on the structure of the protein could be seen (The shift of the Cα of residue 133 is only 0.34 Å). Furthermore, Leu133 is a nonconserved residue far away from the DNA binding site, leucine is very similar to isoleucine, and, in fact, some bacterial UVDE have isoleucine in this position. Thus, the mutation L133I is likely to have no impact on the behavior of K229R.
Figure 7.
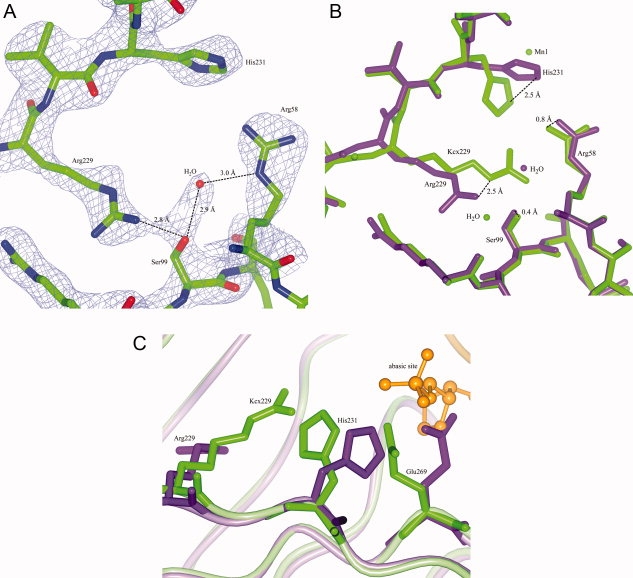
Structure of UVDE K229R and model with DNA. Panel A: Model and map (contoured at 1.5 σ) of UVDE K229R. Distances between a water molecule and Arg58 and Ser99 are indicated. Panel B: Detail of the superposition of the original structure of UVDE (in light green) and UVDE K229R (in dark purple) showing the position of Arg229 compared with the carboxylated lysine and the large shift in the position of His231. Also the shifts in the position of Arg58 and Ser99 are indicated. Panel C: Detail of superposition of the original structure of UVDE (in light green) and the structure of UVDE K229R (in dark purple) in which DNA with an abasic site was modeled (orange) based on a comparison with the structure of endonuclease IV with damaged DNA. Arg229/Kcx229, His231, and Glu269 are depicted in cylinder representation, the abasic site of the DNA is depicted in ball-and-stick representation whereas the rest of the protein and the DNA is depicted in ribbon representation. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Metal ions were not observed in the K229R structure, which is probably caused by the low pH of the crystallization condition (below the pKa of histidine side-chains). Although two of the metal-coordinating residues have shifted considerably compared with the wild type structure (His231, Glu269), the coordination environments for the three metal ions seem to be intact. Thus, the metals probably can bind stably in all three sites (as the activity also suggests) if the pH permits it, although Mn1 would have to shift over about 1 Å to get into a proper coordination environment.
To get insight into the possible influence of the shifts of His231 and Glu269 on the activity of UVDE K229R, DNA was modeled into K229R based on the crystal structure of endonuclease IV with DNA12. In this model, it can be seen that the observed shifts are near the part of the DNA where the damage is expected to be located. In contrast to wild type, in K229R the shifted Glu269 clashes with the abasic site (damaged site) of this model [see Fig. 7(C)], suggesting that the part of DNA near the damage might have to bind slightly different in UVDE K229R compared with wild type.
