

Abstract
Pseudomonas aeruginosa is the prototypical biofilm-forming gram-negative opportunistic human pathogen. P. aeruginosa is causatively associated with nosocomial infections and with cystic fibrosis. Antibiotic resistance in some strains adds to the inherent difficulties that result from biofilm formation when treating P. aeruginosa infections. Transcriptional profiling studies suggest widespread changes in the proteome during quorum sensing and biofilm development. Many of the proteins found to be upregulated during these processes are poorly characterized from a functional standpoint. Here, we report the solution NMR structure of PA1324, a protein of unknown function identified in these studies, and provide a putative biological functional assignment based on the observed prealbumin-like fold and FAST-NMR ligand screening studies. PA1324 is postulated to be involved in the binding and transport of sugars or polysaccharides associated with the peptidoglycan matrix during biofilm formation.
Keywords: Pseudomonas aeruginosa PA1324, NMR, functional genomics, NMR high-throughput screens, protein-ligand binding, protein-ligand co-structures, structural biology, structural genomics, chemical proteomics, protein structure initiative, hypothetical proteins, FAST-NMR, Northeast Structural Genomics Consortium
Introduction
Genome sequencing projects conducted in the past decade have enabled many novel areas of research in biology, chemistry, and drug discovery. With more than 686 completed genomes and 1270 on-going projects, ∼6.5 million protein sequences have been identified, but ∼30–50% of these proteins are not functionally annotated.1 The protein structure initiative (PSI) was initiated to determine a representative structure for each protein in the proteome with an expectation that structural information will assist in solving the functional annotation problem.2 PSI protein selection is based on the goal of maximizing the coverage of structural space by choosing representative targets from protein families that lack a known structure.3 A single structure may serve as a model for the other member in the protein family.4
Since 2004, PSI has been shown to contribute 50% of the novel structures deposited in the protein database (PDB).5 This coincides with an increase in the number of proteins classified as unknown function, where there are currently 2755 such proteins in the PDB.6 A number of computational methods have been developed to aid in the annotation of proteins of unknown function.7 These methods use protein structures and a range of techniques to compare global folds, local structural features, or matches to 3D templates to assign putative biological functions.8 Although these computational approaches are extremely efficient in analyzing large data sets, the results for a specific protein may be ambiguous or absent.9,10
Functional Annotation Screening Technology by NMR (FAST-NMR) provides experimental data for the functional annotation of novel proteins by combining ligand affinity screens and structural biology with bioinformatics.7,11 Functional ligands that bind to the protein are identified using a tiered NMR ligand screen.12 The protein's functional epitope is determined by mapping the ligand induced chemical shift perturbations (CSPs) onto the surface of the protein. A co-structure is rapidly determined using this CSPs data,13 which is then used to drive the bioinformatics analysis. The Comparison of Protein Active Site Structures (CPASS) program and database compares the experimental ligand-defined active site determined from the FAST-NMR screen to all the ligand-defined active sites available in the PDB.7,11,14 A general biological function is assigned through the comparative similarity of the experimentally determined ligand binding site with a protein of known function.
The sequence of the Pseudomonas aeruginosa PA01 genome was completed in 2000, but as of January 2008 > 43% of the proteome lacks a functional annotation.15 P. aeruginosa is one of the primary bacteria that forms biofilms16,17 and causes serious problems with individuals with chronic lung problems, such as cystic fibrosis.17,18 In the past decade, the prevalence of nosocomial infections has risen significantly, particularly in developing countries.19,20 Biofilms are often the cause of these infections, and their formation is poorly understood.21 This situation is further exacerbated by the increasing rate of antibiotic resistance for P. aeruginosa, which has reached upwards of 40% for some strains. Resistance to imipenem, ciprofloxacin, and ceftazidine have all been observed in the past year.22 Therefore, developing new antibiotics against P. aeruginosa biofilms is of great importance, requiring the identification of new drug targets.23,24
P. aeruginosa genes that are upregulated in biofilms may represent potential therapeutic targets. PA1324 is an example of such a gene that was identified by microarray analysis of gene expression associated with biofilms and quorum sensing in P. aeruginosa. PA1324 and PA1323 were identified in two expression profiling studies in association with quorum sensing: transcription of both genes appears to increase similarly in quorum sensing conditions.25,26 One of these studies indicated that PA1324 and PA1323 expression was not repressed upon treatment of cells with furanone, a repressor of quorum sensing.25 A study of biofilm formatation found PA1324 to be significantly upregulated with expression levels 10–30 times higher versus planktonic cultures.27 PA1323 was upregulated as well. Another study of biofilm formation in P. aeruginosa included microarray data indicating that PA1324 is not regulated by the SadARS three-component regulatory system required for biofilm maturation.28 To better understand the role of PA1324 in biofilms, the structure and putative biological function of the protein was determined using NMR spectroscopy and FAST-NMR.
Results
PA1324 sequence analysis
PA1324 (21–170) is target PaP1 of the Northeast Structural Genomics Consortium (NESG; http://www.nesg.org)29,30 and is currently listed as a hypothetical protein in KEGG31 and TIGR.32 PA1324 occurs in at least five strains of P. aeruginosa and has several homologues from a diverse assortment of proteobacteria, all with 26–28% sequence identity (Fig. 1). The proteins identified in the BLASTp sequence analysis as being most similar to PA1324 are hypothetical proteins and putative lipoproteins. Residues 1–18 in PA1324 are predicted to be a hydrophobic signal peptide by SOSUI.33 Thus, PA1324 was expressed with a 20 residue His-tag (GSSHHHHHHSSGLVPRGSHM) in place of the signal peptide plus two residues at the N-terminus, such that the numbering of residues 21–170 corresponds to the natural PA1324 sequence.
Figure 1.

Multiple sequence alignment of PA1324 and six homologues. Gene names and NCBI RefSeq numbers are indicated by three letter abbreviations as follows: Pae, Pseudomonas aeruginosa PA1324, NP_250015; Csa, Chromohalobacter salexigens Csal_0658, YP_572718; Sde, Saccharophagus degradans Sde_0431, YP_525907; Dvu, Desulfovibrio vulgaris Dvul_1060, YP_966507; She, Shewanella sp. W3-18-1 Sputw3181_2461, YP_963839; Sul, Sulfurovum sp. NBC37-1 SUN_1567, YP_001358874.
The gene for PA1323 is in the same reading frame as PA1324 and terminates just 60 base pairs upstream from the start of PA1324, suggesting the two genes may form an operon. PA1323 is a conserved hypothetical protein (COG4575, Pfam05957, DUF883). On the same strand and upstream from PA1323 is the gene for PA1322, which is a putative TonB-dependent receptor-like transporter for catechols/ferric iron uptake (FIU). Interestingly, Psi-BLAST queries with the PA1324 sequence report low similarity hits with E scores of >0.2 to the N-terminal region of numerous TonB-dependent receptor-like proteins (though not any of the FIU class). The N-terminal 150–200 amino acids of many TonB receptors form a plug on the periplasmic side of the membrane that helps control transport through the barrel-like receptor-transporter. This may suggest a role for PA1324 and PA1323 in mediating the activity of PA1322; however, in the absence of structural information, the sequence similarity may be too low to be reliable.
PA1324 structure description and analysis
The solution structure of the globular portion (residues 21–170) of protein PA1324 from P. aeruginosa was determined by NMR. Table I summarizes key details of the structural ensemble. PA1324 forms an elongated barrel-like sandwich of two β-sheets, open on one side of the sandwich, and partially closed on the other, but not to the extent that the edge strands form hydrogen bonds to each other (Fig. 2). Around the barrel the strands occur in the order 7-1-4-3-2-5-6. A prominent loop of 37 residues between strands 2 and 3 (from residue 71 to 107) folds back upon the outside of the barrel and appears to form a pocket. Several polar residues occupy this pocket: E134, R91, G84, T153, Y79, S132, E70, R104. Near the N-terminal purification tag (residues 1–20), residues 27 to 41 form a loose region that contacts (NOEs observed) the prominent loop, but does not form a regular structure that is converged in the ensemble. The fold of the barrel-like sandwich is clearly that of the prealbumin-like fold (SCOP), which contains several superfamilies, including that of the thyroxine transport protein transthyretin (pre-albumin) and of a conserved accessory domain of membrane-type carboxypeptidases. One subfamily of the transthyretin superfamily is a starch-binding domain, and recent evidence from the Gene Ontology database suggests that PA1324 has a carbohydrate-binding domain.38
Table I.
