

Abstract
The molecular mechanisms underlying antiproliferative actions of the steroid 1α,25-dihydroxy vitamin D3 (1,25D) in human osteosarcoma cells are known only partially. To better understand the signaling involved in 1,25D anti-tumorigenic properties in bone, we stably silenced vitamin D receptor (VDR) expression in the human osteosarcoma SaOS-2 cell line. We found that 1,25D treatment reduced cell proliferation by approximately 25% after 3 days only in SaOS-2 cells expressing native levels of VDR protein, and involved activation of MAPK/AP-1/p21waf1 pathways. Both sustained (3 days) and transient (15 min) 1,25D treatment activated JNK and ERK1/2 MAPK signaling in a nongenomic VDR-dependent manner. However, only sustained exposure to hormone led to upregulation of p21 and subsequent genomic control of the cell cycle. Specific blockade of MEK1/MEK2 cascade upstream from ERK1/2 abrogated 1,25D activation of AP-1 and p21, and subsequent antiproliferative effects, even in the presence of a nuclear VDR. We conclude that 1,25D-induced inhibition of human osteosarcoma cell proliferation occurs via sustained activation of JNK and MEK1/MEK2 pathways downstream of nongenomic VDR signaling that leads to upregulation of a c-Jun/c-Fos (AP-1) complex, which in turn modulates p21waf1 gene expression. Our results demonstrate a cross-talk between 1,25D/VDR nongenomic and genomic signaling at the level of MAP kinase activation that leads to reduction of cell proliferation in human osteosarcoma cells.
Keywords: 1α25(OH)2 vitamin D3, Antiproliferative effects, AP-1, Osteosarcoma, p21waf1, Vitamin D Receptor
1. Introduction
Among natural compounds with strong antiproliferative effects on different cancer cell systems including osteosarcoma is the steroid hormone 1α,25-dihydroxy vitamin D3 (1,25D) (1). A significant amount of research has been carried out on anti-cancer properties of 1,25D and synthetic analogs (2). However, the precise molecular mechanisms underlying tissue-specific anti-tumorigenic effects of the hormone remain partially understood. Precise identification of tissue specific 1,25D-regulated cascades leading to control of the cell cycle is the basis for proper management of anti-cancer benefits of the hormone.
Previous studies in osteosarcoma cells have shown that 1,25D inhibits cell proliferation in a manner that appears to be time- and dose-dependent (3-5). Although remarkable progress has been made in understanding the biology of osteosarcomas, their molecular etiology remains obscure, and specific targets for the action of anti-tumorigenic agents have not yet been discovered. With the aim to identify molecular players that can potentially be used as targets in the treatment of bone cancer, we studied here a mitogen activated protein kinase (MAPK) cascade involved in 1,25D antiproliferative effects in human osteosarcoma cells. In particular, we investigated the requirement of a vitamin D receptor (VDR) in 1,25D-induction of MAPK activities and further upregulation of genes involved in the control of cell cycle progression, and we discerned between biological effects promoted by transient versus sustained hormone stimulus.
Osteosarcomas, the most common type of bone cancer in children and adolescents, are in part characterized by insensitivity to typical growth inhibitory signals, and evasion of apoptosis. Loss of control of cell proliferation and survival appears to be a key event in the etiology of the disease (6). 1,25D, however, has been previously proposed to be an effective growth inhibitory stimulus in osteoblasts (3;7;8). In general terms, 1,25D antiproliferative mechanisms can be explained by its binding to a nuclear vitamin D receptor (VDR) that functions as a transcription factor and modulates the expression of genes involved in cell cycle progression (9). In addition, 1,25D appears to activate a cytoplasmic or membrane-associated VDR that triggers rapid signaling pathways that involve kinase activation and regulate cellular functions that prime the cell for latter 1,25D/VDR modulated response at the genomic level (10;11). In osteoblasts, cytoplasmic 1,25D signaling initiates nongenomic pathways via a classic VDR that include modulation of the electrical state of the plasma membrane (12-14), elevation of intracellular calcium (15), and activation of protein kinases including MAPKs (11). A cross-talk between 1,25D nongenomic and genomic actions leading to control of the cell cycle has been suggested at the point of MAPK activation in some cell types (16;17). However, there is no clear description of the molecular pathways and key players involved in this VDR-dependent cross-talk in human osteosarcoma up to date.