Discussion
The results from mass spectrometry and crystallization at low pH showed that the modification we initially observed on Lys229 is (acid) labile. Together with the observed hydrogen bonding pattern of the modification in the original structure, we conclude that the modification is a carboxyl group. Carboxylated lysines have been observed before in prokaryotic proteins and carboxylation of a lysine does not require an enzyme. Therefore, it is a likely possibility for a protein (TthUVDE) overexpressed in a foreign host (E. coli). With activitiy assays and structural analysis on mutants, we conclude the carboxyl group might be involved in the stable binding of Mn1 by donating some of its negative charge to the His231. Reduced stability of metal binding explains the reduced activity of K229A and K229L. For K229L, this reduction in activity is stronger on CPD than on 6-4PP, possibly because the presence of DNA with a 6-4PP might facilitate metal binding and thus allow activity on 6-4PP. Notably, also UVDE E175A (in which also reduced binding of Mn1 is involved) has a more severe phenotype on CPD than on 6-4PP.4
The mutant K229R, however, does not have a negative charge near His231. Quite the contrary: the protein has a positive charge at position 229 (an arginine). Still, the mutant is active on CPD, 6-4PP and abasic sites with a higher incision efficiency than wild type on these substrates. This increased activity of K229R might be explained by considering a potential mechanism for UVDE where, the protein first recognizes a general distortion in the DNA and binds to it (proposed previously based on the broad substrate specificity of SpUVDE3). K229R performs this step similar to UVDE wild type, since overall DNA binding was seen not to be affected by the mutation. Then, the DNA gets into the right conformation for incision and the incision takes place. This step is performed more efficiently in K229R than wild type. Structural changes were observed in the active site of K229R and the induced changes block the previously proposed damage-binding pocket. However, the structure of UVDE in complex with DNA is needed to clarify the relevance of the latter observation. The structural changes in the active site of K229R may cause the portion of the DNA near the damage to bind slightly different in the active site, perhaps in a conformation more favorable for incision.
The results on mutant UVDE K229R raise an interesting question. Why did T. thermophilus not use an arginine at position 229 if this protein is more efficient than wild type? An answer to this may be found in the activity of UVDE K229R on undamaged DNA. Both UVDE wild type and K229R were found to incise undamaged DNA which has not been reported for UVDE from S. pombe. Apparently, TthUVDE has more difficulties in discriminating damaged and undamaged DNA. The high temperature used for incision assays with this thermophile, might play a role in the incision of undamaged DNA, since the DNA can be more readily distorted at higher temperature. Interestingly, we found that the activity on undamaged DNA was much higher for K229R than for UVDE wild type. Perhaps Arg229 is not favorable for T. thermophilis because the incision on undamaged DNA in the cell might be too high.
UVDE is present in several organisms, both prokaryotic and eukaryotic. Is carboxylation a general phenomenon in all these proteins? Two of the residues near the carboxylated lysine, Arg58 and His231, are fully conserved in all UVDE proteins.8 In eukaryotic UVDEs, Lys229 itself is also conserved and these proteins have a threonine instead of a serine at the position corresponding to Ser99. Thus, these proteins might have a similar site at this position and therefore, they might be carboxylated like TthUVDE.
In the prokaryotic UVDEs, however, Lys229 is only partially conserved. Most UVDE proteins have a lysine at this position as well as a serine at the position corresponding to Ser99 and thus might be carboxylated like TthUVDE. A few UVDEs, however, have a leucine, isoleucine, methionine, glutamic acid, threonine, or valine at the position corresponding to Lys229. Thus, these UVDE homologues cannot have the same modification.
We have shown here that a leucine at position 229 results in an inactive enzyme for TthUVDE while some of the other UVDEs do have a leucine at that position, such as Desulfotelea psychrophila UVDE.17 Perhaps in these UVDEs the environment near His231 is such that a negative charge is provided to His231 by other residues nearby so that stable metal binding is assured in these proteins as well. Indeed, the UVDEs with a leucine at position 229 all have a glutamic acid or an aspartic acid at the position of Met267, which is spatially close to His231 (the side chain of glutamic acid is 2.5 Å away from the side-chain of His231 if glutamic acid is modeled at this position in the structure of TthUVDE). Such a negatively charged residue is not present in the UVDEs that do have a lysine at position 229; those all have a methionine at that position. Moreover, most of the other UVDEs that do not have a lysine at the position of Lys229, also have a glutamic acid near or at position 229. Thus, also these UVDEs might have a negative charge near His231 for assuring proper metal binding, though no definite conclusions can be made in absence of structural data on these proteins.