Structural Statistics for the 20-member Ensemble of Structures for PA1324
| Distance restraints | |||
|---|---|---|---|
| Total | 1494 | ||
| Intraresidue | 231 | ||
| Sequential | 457 | ||
| Medium range (1 < |i−j| < 5) | 189 | ||
| Long range | 617 | ||
| Hydrogen bonds (2 per H-bond) | 70 | ||
| Dihedral angle restraints | |||
| Total | 132 | ||
| Phi | 65 | ||
| Psi | 67 | ||
| Total restraints | 1696 | ||
| Restricting restraints per restrained residue (144 residues) | 11.8 | ||
| Restricting restraints, long range, per restrained residue (144 residues) | 4.7 | ||
| Average restraint violations per structure | |||
| Distance restraints (all > 0.0 Å) | 60.1 ± 5.1 | ||
| Maximum violation (Å) | 0.05 | ||
| Dihedral restraints (all > 0.00) | 6.3 ± 3.8 | ||
| Maximum violation (°) | 1.02 | ||
| RMSD to average structures (Å): | |||
| Residues 27–170 (144 residues) | |||
| Backbone atoms (N,C,C′) | 0.92 | ||
| All heavy atoms | 1.42 | ||
| Residues 42–70, 108–170 (92 residues)a | |||
| Backbone atoms (N,C,C′) | 0.66 | ||
| All heavy atoms | 1.08 | ||
| Residues 33–37, 43–57, 60–82, 86–113, 116–140, 144–159, 161–168b | |||
| Backbone atoms | 0.80 | ||
| All heavy atoms | 1.20 | ||
| Ramachandran plotc: | |||
| Protein residues | 27–170 inclusive (144) | Barrel only (92) | Ordered (120) |
| Most favored region (%) | 84.0 | 86.6 | 89.6 |
| Additional allowed region (%) | 14.3 | 12.6 | 10.3 |
| Generously allowed region (%) | 1.4 | 0.2 | 0.1 |
| Unallowed region (%) | 0.3 | 0.4 | 0.0 |
| Global quality scoresd: | Raw | Z-Score | |
| PROCHECK(all) | −0.46 | −2.72 | |
| PROCHECK(phi-psi) | −0.60 | −2.05 | |
| MolProbitye clash score | 18.41 | −1.63 | |
| RPF Scoresf | |||
| Recall | 0.84 | ||
| Precision | 0.77 | ||
| F-measure | 0.81 | ||
| DP-score | 0.62 | ||
Figure 2.
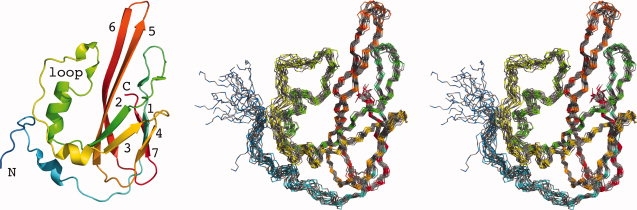
Structure of PA1324 21-170. Left: ribbon cartoon structure with rainbow coloring. The N- (blue) and C- (red) termini, the prominent loop (residues 71–107), and β-strands of the barrel (numbered 1–7) are indicated. Right: stereo view of backbone atoms for all 20 superimposed structures comprising the ensemble.
Dali analysis of PA1324 revealed several proteins of modest structural similarity, with Z-scores of between 2 and 5.39 Despite sequence similarities less than 20%, these proteins clearly adopt the same fold as PA1324. The top protein identified was transthyretin. Other proteins identified have functions corresponding to sugar and polysaccharide-binding proteins, extracellular matrix and cell adhesion proteins and structural proteins. Because the Dali and primary sequence analysis were unsuccessful for definitive functional annotation, further investigation was needed to uncover a putative biological function of PA1324.
FAST-NMR results for PA1324
FAST-NMR was used to screen ligands for possible binding by PA1324 to leverage a functional hypothesis.11 FAST-NMR provides functional information for hypothetical proteins by experimentally identifying ligands and ligand binding sites. After ligands are identified and the binding site is mapped by CSPs, a co-structure is obtained in ∼30–45 min by using the CSPs to direct an AutoDock simulation where our ADF program selects the best conformer based on consistency with the magnitude of chemical shift changes.13 A functional hypothesis can be developed by comparing the ligand-defined active-site from FAST-NMR to a database of protein-ligand binding sites for proteins of known function using CPASS (Comparison of Protein Active-Site Structures).7,11,14 This information provides a starting point for further functional investigations, additional screening of related ligands, ligand modification or optimization, or even identification of drug leads.
FAST-NMR consists of a two-stage screening methodology. The primary 1D 1H line-broadening screen yielded 20 hits from the functional compound library (5.46% hit rate) that bind PA1324.12 Figure 3 shows a representative example of the typical results obtained from these line broadening experiments, where the increase of line width (decrease in peak intensity) indicates a binding event. Seven of these 20 compounds contain aryl-sulfonate, sulfonyl fluoride, or sulfone functional groups. This suggests potential chemical characteristics that may be present in the native ligand. Some of the compounds found to bind PA1324 in the 1D 1H line-broadening screen are shown in Figure 4.
Figure 3.
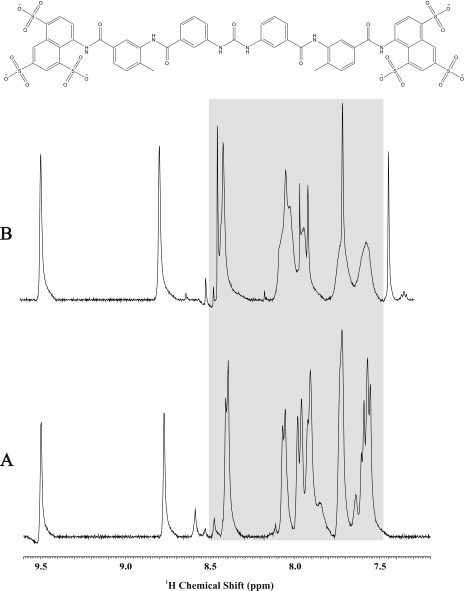
A 1D 1H NMR line-broadening experiment showing an expanded reference mixture spectrum (A) and the mixture with PA1324 (B). The highlighted region indicates the observed line broadening and binding of suramin to PA1324.
Figure 4.
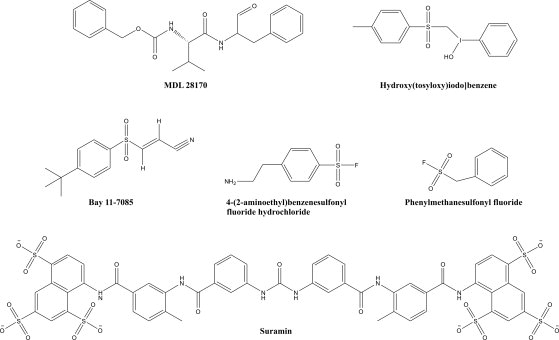
A subset of the 20 compounds found to bind to PA1324 by 1D 1H NMR line-broadening experiments was Aryl or benzyl sulfonyl, and sulfonate moieties are present in 7 of the 20 compounds.