It has been demonstrated in different cell systems that 1,25D inhibition of cell proliferation at the gene level involves upregulation of the cyclin-dependent kinase (cdk) inhibitor p21(waf1/cip1) (3;18;19). At least three nuclear VDR responsive regions have been identified in the p21 promoter of breast cancer cells (19), providing a site for 1,25D genomic control of the cell cycle. Increased p21expression causes cell cycle arrest and suppression of cell growth in response to hormone stimulation. In addition, it is well known that the p21 gene promoter region has a binding site for activator protein 1 (AP-1), which links cytoplasmic MAP kinase activation to p21 upregulation (20). AP-1, one of the first identified transcription factors (21), regulates a wide range of cellular processes, including cell proliferation, death, survival, and differentiation (22). AP-1 is composed of a variety of combinations of dimerized proteins that belong to the Jun, Fos, Maf and ATF sub-families. C-Jun, the most potent transcription factor in the group, forms stable heterodimers with c-Fos proteins, another ubiquitous transcription factor. The regulation of AP-1, which occurs via members of the MAPK family that activate both c-Jun and c-Fos, is complex. For example, the mitogen activated Jun N-terminal protein kinase (JNK) activates c-Jun protein, a signal transducing transcription factor, by means of N-terminal phosphorylation at serines 63 and 73 (23). It has been shown previously that 1,25D induces c-Jun phosphorylation, and this has been linked to decreased cell proliferation (24;25). In addition, c-Fos upregulation occurs as a result of cytoplasmic activation of the extracellular signal-regulated kinase (ERK) MAP kinase, which has also been shown to be a target of 1,25D/VDR rapid signaling in osteoblasts (11). Here, we investigated the hypothesis that nongenomic 1,25D/VDR signaling induces AP-1 activity in osteosarcoma cells upstream of p21 genomic upregulation. We verified that long-term 1,25D cytoplasmic induction of MAP kinases, and consequent accumulation of activated JNK, AP-1 and p21 lead to cell cycle arrest and tumor growth inhibition in the presence of a classic VDR. More specifically, we found that this cascade initiates as a rapid, nongenomic VDR-dependent signaling, but only sustained accumulation of the stimulus cross-talks with VDR genomic pathways to reduce cell proliferation in human osteosarcoma.
Our study contributes to the current understanding on the signaling that controls cell proliferation in human osteosarcoma and its modulation by the anti-tumorigenic hormone 1,25D. In addition, it sets the foundation for the molecular identification of cell targets involved and future design of therapeutic agents for the treatment of bone cancer diseases.
2. Materials and methods
2.1. Chemicals and constructs
1α,25(OH)2 vitamin D3 (1,25D) was purchased from Biomol International L.P. (Plymouth, PA). A stock 1,25D solution was dissolved in pure ethanol and kept at -20° C in the dark. In all experiments, the final ethanol concentration was kept below 0.01% (v/v). The GenEclipse VDR siRNA and control (empty) vector constructs were purchased from Chemicon International Co. (Temecula, CA). The pRSV-β-Galactosidase plasmid and p21 promoter luciferase reporter construct were provided by Dr. A. Walker (University of California-Riverside). The AP-1 (7×) luciferase enhancer reporter construct was obtained from Stratagene (La Jolla, CA). ERK pathway blocker U0126 was purchased from CalBiochem (San Diego, CA). Unless specified, all other chemicals were purchased from Sigma (Saint Louis, MO).
2.2. Cell culture
Human osteosarcoma SaOS-2 cells were purchased from the American Type Culture Collection (Manassas, VA). Cells were maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 1 mM GlutaMAX(TM) (Invitrogen, Carlsbad, CA) and 25 mM Hepes buffer, with the addition of 10% (v/v) fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Rat osteosarcoma ROS 17/2.8 cells (kindly provided by A.W. Norman, University of California-Riverside) were cultured in Ham F-12 nutrient mixture supplemented with 1 mM GlutaMAX, 5% (v/v) fetal bovine serum and 5% (v/v) Serum Plus (JHR Biosciences, Woodland, CA), antibiotics, and 1.1 mM CaCl2. Cultures were kept at 37°C in a humidified incubator with 5% CO2 in air. Typically, cells were seeded in 6-well plates at low density (1,000 cells/ml), and used at 70-85% confluence.