In conclusion, we think that UVDE from T. thermophilus is carboxylated at Lys229. The carboxyl group might be required for stable metal binding and perhaps also for preventing an unfavorably high incision of undamaged DNA.
Methods
Proteins
The T. thermophilus UVDE protein used in this study was expressed and purified as described before.4 Mutants K229A, K229L, and K229R were constructed by site-directed mutagenesis using PCR and purified in the same way as the wild type enzyme.
DNA substrates
DNA substrates used in all activity assays are 30-bp DNA containing either a CPD or a 6-4PP in the following sequence: 5′ CTCGTCAGCATCTTCATCATACAGTCAGTG 3′ with TT representing the position of the UV lesion. In case of the abasic site the same DNA sequence has been used: 5′ CTCGTCAGCATCXTCATCATACAGTCAGTG 3′, with X representing the position of the abasic site. The oligonucleotides containing CPD or 6-4PP lesions were synthesized as described.18 The 30 bp substrate containing an abasic site lesion has been obtained commercially (Eurogentec, Belgium).
Incision assay
The DNA substrates were labeled at the 5′ side of the top strand using polynucleotide kinase as described.19 The DNA substrates (0.2 nM) were incubated with 5 nM UVDE in 20 μL reaction mix (20 mM HEPES pH 6.5, 100 mM NaCl, 1 mM MnCl2). After 15 min incubation at 55°C the reaction was terminated by adding 3 μL EDTA/SDS (0.33M EDTA, 3.3% SDS) and 2.4 μL glycogen (4 μg/μL) followed by ethanol precipitation. The incision products were loaded on a 15% denaturing polyacrylamide gel and visualized by irradiation of a photographic film after which they were quantified.
Incision of supercoiled plasmid DNA
Supercoiled plasmids pUC18 (2686 bp; 5 ng/μL; UV-irradiated at 300 J/m2 or not UV-irradiated) and pNP228 (4686 bp; 5 ng/μL) were incubated with 10 nM UVDE (unless stated otherwise) in a 10 μL reaction mix (20 mM HEPES pH 6.5, 100 mM NaCl, 1 mM MnCl2). After 15 min incubation at 55°C, the reactions were terminated by addition of 3 μL Ficoll/dyes/SDS/EDTA (0.05M EDTA, 3% SDS). Samples were loaded on a 0.7% agarose gel and visualized by staining with ethidiumbromide.
Filter binding assay
The filter binding assays were conducted in 10 μL samples containing 0.1 μM UVDE and 8 nM of 32P-labeled DNA in a reaction buffer containing 20 mM Tris pH 6.5, 100 mM NaCl, and 1 mM MnCl2. Samples were incubated for 10 min at 55°C. At the end of the incubation time 0.5 mL reaction buffer (preheated at 55°C) was added. The mixture was poured over a nitrocellulose filter and the incubation vial was rinsed with 0.5 mL of the preheated reaction buffer. Finally the filters were washed with 0.5 mL incubation buffer. Each sample was corrected for the amount of DNA retained on a filter in the absence of protein. Binding is expressed as the percentage of the input DNA retained on the filter.
Mass spectrometry
TthUVDE was precipitated in 10% TCA and subsequently washed with acetone. After drying, it was resuspended in 8M urea and 0.4M NH4HCO3. After 15 min incubation with dithiothreitol at 50°C and addition of iodoacetamide, trypsin digestion was carried out for 24 h at 37°C. The trypsinated sample was loaded on a LTQ-orbitrap mass spectrometer and MS and MSMS was run.
Crystallization
The purified protein was dialysed against 1× PBS (25 mM Phosphate, 150 mM NaCl, pH 7.4) and concentrated to 3–5 mg/mL by centrifugation using an Ultrafree Filter Device (Millipore). Crystals were grown by sitting-drop vapor diffusion at 22°C using as precipitant: 0.1M sodium acetate buffer pH 4.4, 2M sodium formate and 1 mM MnCl2 (UVDE pH 4.4; 1 μL protein and 1 μL precipitant) or 0.5M (NH4)2SO4, 0.1M sodium acetate buffer pH 6.0, 1M Li2SO4 (UVDE K229L and UVDE E175A; 1 μL protein and 0.5 μL precipitant) or 0.1M sodium acetate buffer pH 5.6, 1M sodium formate (UVDE K229R; 1 μL protein and 1 μL precipitant). The crystals grew like diamonds (40–100 μm) in 1–2 days. Crystals were transferred to precipitant solution with 10% glycerol prior to data collection.