The 20 compounds were further evaluated for PA1324 binding with a secondary screen using the 2D 1H-15N HSQC experiment. CSPs were identified through the comparison of a reference 2D 1H-15N HSQC spectrum for free PA1324 to the spectrum in the presence of the compound. Suramin was identified as the primary lead compound based on the larger quantity and greater magnitude of CSPs observed relative to the other compounds [Fig. 5(A)]. Suramin is a known analogue of heparin, a highly negatively charged sulfated biopolymer from the glycosaminoglycan family of carbohydrates.41 Titration of suramin into a sample of PA1324 was conducted to determine the suramin binding site. 2D 1H-15N HSQC spectrum of the PA1324-surmain complex showed the backbone amide chemical shifts of residues N23, T38, L52, M76, K77, G84, L86, D101, Y130, T153, K154, and S159 were perturbed upon the addition of suramin. Interestingly, this putative suramin binding site resembles a heparin binding site because it contains regions of positively charged lysine and arginine residues.42,43 Figure 5(B) shows an electrostatic potential surface of the binding site of suramin. A titration with a heterogeneous sample of heparin (Sigma Aldrich, St. Louis, MO) was also completed [Fig. 1(S)]. Heparin appears to bind predominately in the same cleft occupied by suramin.
Figure 5.
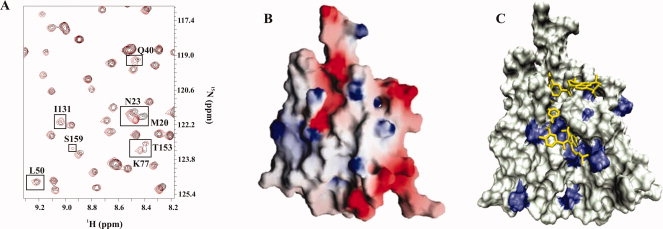
Suramin binding to PA1324. (A) An expanded region of an overlay of the 2D 1H-15N HSQC spectrum of free PA1324 (black) and PA1324 in the presence of suramin (red). (B) A GRASP40 electrostatic surface of PA1324 demonstrating the positive electrostatic potential of the suramin binding site. Blue and red indicate positively charged and negatively charged surfaces, respectively. (C) A docked model of suramin in the PA1324 binding site identified by FAST-NMR. Residues that incurred a CSP upon binding suramin are colored blue. The distal CSPs could be long-range effects of the binding interaction or potential suramin conformational exchange.
Possible structures of PA1324 bound to suramin were modeled using the NMR-defined binding site to guide an AutoDock simulation.13 Figure 5(C) shows a co-structure of suramin in the FAST-NMR defined PA1324 binding site, which was compared with all other ligand-defined binding sites in the PDB using CPASS. The highest CPASS scoring comparisons (∼40–45% similarity) were from proteins involved in polysaccharide biosynthesis, the cellulosome (scavenger of plant cell-wall polysaccharides), transport, and DNA binding proteins (segregation proteins). Many of these proteins bind sugars and polysaccharides. A comparison of the FAST-NMR defined PA1324 binding site to the protein with the highest CPASS score, the spore coat polysaccharide biosynthesis protein SpsA from Bacillus subtilis (PDB ID 1H7L) bound to thymidine-5′-phosphate (TMP),44 shows many similar features and interactions (Fig. 6). SpsA is a glycosyltransferase, but although the PA1324 has a similar binding site to SpsA, the functional analysis by FAST-NMR does not indicate any enzymatic activity for PA1324. The 1D 1H NMR spectrum of suramin in the presence of PA1324 does not undergo any changes with time. The CPASS results do, however, support the DALI inference relating the putative biological function of PA1324 with general sugar binding proteins and transport.
Figure 6.
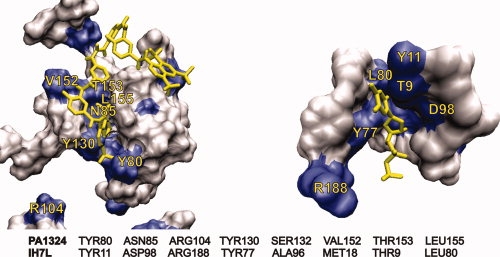
The CPASS comparison of PA1324 with suramin (left) to 1H7L, a spore coat polysaccharide biosynthesis protein bound to thymidine-5′-phosphate (right). The sequence alignment below the figures illustrates the similarities in the ligand binding sites.
Discussion
Functional analysis of PA1324
Chronic pulmonary infection with P. aeruginosa is the major cause of mortality among cystic fibrosis patients.45 P. aeruginosa virulence results from its antibiotic resistance and the ability of P. aeruginosa to form biofilms. PA1324 was identified as being significantly upregulated in P. aeruginosa biofilms along with PA1323.46 P. aeruginosa biofilms are controlled by known quorum-sensing systems47–49 that coordinate the regulation of gene transcription through the secretion of signaling molecules or autoinducers.50 The transcription of genes for both PA1323 and PA1324 have been demonstrated to increase similarly under quorum sensing conditions,25,51 but are not affected by furanone, a repressor of quorum sensing.25 A number of environmental factors have also been shown to induce biofilm formation,52,53 which includes anaerobic conditions,54 Fe limitation,55–57 high osmolarity,58 and high temperature.58 Interestingly, sub-inhibitory concentrations of some antibiotics were found to enhance biofilm formations.59 The addition of ethanol,60 oleic acid,61 glucose,62 and UDP-N-acetylglucosamine63 also stimulate biofilms.
An important component of biofilm formation is the production of an extracellular polysaccharide called polysaccharide intercellular adhesin (PIA) that mediates cell–cell adhesion.64 Bacteria within a biofilm are embedded in this hydrated extracellular matrix composed of β (1-6)-linked N-acetylglucosamine polymer.65,66 Glucose is required for PIA production,67 where UDP-N-acetylglucosamine is the precursor of the biofilm's polysaccharide matrix.63 The addition of both glucose and UDP-N-acetylglucosamine stimulate PIA production and biofilm formation.62,63 Interestingly, N-acetylglucosamine is also a repeating unit within heparin, which also has been shown to stimulate biofilm formation.68 Furthermore, heparin plays an important role in P. aeruginosa adherence to epithelial respiratory cells through an interaction with P. aeruginosa outer-membrane proteins.69
The tertiary structure of PA1324 determined by NMR indicates a barrel-like sandwich topology corresponding to a prealbumin fold. This fold contains a carbohydrate-binding superfamily, where the highest similarity from a Dali analysis identified a protein involved in transport. Other low-similarity proteins from the Dali analysis include sugar or heparin binding proteins and proteins involved in the extracellular matrix or cell adhesion. Recent evidence from the Gene Ontology database also suggests PA1324 is a carbohydrate-binding domain.38 The FAST-NMR screen found only one significant compound that bound to PA1324—suramin, a heparin analog. The suramin PA1324 binding site was compared with all ligand binding sites in the PDB using CPASS, where the highest CPASS scores were to proteins that interact with carbohydrates or carbohydrate-like molecules. Site-directed mutational studies for a number of carbohydrate binding domains (CBD) indicate some common features for carbohydrate affinity. Specifically, carbohydrate binding involves a combination of hydrogen-bond and aromatic interactions, where replacing conserved hydrogen bonding (D, N, E, Q, K, R) or aromatic residues (W, Y) significantly diminished or eliminated carbohydrate binding.70–72 These specific amino acid interactions have been corroborated by NMR and x-ray structures of CBD bound to carbohydrates.73–75 Importantly, the experimentally identified PA1324 suramin binding site contains these typical carbohydrate binding residues (D, N, K, Y). The ability of PA1324 to bind carbohydrates was further confirmed by the observation that PA1324 binds heparin in a manner similar to suramin.