2.3. Establishment of a stable siRNA VDR cell line
Native SaOS-2 cells were grown in 6-well dishes until they reached 70-85% confluence. Cells were then transfected with 0.5-1 μg GenEclipse VDR siRNA construct or control vector construct DNA in 100 μl DMEM serum free medium containing 6 μl Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according with the manufacturer's protocol. After 5 hours, the transfection medium was replaced by serum free medium, and cultures were maintained for additional 24 h. Cells were then trypsinized, diluted 1:10, and re-seeded in medium supplemented with 200 μg/ml geneticin (Gibco, Auckland, NZ) to select for stably transfected cells. Stable transfectants developed after two weeks. Colonies were collected separately, and maintained in geneticin-containing medium until no significant cell death was observed. Surviving colonies were assayed for VDR protein levels and transcriptional activity, expanded and frozen for future use.
2.4. Immunofluorescence
Stable vector transfected (V-T) and siRNA VDR transfected (siRNA-T) SaOS-2 cells were grown on glass cover slips for 3 days. Cells were fixed with 3.7% (v/v) formaldehyde (Sigma) in PBS (pH 7.4) for 20 min at room temperature, and permeabilized with ice cold ethanol for 5 min. Next, cells were blocked with 5% (v/v) goat serum in PBS for 1 h, and incubated with a primary monoclonal anti-VDR antibody (C-20, Santa Cruz Biotechnology, CA, 1:500 dilution in 5% goat serum in PBS) overnight at 4°C. An Alexa 488 conjugate goat anti-rabbit secondary antibody (Invitrogen) was used at a 1:500 dilution in PBS, for 2 h at room temperature. Stained cells were visualized with an Olympus IX50 epifluorescence microscope. Images were obtained with a Spot Pursuit digital camera (Diagnostic Instruments, Sterling Heights, MI), and processed with Simple PCI C-Imaging Systems software (Compix Inc., Cranberry Township, PA)
2.5. Alkaline phosphatase (ALP) assay
SaOS-2 cells were grown for 3 days in the presence of 10 nM 1,25D. The ALP fluorimetric detection kit (Sigma) was used on whole cell lysates according to the manufacturer's protocol. Intensity of fluorescence was measured at 360 nm excitation and 460 nm emission with a Synergy HT Multi-Detection Microplate Reader (Bioteck, Winooski, VT).
2.6. Reporter gene assays
Native non-transfected (Non-T), control vector transfected (V-T), and siRNA VDR SaOS-2 cells were grown to 70-85% confluency, and co-transfected with either a AP-1 (x7) enhancer-luciferase (1μg/5ml) or a p21 promoter-luciferase (1μg/5ml) construct. Transfection was performed with 6 μl Lipofectamine 2000 as described in previous paragraphs. Each transfection was performed in triplicate. Whenever necessary, empty control plasmid was added to ensure that each transfection received the same amount of total DNA. To normalize for transfection efficiency, 0.2 μg of pRSV-β-galactosidase reporter plasmid was added to each transfection. Approximately 16 h after transfection, luciferase reporter assays were performed using a luciferase assay kit (Stratagene) following the manufacturer's protocol. β-galactosidase activity was measured using the Galacto-Light chemiluminescence kit (Tropix Inc., Bedford, MA). Luciferase activity was normalized relative to β-galactosidase activity. Luminescence and fluorescence measurements were performed with a Synergy HT multiplate reader as indicated previously.
2.7. Western blot analysis
Cells were rinsed with Dulbecco's phosphate-buffered saline (DPBS, Gibco, Grand Island, NY, USA) and lysed with a buffer containing 20 mM Tris-HCl, 140 mM NaCl, 0.05 mM EDTA, 10 μg/mL leupeptin, 10 μg/mL aprotinin, 25 μg/mL pepstatin, 1 mM PMSF, 1 mM Na3VO4, 10 nM NaF, 1 mM EGTA, and 1% NP-40 (pH 7.4). After centrifugation at 12,000×g for 10 min, the supernatant was collected as whole cell lysate. Twenty micrograms of protein were loaded on a 12% (w/v) reducing SDS-PAGE gel. After electrophoresis, protein was transferred to a nitrocellulose membrane in transfer buffer containing 25 mM Tris, 192 mM glycine, 0.1% SDS, and 20% methanol (pH 8.3). Membranes were blocked with 5% nonfat milk in wash buffer (DPBS containing 0.1% Tween 20), and incubated with a monoclonal antibody against VDR (C-20, Santa Cruz Biotechnology, CA), or polyclonal antibodies against active (p)-JNK (dually phosphorylated Thr183-Tyr185, Promega, Madison, WI), p-c-Jun (phosphorylated Ser63, Cell Signaling Technology, Danvers, MA), JNK, and c-Fos (Santa Cruz) (diluted 1:1,000) overnight at 4 °C. Incubation with a secondary antibody (1:10,000) conjugated to horseradish peroxidase (Sigma) was done for 45 min at room temperature. Membranes were then exposed to SuperSignal West Pico Chemiluminescent agent (PIERCE, Rockford, IL) followed by autoradiography and image analysis. Protein expression levels were estimated from Western blot density measurements, and analyzed with the Un-Scan-It gel software (Silk Scientific Inc., Oren, UT). β-Actin and vinculin were used to normalize for gel loading and transfer.