Data collection and processing
Datasets were collected on beam-line ID14-2 at the European Synchrotron Radiation Facility at a wavelength of 0.933 Å. The crystals were flash-frozen and kept at 100 K during data collection. Data collection strategy was determined with the program BEST.20 Reflections were indexed and integrated with iMosflm.21 Scaling and merging was performed with SCALA from the CCP422 suite. For data statistics, see Table I.
Structure solution and refinement
Structure solution was performed by molecular replacement with as search model the structure of TthUVDE wild type (PDB-code: 2j6v) with an unmodified lysine using MOLREP23 from the CCP4 suite, though for K229R, the structure of UVDE pH 4.4 was used as input directly into refinement. Manual adjustments to the model were done with COOT.24 Refinement was done using REFMAC16 and water molecules were added using ARP/wARP25 and COOT. TLS refinement was used in refinement for structures UVDE pH 4.4, UVDE K229L, and UVDE E175A. Illustrations were prepared using ccp4mg.26
Acknowledgments
The authors thank Marian van Kesteren for technical assistance and Hans van Elst, Bobby Florea, Wilbert Vermeij, and Patrick Voskamp for their technical assistance with the mass spectrometry. They acknowledge the European Synchrotron Radiation Facility for provision of synchrotron radiation facilities, and Ganesh Natrajan for assistance in using beamline ID14-2. They also thank Anita Coetzee and Bram Schierbeek for help in screening crystals. Coordinates and structure factor amplitudes have been deposited in the Protein Data Bank under ID code 3bzg for UVDE pH 4.4, 3bzj for UVDE K229L, 3c0l for UVDE K229R, and 3c0q for UVDE E175A.
References
- 1.Sidik K, Lieberman HB, Freyer GA. Repair of DNA damaged by UV-light and ionizing-radiation by cell-free-extracts prepared from Schizosaccharomyces pombe. Proc Natl Acad Sci USA. 1992;89:1212–1216. doi: 10.1073/pnas.89.24.12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bowman KK, Sidik K, Smith CA, Taylor JS, Doetsch PW, Freyer GA. A new ATP-independent DNA endonuclease from Schizosaccharomyces pombe that recognizes cyclobutane pyrimidine dimers and 6-4-photoproducts. Nucleic Acids Res. 1994;22:3026–3032. doi: 10.1093/nar/22.15.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avery AM, Kaur B, Taylor J, Mello JA, Essigmann JM, Doetsch PW. Substrate specificity of ultraviolet DNA endonuclease (UVDE/Uve1p) from Schizosaccharomyces pombe. Nucleic Acids Res. 1999;27:2256–2264. doi: 10.1093/nar/27.11.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paspaleva K, Thomassen E, Pannu NS, Iwai S, Moolenaar GF, Goosen N, Abrahams JP. Crystal structure of the DNA repair enzyme UV damage endonuclease. Structure. 2007;15:1316–1324. doi: 10.1016/j.str.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 5.Abendroth J, Niefind K, Schomburg D. X-ray structure of a dihydropyrimidinase from Thermus sp. at 1.3 Angstrom resolution. J Mol Biol. 2002;320:143–156. doi: 10.1016/S0022-2836(02)00422-9. [DOI] [PubMed] [Google Scholar]
- 6.Cha J, Mobashery S. Lysine N-zeta-decarboxylation in the BlaR1 protein from Staphylococcus aureus at the root of its function as an antibiotic sensor. J Am Chem Soc. 2007;129:3834–3835. doi: 10.1021/ja070472e. [DOI] [PubMed] [Google Scholar]
- 7.Golema-Kotra D, Cha JY, Meroueh SO, Vakulenko SB, Mobashery S. Resistance of beta-lactam antibiotics and its mediation by the sensor domain of the transmembrane BlaR signalling pathway in Staphylococcus aureus. J Biol Chem. 2003;278:18419–18425. doi: 10.1074/jbc.M300611200. [DOI] [PubMed] [Google Scholar]