The binding affinity of carbohydrates to CDBs tend to range from low micromolar to 100s of micromolar, where the magnitude of the dissociation constant (KD) is strongly dependent on the carbohydrate.71–78 For example, the affinity of cyanovirin-N to Man-2 and Man-9 were determined to be 757 ± 80 μM and 3.4 ± 0.05 μM, respectively.78 Similarly, the human and mouse galectin-9 N-terminal carbohydrate recognition domains were shown to bind 42 different carbohydrates with KDs ranging from 0.3 μM to 109 μM.75 The PA1324 NMR chemical shift titration studies provides a KD estimate of 152 ± 18 μM and 51 ± 14 μM for suramin and heparin, respectively. This is consistent with other CBDs, especially considering that suramin is an unoptimized structural homolog of the natural ligand and heparin is a heterogeneous mixture.
The gene for PA1322, a putative TonB-dependent receptor-like transporter,79 is on the same strand and upstream from PA1323 and PA1324. P. aeruginosa TonB1 was shown to be essential in both biofilm formation and quorum sensing.80 This activity is separate from its known role in iron uptake. Furthermore, a TonB-dependent receptor was identified that transports sucrose in the phytopathogenic bacterium Xanthomonas campestris pv. campestris and is conserved in other bacteria.81 Similarly, a TonB-dependent receptor that transport maltodextrins across the outer membrane were identified in Caulobacter crescentus.82 These observations suggest that TonB-dependent receptors transport other nutrients besides iron and vitamin B12 and may play a role in PIA production.83 This is especially relevant because heparin and UDP-N-acetylglucosamine stimulate PIA production and biofilm formation, where UDP-N-acetylglucosamine is also a PIA precursor.
The genetic organization of TonB-dependent receptors implies that functionally related proteins (σ and anti-σ) are neighboring genes,79 which infers an association between PA1322, PA1323, and PA1324. PA1323 is predicted by Phobius84 to contain at least one transmembrane helix and exhibits sequence similarity to other putative membrane proteins. The TonB dependent regulatory systems consist of a number of membrane bound proteins (anti-σ, TonB, ExbB, ExbD) suggesting a potential biological role for PA1323. PA1324 has a predicted signal peptide suggesting transport to the membrane. Also, PA1324 has shown low-sequence similarity (E scores of >0.2) to the N-terminal region of TonB-dependent receptor-like proteins, which forms a plug that controls substrate transport through the barrel-like receptor-transporter. Deleting the plug domain inactivates substrate transport where activity is regenerated by cosynthesis of the separate plug and β-barrel domains.85 The plug structure consists of a twisted β-sheet with loops and helices connecting the strands, where the substrate binds the outer surface of the plug.86 Interestingly, an isolated plug domain does not adopt a folded structure.87 Although there appears to be some gross similarities between the PA1324 NMR structure and the general description of a plug domain, Dali did not predict any similarity between the two structures. Also, a sequence alignment of PA1322 with other known TonB-dependent receptor proteins implies an intact N-terminus in PA1322. Thus, does PA1324 represent an alternative mechanism of regulating PA1322, the putative TonB-dependent receptor-like transporter by mimicking a folded plug domain complexed to a substrate?
Combining the sequence and structural analysis with the functional screening, we suggest a role for PA1324 in carbohydrate binding or transport together with PA1323, the protein predicted to co-express with PA1324. PA1324 and PA1323 might be involved in the polysaccharide secretion or scavenging process required for PIA formation in P. aeruginosa biofilms. This biological function may also involve the regulation of PA1322, a putative TonB-dependent receptor-like protein that may transport carbohydrates across the P. aeruginosa membrane.
Materials and Methods
Sample preparation
Chemicals were purchased from Sigma (St. Louis, MO). Stable isotope-labeled compounds were purchased from Cambridge Isotope Laboratories (Andover, MA). Genomic DNA from P. aeruginosa strain PA01 was obtained from the American Type Culture Collection (Manassas, VA). Primers were designed for PCR amplification of the PA1324 21-170 gene fragment, so that residues 1–20 (MLSIKKNLGLLAMTAALAAC) would be absent in the expressed protein. Residues 1–18 are predicted to be a hydrophobic signal peptide by SOSUI.33 Thus, the 170 residue expressed protein is PA1324 21–170 with a 20 residue His-tag (GSSHHHHHHSSGLVPRGSHM) in place of the signal peptide sequence at the N-terminus. A M21A mutation occurs as a cloning artifact at the insertion point in the expression vector, pET28b. Escherichia coli [Rosetta BL21(DE3)] cells were transformed by heat shock with this vector and plated on LB agar plates containing 30 μg/mL kanamycin. Single colonies from the plate were used to inoculate 4 mL overnight seed cultures in M9 minimal medium made with U-15N NH4Cl (1 g/L) and U-13C glucose (2 g/L) and supplemented with 7.5 μM FeCl3, 10 μM ZnSO4, and 10 μM MnSO4; seed cultures were shaken overnight at 37°C, then used to inoculate 50 mL starter cultures; these were grown to OD600 0.7 and diluted into 0.5 L media in 2L glass flasks. When the cultures reached OD600 0.6–0.8, expression of PA1324 was induced with 1.1 mM IPTG, the flasks were moved to a 25°C shaking incubator for overnight growth. After 15 h, cells were pelleted with gentle centrifugation and resuspended in 25 mL lysis buffer (50 mM sodium phosphate, 500 mM NaCl, 10 mM imidazole, pH 8.0). Cells were lysed by passage four times through a French Press. After insoluble material was removed by centrifugation, protamine sulfate (12.5 mg) was added to precipitate nucleic acids, and the supernatant was ultracentrifuged for 1 h. The His-tagged protein was loaded onto a gravity column containing 10 mL Ni-NTA affinity resin (Qiagen, Valencia, CA) equilibrated with lysis buffer, washed with 50 mL lysis buffer, and removed with elution buffer (50 mM phosphate, 500 mM NaCl, 400 mM imidazole). Protein in elution buffer was exchanged into NMR buffer (20 mM BisTris Propane, 300 mM NaCl pH 6.8) on a PD10 column. The yield of protein was >100 mg/L of culture. For NMR samples, 7% (v/v) D2O was added, and 250 μL of sample was put into a Shigemi NMR tube. In addition, a U-15N and 5% biosynthetically directed fractionally 13C-labeled sample was generated for stereospecific assignment of isopropyl methyl groups in leucine and valine residues by growing cells as described above but with 5% of the carbon as U-13C-glucose and 95% as unenriched glucose.88
NMR structure determination
NMR experiments were conducted at 298 K using standard triple-resonance experiments.89,90 NMR experiments were conducted at the Environmental Molecular Sciences Laboratory of Pacific Northwest National Laboratory, Richland, WA. Experiments were collected on Varian Inova 600, 750, and 800 instruments. The following experiments were recorded: 1H-15N-HSQC, 1H-13C-HSQC, HNCO, HNCA, HNCOCA, HNCACB, CBCACONNH, CBCACOCAHA, CCC-TOCSY-NNH, HCC-TOCSY-NNH, HCCH-TOCSY, HCCH-COSY, 3D-15N-NOESY (100 ms mixing time), 4D-13C-13C-HMQC-NOESY-HMQC (80 ms mixing time), two 3D-13C-NOESYs (80 ms mixing time) optimized for either aliphatic or aromatic carbons, HNHA, and aromatic ring side chain correlation experiments (HBCBCG-CDHD/-CEHE). All pulse sequences were from the BioPack library (Varian) except 4D-13C-13C-HMQC-NOESY-HMQC, and HBCBCG-CDHD/-CEHE-aro, which were from Lewis Kay (University of Toronto). Amide proton exchange was monitored by acquiring 1H-15N-HSQC spectra following dissolution of lyophilized protein in D2O. Stereospecific Leu and Val side chain assignments were obtained from a 1H-13C-HSQC experiment recorded on the 5% 13C-sample.88 Chemical shift assignments were deposited to BioMagResBank (bmrb-id 6343).