2.8. Cell proliferation assay
SaOS-2 cells were seeded at 1,000 per well in a 96-well plate, and treated with 10 nM 1,25D for 15 min (for transient studies) or 1-5 days (for sustained studies) in DMEM medium. We utilized the CellTiter 96 Aqueous non-radioactive cell proliferation assay kit (Promega) according to the manufacturer's protocol. Cell proliferation was estimated with the use of [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2- (4-sulfophenyl)-2H-tetrazolium salt (MTS), which is reduced by live cells into a formazan product. The absorbance (optical density) of formazan was measured at 490 nm and plotted over time as an indirect measure of cell density.
2.9. Statistical analysis
Data were expressed as average values ± S.E., and compared by means of ANOVA t-Student test. All experiments were repeated a minimum of three times.
3. Results
3.1. 1,25D activates MAPK cascades in osteosarcoma cells
Precise molecular mechanisms of antiproliferative actions of 1,25D in osteosarcoma cells are only partially understood (7;8;26). Here we investigated rapid effects of the steroid hormone on specific molecular players in the Jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK) cascades, both members of the MAPK family broadly implicated in the control of cell growth. JNK has been previously shown to be activated by 1,25D in colon carcinoma and leukemia cells (24;25). We report here for the first time that 10 nM 1,25D added to the culture medium significantly elevated levels of dually phosphorylated (activated, p-) JNK in human SaOS-2 and rat ROS 17/2.8 osteosarcoma cell lines (Figure 1). Although in vivo physiological levels of 1,25D are around 1 nM, we reported previously that a concentration of 10 nM appears to be optimal for the stimulation of membrane-initiated actions by the hormone in in vitro systems (12-14).
Fig. 1.
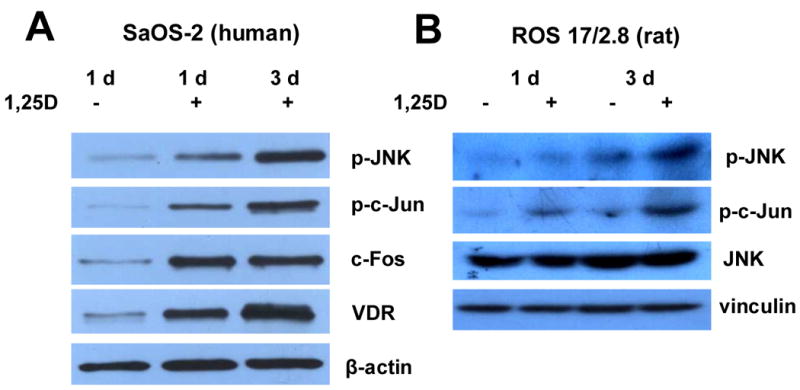
1,25D induces activation of MAPK cascades in human SaOS-2 and rat ROS 17/2.8 cells. Osteosarcoma cells were grown in the absence (-) or presence (+) of 10 nM 1,25D for 1 and 3 days. Western blots from whole cell lysates show increasing levels of phosphorylated (p-) JNK and c-Jun (A and B), and higher expression of c-Fos protein (A) in hormone-treated cell cultures. Beta-actin and vinculin were used for normalization of gel loading and transfer. Increasing levels of VDR protein were also detected over time under sustained treatment with hormone.