- 8.Golemi D, Maveyraud L, Vakulenko S, Samama J, Mobashery S. Critical involvement of a carbamylated lysine in catalytic function of class D beta-lactamases. Proc Natl Acad Sci USA. 2001;98:14280–14285. doi: 10.1073/pnas.241442898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Cross JB, Vreven T, Meroueh SO, Schlegel HB. Lysine carboxylation in proteins: OXA-10 beta-lactamase. Proteins. 2005;61:246–257. doi: 10.1002/prot.20596. [DOI] [PubMed] [Google Scholar]
- 10.Thoden JB, Philips GN, Neal TM, Raushel FM, Holden HM. Molecular structure of dihydroorotase: a paradigm for catalysis through the use of a binuclear metal center. Biochem. 2001;40:6989–6997. doi: 10.1021/bi010682i. [DOI] [PubMed] [Google Scholar]
- 11.Thoden JB, Marti-Arbona R, Raushel FM, Holden HM. High-resolution X-ray structure of isoaspartyl dipeptidase from Escherichia coli. Biochemistry. 2003;42:4874–4882. doi: 10.1021/bi034233p. [DOI] [PubMed] [Google Scholar]
- 12.Hosfield DJ, Guan Y, Haas BJ, Cunningham RP, Tainer JA. Structure of the DNA repair enzyme endonuclease IV and its DNA complex: double-nucleotide flipping at abasic sites and three-metal-ion catalysis. Cell. 1999;98:397–408. doi: 10.1016/s0092-8674(00)81968-6. [DOI] [PubMed] [Google Scholar]
- 13.Weiss MS, Hilgenfeld R. On the use of the merging R factor as a quality indicator for X-ray data. J Appl Cryst. 1997;30:203–205. [Google Scholar]
- 14.Diederichs K, Karplus PA. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat Struct Biol. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]
- 15.Laskowski RA, MacAuthor MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 16.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst D. 1997;50:760–763. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 17.Goosen N, Moolenaar GF. Repair of UV damage in bacteria. DNA repair. 2008;7:353–379. doi: 10.1016/j.dnarep.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 18.Iwai S. Chemical synthesis of oligonucleotides containing damaged bases for biological studies. Nucleosides Nucleotides Nucleic Acids. 2006;25:561–582. doi: 10.1080/15257770600685826. [DOI] [PubMed] [Google Scholar]
- 19.Verhoeven EE, Van Kesteren M, Turner JJ, Van der Maarel GA, Van Boom JH, Moolenaar GF, Goosen N. The C-terminal region of Escherichia coli UvrC contributes to the flexibility of the UvrABC nucleotide excision repair system. Nucleic Acids Res. 2002;30:2492–2500. doi: 10.1093/nar/30.11.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Popov AN, Bourenkov GP. Choice of data-collection parameters based on statistical modelling. Acta Cryst D. 2003;59:1145–1153. doi: 10.1107/s0907444903008163. [DOI] [PubMed] [Google Scholar]
- 21.Leslie AG. Integration of macromolecular diffraction data. Acta Cryst D. 1999;55:1696–1702. doi: 10.1107/s090744499900846x. [DOI] [PubMed] [Google Scholar]
- 22.Collaborative Computational Project 4. The CCP4 suite: programs for protein crystallography. Acta Cryst D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 23.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 24.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 25.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 26.Potterton L, McNicholas S, Krissinel E, Gruber J, Cowtan K, Emsley P, Murshudov GN, Cohen S, Perrakis A, Noble M. Developments in the CCP4 molecular graphics project. Acta Cryst D. 2004;60:2288–2294. doi: 10.1107/S0907444904023716. [DOI] [PubMed] [Google Scholar]