NOESY peak assignments and preliminary structure ensembles were determined with AutoStructure using peak-picked NOESY data and chemical shift assignments as input.37 Manual inspection of the data clearly indicated the correct fold was calculated, and manual refinement of the preliminary ensemble and error-checking of the restraint lists was conducted using XPLOR-NIH, using standard sa.inp and dgsa.inp protocols modified to use 20,000 high temperature steps at 2000 K, followed by 200,000 cooling steps (3 fs steps). Sum averaging of NOE restraints was used. Final refinement in explicit water solvent models with Lennard-Jones and electrostatic potentials included was accomplished with CNS using a modification of the procedure and forcefield of Nilges.91 NOE distance restraints had uniform lower bounds of 1.8 Å and upper bounds of either 2.8, 3.0, 3.5, 4.0, 4.5, or 5.0 Å. Hydrogen bond restraints were derived from amide proton D2O exchange data. Amide 1H-15N-HSQC cross peaks still present 30 min. after dissolution of a lyophilized sample in D2O were given bounds of 1.8–2.5 Å for the HN-O distance and 2.8–3.5 Å for the N-O distance, provided preliminary structural ensembles clearly indicated the probable acceptor atom. Dihedral restraints were obtained from TALOS,92 only those dihedrals with a score of 10 were used. TALOS reported restraints were given bounds of ± 30° for phi and ± 40° for psi, and only dihedral angles in regions of regular secondary structure except in regions where the angle reported by TALOS was consistent with the HNHA experiment for phi angles or with the ratio of sequential interresidue and intraresidue amide NH to CαH cross peak intensities in the 15N NOESY data.93 The entire ensemble of 20 structures calculated, together with the distance and dihedral restraints, was deposited to the Protein Data Bank (pdb-id 1XPN).
FAST-NMR
The 1D 1H NMR line-broadening samples consist of 100 μM of each compound and 25 μM protein in a 20 mM D-Bis-Tris buffer with 11.1 μM TMSP as a NMR reference in 100% D2O at pH 7.0 (uncorrected). The NMR spectra were collected on a Bruker 500 MHz Avance spectrometer equipped with a triple-resonance, Z-axis gradient cryoprobe, a BACS-120 sample changer, and Icon NMR software for automated data collection. 1H NMR spectra were collected with 128 transients at 298 K with solvent presaturation of the residual HDO, a sweep-width of 6009 Hz and 32 K data points and a total acquisition time of 6 min. The NMR spectra were processed automatically using a macro in the ACD/1D NMR manager (Advanced Chemistry Development, Toronto, ON). The NMR data was Fourier transformed, zero-filled, phased, and baseline corrected. Each spectrum was referenced with the TMSP peak set to 0.0 ppm and peak-picked. Compounds were identified as binding by a visual comparison of the free and protein spectra for all 113 mixtures.
Only the 20 compounds that exhibited line broadening in the 1D 1H experiment were used in the 2D 1H-15N HSQC experiments to verify specific binding and identify the ligand binding site. Twenty-one 2D 1H-15N HSQC spectra were collected with 16 transients at 298 K with a sweep-width of 6009 Hz and 1 K data points in the direct dimension and 1612 Hz and 256 data points in the indirect dimension for a total acquisition time of 2.5 h. Each NMR sample consists of 100 μM protein and 400 μM compound in a Bis-Tris buffer in 95% H2O, 5% D2O at pH 7.0 (uncorrected). The spectra were processed using NMRPipe94 on a Intel Xeon 3.06 GHz dual processor Linux Workstation. Chemical shift differences were identified by comparing a reference 2D 1H-15N HSQC spectrum of the free protein to the spectrum of the compound-protein samples. CSPs were then mapped onto the surface of the PA1324 structure (PDB ID: 1XPN) and visualized using VMD-XPLOR.95 Only one compound, suramin, exhibited a definitive binding site. A series of titration experiments were done with increasing amount of suramin to determine the binding site. The PA1324 concentration was approximately constant at 100 μM in a single tube whereas the suramin concentrations were increased from 0 to 5 μM, 10 μM, 15 μM, 20 μM, 25 μM, 50 μM, 100 μM, 200 μM, 300 μM, 400 μM, 500 μM, and 600 μM. A titration experiment was also done with a heterogeneous sample of heparin (Sigma Aldrich, St. Louis, MO) to verify suramin binding to PA1324 was analogous to PA1324 binding heparin. Because the heterogeneous heparin sample has a molecular weight range of 6–12 kDa, an average MW of 9 kDa was used for determining the heparin concentration for the titration experiment. The PA1324 was again held constant at 100 μM whereas the heparin concentrations were increased from 0 to 5 μM, 10 μM, 15 μM, 20 μM, 25 μM, and 50 μM. At higher heparin concentrations, PA1324 began to precipitate out of solution. The addition of suramin and heparin caused chemical shift changes and peak broadening, where some peaks were broaden beyond detection at higher ligand concentrations. KD were measured by fitting chemical shift changes [Δ(ppm)] versus ligand concentration (L) using a standard Langmuir binding isotherm:
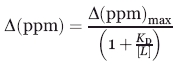 |
(1) |
The chemical shift changes were measured as:
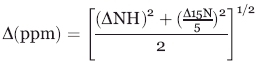 |
(2) |
where ΔNH is the difference between free and bound 1H amide chemical shifts (ppm) and Δ15N is the difference between free and bound 15N chemical shifts (ppm). The chemical shift changes from the suramin and heparin titrations for eleven and eight PA1324 residues, respectively, were superimposed based on normalizing the average chemical shift changes. A KD was measured by simultaneously fitting a single binding curve to this data using KaleidaGraph (Synergy Software, Reading, PA). A KD of 152 ± 18 μM with an R2 of 0.89 was determined for suramin binding to PA1324 and a KD of 51 ± 14 μM with an R2 of 0.84 was determined for heparin binding to PA1324 [Fig. 2(S)].
AutoDock96 was used to model the interaction between suramin and PA1324. Amides found to undergo CSPs following suramin binding were used to define a binding site region encompassing all the perturbed residues. A grid was manually defined within AutoDock to encompass the determined ligand-binding site by adjusting the x,y,z coordinates for the center of the grid box to position the grid in the binding pocket. The grid size is determined by the number of points in the x,y,z dimensions and is just visibly large enough to fit the entire ligand. The co-structures are selected based on a consistency with the observed CSPs in the 2D 1H-15N HSQC spectra.
CPASS database and software was developed to aid in the functional annotation of hypothetical proteins by using a protein-ligand co-structure.14 The CPASS database contains ∼27,000 ligand-defined active-sites identified from structures deposited in the PDB. CPASS returns a similarity score (0–100%) and an interactive 3D graphical display of the structural alignment for each active-site comparison. The CPASS software runs on 16-node Beowulf Linux cluster with a simple web-based interface. Each comparison averages ∼40s requiring ∼18 h to complete a comparison against the entire database.
Acknowledgments
A portion of the research was performed using the Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility sponsored by the Department of Energy's Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory. Montelione laboratory and other members of the NESG Consortium for shared NMR technologies.