Figure 1 shows Western blot results for the detection of p-JNK after 1 and 3 days of uninterrupted hormone treatment. Increasing levels of p-JNK were detected in both cell lines over this time period relative to p-JNK levels obtained in the absence of hormone treatment, and unphosphorylated JNK. In addition, we measured a significant increase of phosphorylated (activated, p-) c-Jun, a major downstream signaling target of p-JNK, upon 1,25D treatment within the same time frame and concomitantly with p-JNK increment. We found an elevation of c-Fos levels, a ubiquitous transcription factor downstream from rapid MEK1/MEK2 activation also involved in the control of cell cycle progression within the first day of 1,25D treatment in SaOS-2 cells (Figure 1A). However, we did not find equivalent increased levels of c-Fos in rat ROS 17/2.8 cells upon 1,25D treatment (not shown). Levels of c-Fos are known to be upregulated via ERK1/2 MAPK activation (27), which in turn has been shown to be a target for 1,25D cytoplasmic actions (11). Levels of c-Fos remained elevated during 3 days of hormone treatment in human SaOS-2 osteoblasts.
SaOS-2 cells express relatively high amounts of the VDR molecule. Here we show that JNK activation by 1,25D synchronizes with VDR upregulation by the hormone in the time frame of 1-3 days (Figure 1A), suggesting that upregulated VDR could potentiate 1,25D effects on JNK activation. In the present study, we investigated the hypothesis that 1,25D induction of JNK phosphorylation requires a functional VDR and leads to increased p-c-Jun, which acts as a transcription factor complex with c-Fos to upregulate the expression of genes responsible for reduction of cell proliferation in the tumoral cell line.
3.2. A classic VDR is required for antiproliferative effects of 1,25D in SaOS-2 cells
Rapid, nongenomic actions of 1,25D are believed to initiate in the cell cytoplasm in close proximity to the plasma membrane (10;14;28;29). Whether or not a classic VDR is required is still a field of controversy. To investigate the requirement of a VDR in 1,25D-induced JNK activation measured in Figure 1, we stably knocked down VDR expression in SaOS-2 cells by using vector-based siRNA technology (GeneEclipse, Invitrogen). We transfected native SaOS-2 cells with a siRNA VDR vector construct, on one hand, and a control empty vector construct, on the other, and selected for stable clones with 200 μg/ml geneticin added to the culture medium in the time course of 3 months. By these means, we created a stable siRNA-T SaOS-2 subclone in which VDR protein expression was significantly reduced, and a control vector-transfected (V-T) SaOS-2 subclone in which VDR levels remained unaltered, as shown in Figure 2A-C. We verified that the siRNA-T SaOS-2 subclone carried a functional VDR deficiency by measuring a significant decrease in 1,25D-induced endogenous alkaline phosphatase (ALP) activity compared to non-transfected (Non-T) and control V-T cells (Figure 2D). We used ALP activity as a native reporter gene known to be upregulated by 1,25D via transcriptional mechanisms that involve the VDR (30). The ability of 1,25D to induce ALP synthesis and activity was significantly reduced in the siRNA-T VDR subclone, as shown in Figure 2D.
Fig. 2.
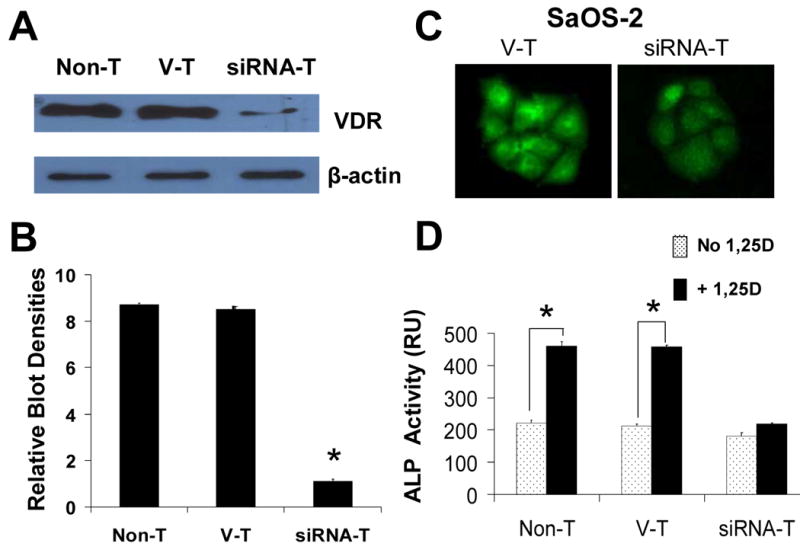
Stable VDR silencing in SaOS-2 cells. (A) VDR protein expression levels in native non-transfected (Non-T), control (vector transfected, V-T), and siRNA VDR transfected (siRNA-T) Saos-2 cells. VDR levels are quantitatively shown in (B) as a measure of blot densities. (C) Immunofluorescence detection of VDR in control V-T (left), and VDR silenced (siRNA-T, right). Cells treated with a goat antibody against human VDR were visualized with a secondary anti-goat Cy3-conjugate antibody. VDR was profusely localized in the cell cytoplasm and nucleus of control SaOS-2 cells. Note significantly decreased fluorescence intensity levels corresponding with lower VDR protein levels in VDR silenced versus control V-T cells. Digital images were obtained with identical exposure settings for comparison. (D) 1,25D induction of alkaline phosphatase (ALP) activity requires a VDR. Endogenous ALP enzyme activity was measured in the absence (open bars) and presence (filled bars) of 10 nM 1,25D added to the culture medium for 3 days in native Non-T, control V-T, and siRNA VDR transfected SaOS-2 cells. *, p<0.001, n=3.