References
- 1.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2008;36:D25–D30. doi: 10.1093/nar/gkm929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burley SK, Joachimiak A, Montelione GT, Wilson IA. Contributions to the NIH-NIGMS protein structure initiative from the PSI production centers. Structure. 2008;16:5–11. doi: 10.1016/j.str.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mirkovic N, Li Z, Parnassa A, Murray D. Strategies for high-throughput comparative modeling: applications to leverage analysis in structural genomics and protein family organization. Proteins. 2007;66:766–777. doi: 10.1002/prot.21191. [DOI] [PubMed] [Google Scholar]
- 4.Cort JR, Koonin EV, Bash PA, Kennedy MA. A phylogenetic approach to target selection for structural genomics: solution structure of YciH. Nucleic Acids Res. 1999;27:4018–4027. doi: 10.1093/nar/27.20.4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levitt M. Growth of novel protein structural data. Proc Natl Acad Sci USA. 2007;104:3183–3188. doi: 10.1073/pnas.0611678104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Powers R, Copeland J, Mercier K. Application of FAST-NMR in drug discovery. Drug Discov Today. 2008;13:172–179. doi: 10.1016/j.drudis.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee D, Redfern O, Orengo C. Predicting protein function from sequence and structure. Nat Rev Mol Cell Biol. 2007;8:995–1005. doi: 10.1038/nrm2281. [DOI] [PubMed] [Google Scholar]
- 9.Plewczynski D, Pas J, Von Grotthuss M, Rychlewski L. 3D-Hit: fast structural comparison of proteins. Appl Bioinformatics. 2002;1:223–225. [PubMed] [Google Scholar]
- 10.Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: current status and future challenges. Proteins: Struct Funct Bioinformatics. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- 11.Mercier KA, Baran M, Ramanathan V, Revesz P, Xiao R, Montelione GT, Powers R. FAST-NMR: functional annotation screening technology using nmr spectroscopy. J Am Chem Soc. 2006;128:15292–15299. doi: 10.1021/ja0651759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mercier KA, Germer K, Powers R. Design and characterization of a functional library for NMR screening against novel protein targets. Comb Chem High Throughput Screen. 2006;9:515–534. doi: 10.2174/138620706777935342. [DOI] [PubMed] [Google Scholar]
- 13.Stark J, Powers R. Rapid protein-ligand costructures using chemical shift perturbations. J Am Chem Soc. 2008;130:535–545. doi: 10.1021/ja0737974. [DOI] [PubMed] [Google Scholar]
- 14.Powers R, Copeland JC, Germer K, Mercier KA, Ramanathan V, Revesz P. Comparison of protein active site structures for functional annotation of proteins and drug design. Proteins: Struct Funct Bioinformatics. 2006;65:124–135. doi: 10.1002/prot.21092. [DOI] [PubMed] [Google Scholar]
- 15.Stover C, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 16.Poole K. Pseudomonas aeruginosa. Front Antimicrob Res. 2005:355–366. [Google Scholar]
- 17.Kirisits MJ, Parsek MR. Does Pseudomonas aeruginosa use intercellular signalling to build biofilm communities? Cell Microbiol. 2006;8:1841–1849. doi: 10.1111/j.1462-5822.2006.00817.x. [DOI] [PubMed] [Google Scholar]
- 18.Rejman J, Di Gioia S, Bragonzi A, Conese M. Pseudomonas aeruginosa infection destroys the barrier function of lung epithelium and enhances polyplex-mediated transfection. Hum Gene Ther. 2007;18:642–652. doi: 10.1089/hum.2006.192. [DOI] [PubMed] [Google Scholar]
- 19.Hsueh PR, Chen WH, Luh KT. Relationships between antimicrobial use and antimicrobial resistance in Gram-negative bacteria causing nosocomial infections from 1991-2003 at a university hospital in Taiwan. Int J Antimicrob Agents. 2005;26:463–472. doi: 10.1016/j.ijantimicag.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landman D, Bratu S, Kochar S, Panwar M, Trehan M, Doymaz M, Quale J. Evolution of antimicrobial resistance among Pseudomonas aeruginosa, Acinetobacter baumannii and Klebsiella pneumoniae in Brooklyn, NY. J Antimicrob Chemother. 2007;60:78–82. doi: 10.1093/jac/dkm129. [DOI] [PubMed] [Google Scholar]
- 21.Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 22.Boroumand MA, Esfahanifard P, Saadat S, Sheihkvatan M, Hekmatyazdi S, Saremi M, Nazemi L. A report of Pseudomonas aeruginosa antibiotic resistance from a multicenter study in Iran. Ind J Med Microbiol. 2007;25:435–436. doi: 10.4103/0255-0857.37368. [DOI] [PubMed] [Google Scholar]
- 23.Southey-Pillig CJ, Davies DG, Sauer K. Characterization of temporal protein production in Pseudomonas aeruginosa biofilms. J Bacteriol. 2005;187:8114–8126. doi: 10.1128/JB.187.23.8114-8126.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Li LF, Guan XD, Chen DM, Chen MY, Ouyang B, Huang SW, Wu JF. The drug resistance of pathogenic bacteria of nosocomial infections in surgical intensive care unit. Zhonghua Wai Ke Za Zhi. 2006;44:1189–1192. [PubMed] [Google Scholar]
- 25.Hentzer M, Wu H, Andersen JB, Riedel K, Rasmussen TB, Bagge N, Kumar N, Schembri MA, Song Z, Kristoffersen P, Manefield M, Costerton JW, Molin S, Eberl L, Steinberg P, Kjelleberg S, Hoiby N, Givskov M. Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J. 2003;22:3803–3815. doi: 10.1093/emboj/cdg366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schuster M, Lostroh CP, Ogi T, Greenberg EP. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J Bacteriol. 2003;185:2066–2079. doi: 10.1128/JB.185.7.2066-2079.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waite RD, Paccanaro A, Papakonstantinopoulou A, Hurst JM, Saqi M, Littler E, Curtis MA. Clustering of Pseudomonas aeruginosa transcriptomes from planktonic cultures, developing and mature biofilms reveals distinct expression profiles. BMC Genomics. 2006:7. doi: 10.1186/1471-2164-7-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuchma SL, Connolly JP, O'Toole GA. A three-component regulatory system regulates biofilm maturation and type III secretion in Pseudomonas aeruginosa. J Bacteriol. 2005;187:1441–1454. doi: 10.1128/JB.187.4.1441-1454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J, Hegyi H, Acton TB, Montelione GT, Rost B. Automatic target selection for structural genomics on eukaryotes. Proteins: Struct Funct Bioinformatics. 2004;56:188–200. doi: 10.1002/prot.20012. [DOI] [PubMed] [Google Scholar]
- 30.Wunderlich Z, Acton TB, Liu J, Kornhaber G, Everett J, Carter P, Lan N, Echols N, Gerstein M, Rost B, Montelione GT. The protein target list of the Northeast Structural Genomics Consortium. Proteins. 2004;56:181–187. doi: 10.1002/prot.20091. [DOI] [PubMed] [Google Scholar]
- 31.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peterson JD, Umayam LA, Dickinson T, Hickey EK, White O. The comprehensive microbial resource. Nucleic Acids Res. 2001;29:123–125. doi: 10.1093/nar/29.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirokawa T, Boon-Chieng S, Mitaku S. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics. 1998;14:378–379. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- 34.Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consortia. Proteins. 2007;66:778–795. doi: 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
- 35.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 36.Lovell SC, Davis IW, Arendall WB, III, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 37.Huang YJ, Tejero R, Powers R, Montelione GT. AutoStructure: A topology-constrained distance network algorithm for protein structure determination from NOESY data. Proteins: Struct Funct Bioinformatics. 2006;62:587–603. doi: 10.1002/prot.20820. [DOI] [PubMed] [Google Scholar]
- 38.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heger A, Korpelainen E, Hupponen T, Mattila K, Ollikainen V, Holm L. PairsDB atlas of protein sequence space. Nucleic Acids Res. 2008;36:D276–D280. doi: 10.1093/nar/gkm879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petrey D, Honig B. GRASP 2: visualization, surface properties, and electrostatics of macromolecular structures and sequences. Methods Enzymol. 2003;374:492–509. doi: 10.1016/S0076-6879(03)74021-X. [DOI] [PubMed] [Google Scholar]
- 41.Brown TA, Yang TM, Zaitsevskaia T, Xia Y, Dunn CA, Sigle RO, Knudsen B, Carter WG. Adhesion or plasmin regulates tyrosine phosphorylation of a novel membrane glycoprotein p80/gp140/CUB domain-containing protein 1 in epithelia. J Biol Chem. 2004;279:14772–14783. doi: 10.1074/jbc.M309678200. [DOI] [PubMed] [Google Scholar]
- 42.Botta M, Manetti F, Corelli F. Fibroblast growth factors and their inhibitors. Curr Pharm Des. 2000;6:1897–1924. doi: 10.2174/1381612003398528. [DOI] [PubMed] [Google Scholar]
- 43.Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem Int Ed Engl. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 44.Tarbouriech N, Charnock SJ, Davies GJ. Three-dimensional structures of the Mn and Mg dTDP complexes of the family GT-2 glycosyltransferase SpsA: a comparison with related NDP-sugar glycosyltransferases. J Mol Biol. 2001;314:655–661. doi: 10.1006/jmbi.2001.5159. [DOI] [PubMed] [Google Scholar]
- 45.Lyczak JB, Cannon CL, Pier GB. Lung infections associated with cystic fibrosis. Clin Microbiol Rev. 2002;15:194–222. doi: 10.1128/CMR.15.2.194-222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Overhage J, Schemionek M, Webb JS, Rehm BHA. Expression of the psl Operon in Pseudomonas aeruginosa PAO1 Biofilms: PslA performs an essential function in biofilm formation. Appl Environ Microbiol. 2005;71:4407–4413. doi: 10.1128/AEM.71.8.4407-4413.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vuong C, Gerke C, Somerville GA, Fischer ER, Otto M. Quorum-sensing control of biofilm factors in Staphylococcus epidermidis. J Infect Dis. 2003;188:706–718. doi: 10.1086/377239. [DOI] [PubMed] [Google Scholar]
- 48.Yarwood JM, Bartels DJ, Volper EM, Greenberg EP. Quorum sensing in Staphylococcus aureus biofilms. J Bacteriol. 2004;186:1838–1850. doi: 10.1128/JB.186.6.1838-1850.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kong KF, Vuong C, Otto M. Staphylococcus quorum sensing in biofilm formation and infection. Int J Med Microbiol. 2006;296:133–139. doi: 10.1016/j.ijmm.2006.01.042. [DOI] [PubMed] [Google Scholar]
- 50.De Kievit TR, Iglewski BH. Quorum sensing, gene expression, and Pseudomonas biofilms. Methods Enzymol. 1999;310:117–128. doi: 10.1016/s0076-6879(99)10010-7. [DOI] [PubMed] [Google Scholar]
- 51.Schuster M, Lostroh P, Ogi T, Greenberg EP. Identification, Timing, and Signal Specificity of Pseudomonas aeruginosa Quorum-Controlled Genes: a Transcriptome Analysis. J Bacteriol. 2003:185. doi: 10.1128/JB.185.7.2066-2079.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gotz F. Staphylococcus and biofilms. Mol Microbiol. 2002;43:1367–1378. doi: 10.1046/j.1365-2958.2002.02827.x. [DOI] [PubMed] [Google Scholar]
- 53.Stanley NR, Lazazzera BA. Environmental signals and regulatory pathways that influence biofilm formation. Mol Microbiol. 2004;52:917–924. doi: 10.1111/j.1365-2958.2004.04036.x. [DOI] [PubMed] [Google Scholar]
- 54.Cramton SE, Ulrich M, Gotz F, Doring G. Anaerobic conditions induce expression of polysaccharide intercellular adhesin in Staphylococcus aureus and Staphylococcus epidermidis. Infect Immun. 2001;69:4079–4085. doi: 10.1128/IAI.69.6.4079-4085.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deighton M, Borland R. Regulation of slime production in Staphylococcus epidermidis by iron limitation. Infect Immun. 1993;61:4473–4479. doi: 10.1128/iai.61.10.4473-4479.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Evans E, Brown MRW, Gilbert P. Iron chelator, exopolysaccharide and protease production in Staphylococcus epidermidis: a comparative study of the effects of specific growth rate in biofilm and planktonic culture. Microbiology. 1994;140:153–157. doi: 10.1099/13500872-140-1-153. [DOI] [PubMed] [Google Scholar]
- 57.Elci S, Atmaca S, Guel K. Effect of iron limitation on the amount of slime produced by strains of Staphylococcus epidermidis. Cytobios. 1995;84:141–146. [PubMed] [Google Scholar]
- 58.Rachid S, Ohlsen K, Wallner U, Hacker J, Hecker M, Ziebuhr W. Alternative transcription factor sB is involved in regulation of biofilm expression in a Staphylococcus aureus mucosal isolate. J Bacteriol. 2000;182:6824–6826. doi: 10.1128/jb.182.23.6824-6826.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rachid S, Ohlsen K, Witte W, Hacker J, Ziebuhr W. Effect of subinhibitory antibiotic concentrations on polysaccharide intercellular adhesin expression in biofilm-forming Staphylococcus epidermidis. Antimicrob Agents Chemother. 2000;44:3357–3363. doi: 10.1128/aac.44.12.3357-3363.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Knobloch JKM, Bartscht K, Sabottke A, Rohde H, Feucht HH, Mack D. Biofilm formation by Staphylococcus epidermidis depends on functional RsbU, an activator of the sigB operon: differential activation mechanisms due to ethanol and salt stress. J Bacteriol. 2001;183:2624–2633. doi: 10.1128/JB.183.8.2624-2633.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Campbell IM, Crozier DN, Pawagi AB, Buivids IA. In vitro response of Staphylococcus aureus from cystic fibrosis patients to combinations of linoleic and oleic acids added to nutrient medium. J Clin Microbiol. 1983;18:408–415. doi: 10.1128/jcm.18.2.408-415.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mack D, Siemssen N, Laufs R. Parallel induction by glucose of adherence and a polysaccharide antigen specific for plastic-adherent Staphylococcus epidermidis: evidence for functional relation to intercellular adhesion. Infect Immun. 1992;60:2048–2057. doi: 10.1128/iai.60.5.2048-2057.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gerke C, Kraft A, Sussmuth R, Schweitzer O, Gotz F. Characterization of the N-acetylglucosaminyltransferase activity involved in the biosynthesis of the Staphylococcus epidermidis polysaccharide intercellular adhesin. J Biol Chem. 1998;273:18586–18593. doi: 10.1074/jbc.273.29.18586. [DOI] [PubMed] [Google Scholar]
- 64.Cramton SE, Gerke C, Schnell NF, Nichols WW, Gotz F. The intercellular adhesion (ica) locus is present in Staphylococcus aureus and is required for biofilm formation. Infect Immun. 1999;67:5427–5433. doi: 10.1128/iai.67.10.5427-5433.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mack D, Fischer W, Krokotsch A, Leopold K, Hartmann R, Egge H, Laufs R. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear beta-1,6-linked glucosaminoglycan: purification and structural analysis. J Bacteriol. 1996;178:175–183. doi: 10.1128/jb.178.1.175-183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maira-Litran T, Kropec A, Abeygunawardana C, Joyce J, Mark G, III, Goldmann DA, Pier GB. Immunochemical properties of the Staphylococcal poly-N-acetylglucosamine surface polysaccharide. Infect Immun. 2002;70:4433–4440. doi: 10.1128/IAI.70.8.4433-4440.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dobinsky S, Kiel K, Rohde H, Bartscht K, Knobloch JKM, Horstkotte MA, Mack D. Glucose-related dissociation between icaADBC transcription and biofilm expression by Staphylococcus epidermidis: evidence for an additional factor required for polysaccharide intercellular adhesin synthesis. J Bacteriol. 2003;185:2879–2886. doi: 10.1128/JB.185.9.2879-2886.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shanks RMQ, Donegan NP, Graber ML, Buckingham SE, Zegans ME, Cheung AL, O'Toole GA. Heparin stimulates Staphylococcus aureus Biofilm Formation. Infect Immun. 2005;73:4596–4606. doi: 10.1128/IAI.73.8.4596-4606.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Plotkowski MC, Costa AO, Morandi V, Barbosa HS, Nader HB, De Benizmann S, Puchelle E. Role of heparan sulphate proteoglycans as potential receptors for non-piliated Pseudomonas aeruginosa adherence to non-polarised airway epithelial cells. J Med Microbiol. 2001;50:183–190. doi: 10.1099/0022-1317-50-2-183. [DOI] [PubMed] [Google Scholar]