Next, we measured SaOS-2 cell proliferation in native and transfected cells over a period of time of 5 days in culture. When plated at low density (1,000 cells/well), cells reached confluence by day 5. To characterize the effects of transient versus prolonged 1,25D exposure, which trigger nongenomic and genomic steroid actions respectively, cell cultures were treated with 10 nM 1,25D for 15 min (Figure 3A), or uninterruptedly for the entire duration of the experiment (Figure 3B). No change in the growth curve of cultures was detected after brief (15 min) 1,25D treatment compared with untreated cells. We found, however, a significant (approximately 25%) reduction of cell density starting at day 3 in native Non-T, and control V-T SaOS-2 cells in the presence of 10 nM 1,25D added daily to the medium. We verified that siRNA-T SaOS-2 cells were insensitive to continued 1,25D treatment, thus indicating that a VDR is required for anti-proliferative effects of the steroid hormone.
Fig. 3.
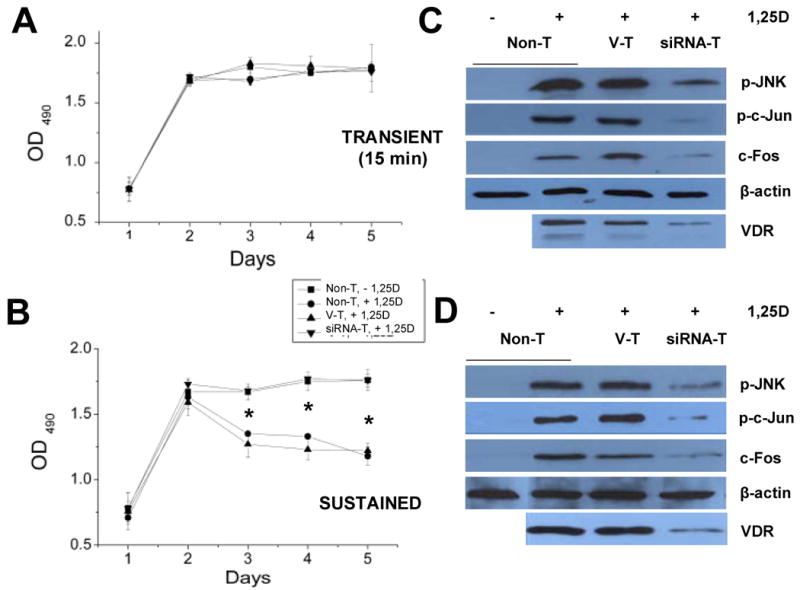
(A,B) Anti-proliferative effects of 1,25D occur in the presence of a VDR. Cell density values, as measured with the CellTiter96 AQueous non-radioactive cell proliferation assay (Promega), were obtained every 24 h over a period of 5 days from SaOS-2 cell cultures treated either with 10 nM 1,25D for 15 min on day 0 (A, transient), or grown in continuous presence of hormone (B, sustained). Squares represent data obtained from control Non-T cells in the absence of 1,25D. Inverse triangles, circles, and upright triangles represent data obtained from siRNA VDR knock-down, control Non-T (3) and control V-T (4) cells in the presence of 10 nM 1,25D; n=5, *, p<0.001. (C,D) Transient and sustained 1,25D treatment induces JNK activation and c-Fos upregulation only in cells expressing a VDR. Western blots obtained for activated (p-) JNK, p-c-Jun, and c-Fos in native Non-T, control V-T, and VDR knock-down (siRNA-T) SaOS-2 cells treated with 10 nM 1,25D for 15 min (transient, C) or 3 days (sustained, D). Activated JNK and c-Jun proteins were detected with commercial antibodies raised against phosphorylated residues. VDR protein expression levels are also shown for the same cell lysates.
3.3. 1,25D activation of c-Jun/c-Fos cascades in SaOS-2 cells occurs under both transient and sustained hormone treatment, and requires a VDR
To investigate if the loss of 1,25D anti-tumorigenic effects in siRNA-T SaOS-2 cells (Figure 3B, inverted triangles) involves VDR-dependent MAPK pathways shown in Figure 1, we measured p-JNK, p-c-Jun, and c-Fos levels in the absence and presence of 10 nM 1,25D in native Non-T, control V-T, and siRNA VDR osteosarcoma cells. To characterize the time frame of these effects, we performed Western blots for these proteins after transient (15 min, Figure 3C) and sustained (3 days, Figure 3D) hormone treatment.
Different cellular responses have been described in a number of cell systems as a consequence of transient versus sustained MAPK activation (31;32). More specifically, transient (minutes to hours) exposure of osteoblasts to 1,25D has been previously associated with anti-apoptotic effects of the hormone (11), while sustained (> 1 day) treatment appears to underlie its anti-tumorigenic actions (3;4). This agrees with our results shown in Figure 3A,B. Here we found significantly lower levels of p-JNK and p-c-Jun, and lack of upregulation of c-Fos in 1,25D-treated siRNA-T SaOS-2 cells compared with levels measured in hormone-treated Non-T and V-T SaOS-2 cells expressing endogenous VDR after both transient (Figure 3C) and sustained (Figure 3D) treatment with hormone. This 1,25D-induced, VDR-dependent JNK activation was measured as rapidly as 15 min of hormone stimulus, indicating that the signaling cascade occurs previous to any transcriptional activity of the VDR. Sustained 1,25D treatment for 3 days activated this MAPK response only in native, non-T SaOS-2 cells (Figure 3D). These results indicate that prolonged stimulus did not compensate for reduced VDR levels in the siRNA-T subclone, thus confirming the requirement of the classic receptor for activation of the MAPK cascade. As also shown in Figure 3C,D, VDR protein levels in siRNA-T cells remained significantly lowered during the entire length of each experiment.
3.4. Sustained but not transient 1,25D treatment upregulates AP-1/p21 downstream MAPK activation in osteosarcoma cells only in the presence of a VDR
In the cell nucleus, AP-1 transcriptional activity results from phosphorylation of c-Jun and its dimerization with c-Fos which, as a complex, regulate the expression of genes involved in cell cycle arrest. It has been described in osteoblasts that the cyclin-dependent kinase inhibitor p21 gene, which controls progression through the cell cycle, carries both a VDR responsive element and an AP-1 binding site in its promoter region (33). To study the hypothesis that antiproliferative actions of 1,25D involve transactivation of p21 as a target gene for both AP-1 and VDR transcription factors in human osteosarcoma, we transiently co-transfected VDR native Non-T, control V-T, and siRNA-T SaOS-2 cells with an AP-1 (x7) enhancer or p21 promoter luciferase reporter construct, and we applied a transient (15 min) or sustained (3 days) 1,25D (10 nM) stimulus. We measured light intensity as a product of luciferase activity associated with our AP-1 enhancer (Figure 4A) and p21 promoter (Figure 4B) activities in each cell group. As seen from our results, AP-1 activation and p21 promoter-induced expression occur only after sustained, but not transient, 1,25D treatment. However, sustained 1,25D treatment did not promote AP-1 or p21 reporter activation in osteosarcoma cells in which VDR protein expression was knocked down. This confirmed the requirement of a VDR in the sustained 1,25D-initiated signaling that leads to reduction of cell proliferation in human osteosarcoma cells. This cascade thus appears to comprise a VDR/JNK/c-Jun/AP-1/p21 pathway likely involved in induction of cell cycle arrest.
Fig. 4.

1,25D-induced upregulation of AP-1 and p21 luciferase reporters occurs only in the presence of a classic VDR. Native (Non-T), control vector transfected (V-T), and siRNA VDR transfected (siRNA-T) SaOS-2cells co-transfected with AP-1 (x7) enhancer (A) or p21-promoter luciferase constructs (B) were cultured in the presence of 10 nM 1,25D for 15 min (open bars) or 3 days (filled bars). Relative light units (RLU) from luciferase activity were measured as a function of AP-1 enhancer and p21 promoter activities. *, p<0.001, n=3. RLU values were normalized for transfection efficiency with β-galactosidase activity.