- 70.Zhu-Salzman K, Shade RE, Koiwa H, Salzman RA, Narasimhan M, Bressan RA, Hasegawa PM, Murdock LL. Carbohydrate binding and resistance to proteolysis control insecticidal activity of Griffonia simplicifolia lectin II. Proc Natl Acad Sci USA. 1998;95:15123–15128. doi: 10.1073/pnas.95.25.15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Simpson HD, Barras F. Functional analysis of the carbohydrate-binding domains of Erwinia chrysanthemi Cel5 (endoglucanase Z) and an Escherichia coli putative chitinase. J Bacteriol. 1999;181:4611–4616. doi: 10.1128/jb.181.15.4611-4616.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chavez CA, Bohnsack RN, Kudo M, Gotschall RR, Canfield WM, Dahms NM. Domain 5 of the cation-independent mannose 6-phosphate receptor preferentially binds phosphodiesters (Mannose 6-Phosphate N-Acetylglucosamine Ester) Biochemistry. 2007;46:12604–12617. doi: 10.1021/bi7011806. [DOI] [PubMed] [Google Scholar]
- 73.Kumar J, Ethayathulla AS, Srivastava DB, Singh N, Sharma S, Kaur P, Srinivasan A, Singh TP. Carbohydrate-binding properties of goat secretory glycoprotein (SPG-40) and its functional implications: structures of the native glycoprotein and its four complexes with chitin-like oligosaccharides. Acta Crystallogr D Biol Crystallogr. 2007;D63:437–446. doi: 10.1107/S0907444907001631. [DOI] [PubMed] [Google Scholar]
- 74.Bae B, Ohene-Adjei S, Kocherginskaya S, Mackie RI, Spies MA, Cann IKO, Nair SK. Molecular basis for the selectivity and specificity of ligand recognition by the family 16 carbohydrate-binding modules from Thermoanaerobacterium polysaccharolyticum ManA. J Biol Chem. 2008;283:12415–12425. doi: 10.1074/jbc.M706513200. [DOI] [PubMed] [Google Scholar]
- 75.Nagae M, Nishi N, Nakamura-Tsuruta S, Hirabayashi J, Wakatsuki S, Kato R. Structural analysis of the human galectin-9 N-terminal carbohydrate recognition domain reveals unexpected properties that differ from the mouse orthologue. J Mol Biol. 2008;375:119–135. doi: 10.1016/j.jmb.2007.09.060. [DOI] [PubMed] [Google Scholar]
- 76.Hachem MA, Karlsson EN, Bartonek-Roxa E, Raghothama S, Simpson PJ, Gilbert HJ, Williamson MP, Holst O. Carbohydrate-binding modules from a thermostable Rhodothermus marinus xylanase: cloning, expression and binding studies. Biochem J. 2000;345:53–60. [PMC free article] [PubMed] [Google Scholar]
- 77.Wandall HH, Irazoqui F, Tarp MA, Bennett EP, Mandel U, Takeuchi H, Kato K, Irimura T, Suryanarayanan G, Hollingsworth MA, Clausen H. The lectin domains of polypeptide GalNAc-transferases exhibit carbohydrate-binding specificity for GalNAc: lectin binding to GalNAc-glycopeptide substrates is required for high density GalNAc-O-glycosylation. Glycobiology. 2007;17:374–387. doi: 10.1093/glycob/cwl082. [DOI] [PubMed] [Google Scholar]
- 78.Matei E, Furey W, Gronenborn AM. Solution and crystal structures of a sugar binding site mutant of cyanovirin-N: no evidence of domain swapping. Structure. 2008;16:1183–1194. doi: 10.1016/j.str.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Koebnik R. TonB-dependent trans-envelope signaling: the exception or the rule? Trends Microbiol. 2005;13:343–347. doi: 10.1016/j.tim.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 80.Abbas A, Adams C, Scully N, Glennon J, O'Gara F. A role for TonB1 in biofilm formation and quorum sensing in Pseudomonas aeruginosa. FEMS Microbiol Lett. 2007;274:269–278. doi: 10.1111/j.1574-6968.2007.00845.x. [DOI] [PubMed] [Google Scholar]
- 81.Blanvillain S, Meyer D, Boulanger A, Lautier M, Guynet C, Denance N, Vasse J, Lauber E, Arlat M. Plant carbohydrate scavenging through TonB-dependent receptors: a feature shared by phytopathogenic and aquatic bacteria. PLoS One. 2007:2. doi: 10.1371/journal.pone.0000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Neugebauer H, Herrmann C, Kammer W, Schwarz G, Nordheim A, Braun V. ExbBD-dependent transport of maltodextrins through the novel MalA protein across the outer membrane of Caulobacter crescentus. J Bacteriol. 2005;187:8300–8311. doi: 10.1128/JB.187.24.8300-8311.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schauer K, Rodionov DA, de Reuse H. New substrates for TonB-dependent transport: do we only see the tip of the iceberg'? Trends Biochem Sci. 2008;33:330–338. doi: 10.1016/j.tibs.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 84.Kall L, Krogh A, Sonnhammer Erik LL. Advantages of combined transmembrane topology and signal peptide prediction—the Phobius web server. Nucleic Acids Res. 2007;35:W429–W432. doi: 10.1093/nar/gkm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Braun M, Endriss F, Killmann H, Braun V. In vivo reconstitution of the FhuA transport protein of Escherichia coli K-12. J Bacteriol. 2003;185:5508–5518. doi: 10.1128/JB.185.18.5508-5518.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wandersman C, Delepelaire P. Bacterial iron sources: from siderophores to hemophores. Ann Rev Microbiol. 2004;58:611–647. doi: 10.1146/annurev.micro.58.030603.123811. [DOI] [PubMed] [Google Scholar]
- 87.Usher KC, Ozkan E, Gardner KH, Deisenhofer J. The plug domain of FepA, a TonB-dependent transport protein from Escherichia coli, binds its siderophore in the absence of the transmembrane barrel domain. Proc Natl Acad Sci USA. 2001;98:10676–10681. doi: 10.1073/pnas.181353398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Neri D, Szyperski T, Otting G, Senn H, Wuthrich K. Stereospecific nuclear magnetic resonance assignments of the methyl groups of valine and leucine in the DNA-binding domain of the 434 repressor by biosynthetically directed fractional 13C labeling. Biochemistry. 1989;28:7510–7516. doi: 10.1021/bi00445a003. [DOI] [PubMed] [Google Scholar]
- 89.Cavanaugh J. Protein NMR spectroscopy: principles and practice. San Diego: Academic Press; 1996. [Google Scholar]
- 90.Ferentz AE, Wagner G. NMR spectroscopy: a multifaceted approach to macromolecular structure. Q Rev Biophys. 2000;33:29–65. doi: 10.1017/s0033583500003589. [DOI] [PubMed] [Google Scholar]
- 91.Linge JP, Nilges M. Influence of non-bonded parameters on the quality of NMR structures: a new force field for NMR structure calculation. J Biomol NMR. 1999;13:51–59. doi: 10.1023/a:1008365802830. [DOI] [PubMed] [Google Scholar]
- 92.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 93.Vuister GW, Bax A. Measurement of two-bond JCOH alpha coupling constants in proteins uniformly enriched with 13C. J Biomol NMR. 1992;2:401–405. doi: 10.1007/BF01874818. [DOI] [PubMed] [Google Scholar]
- 94.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 95.Schwieters CD, Clore GM. The VMD-XPLOR visualization package for NMR structure refinement. J Magn Reson. 2001;149:239–244. doi: 10.1006/jmre.2001.2300. [DOI] [PubMed] [Google Scholar]
- 96.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]