3.5. Inhibition of MEK1/MEK2 activation abolishes 1,25D-induced antiproliferative effects in osteosarcoma
To verify that 1,25D-induced AP-1/p21 activation occurs in part downstream ERK/c-Fos activation, we measured luciferase activity in native VDR Non-T and control V-T SaOS-2 cells co-transfected with AP-1 (x7) enhancer and p21 promoter luciferase reporter constructs in the presence and absence of the specific MEK1/MEK2 blocker U0126 (Figure 5A, B). U0126 inhibits both active and inactive MEK1/MEK2 kinases, two immediate upstream activators of ERK1/2, thus preventing induction of c-Fos. We found that 1,25D-induced luciferase activity was significantly reduced when cells were treated with 10 nM 1,25D in the presence of 25 μM U0126 for 3 days. This confirmed that ERK1/2 activation is required for induction of AP-1 activity and p21 expression in the presence of a functional VDR.
Fig. 5.

Blockade of MEK1/MEK2 activity abolishes 1,25D-induction of AP-1 and p21, and prevents hormone anti-proliferative effects. Native, non-transfected (Non-T) and control vector transfected (V-T) SaOS-2 cells, co-transfected with AP-1 (x7) enhancer (A) or p21 promoter (B) luciferase constructs, were cultured for 3 days in the presence of 10 nM 1,25D, with (filled bars) or without (open bars) specific MEK1/MEK2 blocker U0126 (25 μM). Relative light units (RLU) were measured as a function of AP-1 enhancer and p21 promoter activities. Results show that inhibition of MEK1/MEK2 signaling prevented AP-1 and p21 upregulation, confirming that p21 gene transactivation occurs downstream 1,25D-induced MAPK activity. *, p<0.001, n=4. C: Measurements for cell densities obtained for native (non-transfected) SaOS-2 cell cultures treated with 10 nM 1,25D in the presence or absence of U0126 (25 μM) for 3 days. *, p<0.01, n=3.
Next, we verified that 1,25D induction of MAPK activation leads to a reduction of cell proliferation. As shown in Figure 5C for cell counts obtained from native VDR Non-T SaOS-2 cells treated with 10 nM 1,25D, the presence of the ERK1/2 pathway inhibitor U0126 abolished 1,25D anti-proliferative effects within a 3 day period. This confirmed that a sustained cytoplasmic VDR-dependent MAPK cascade is a required step in anti-cancer effects of the hormone in bone.
4. Discussion
Antiproliferative effects of the steroid hormone 1,25D have been reported previously on different cell types, including osteoblasts (1;4;7;34;35). However, precise pathways involved remained to be elucidated. 1,25D decreases cell growth by promoting apoptosis (8;36) and/or inducing cell cycle arrest (2;34). In osteoblasts, 1,25D and synthetic analogs have been shown to inhibit cell cycle progression at the G1 phase and to promote cell differentiation (7), and this involves p21waf1/cip1 gene upregulation (3). However, 1,25D signaling to p21 in human osteosarcoma remains unknown. Although anti-tumorigenic effects of 1,25D have raised high expectations with regard to its potential in the treatment of different cancer types, this is compromised by calcemic side effects that develop at therapeutic doses (37). Different groups focusing on this problem have proposed the use of 1,25D synthetic analogs with strong antiproliferative and low hypercalcemic actions. We believe that, in addition to this strategy, a detailed understanding of the signaling molecules involved in anti-mitogenic effects of the hormone is necessary to facilitate the appropriate choice and/or design of new pharmacological agents that would target key players in the signaling cascade while avoiding lethal hypercalcemic effects.
Recent studies in the field of steroid hormone research have focused on mechanisms of action that initiate at the plasma membrane and trigger rapid cytoplasmic signaling involved in cell proliferation and death in addition to traditionally known gene transactivation (38-40). In leukemia cells, for example, hormone 1,25D stimulates a JNK mitogen activated protein kinase implicated in cell cycle regulation via a AP-1 pathway (25). Our present study aims at identifying molecular players involved in 1,25D-induced inhibition of cell proliferation in human osteosarcoma, which could potentially be used as therapy targets. Here, we studied the hypothesis that, in SaOS-2 osteosarcoma cells, 1,25D rapidly activates cytoplasmic MAP kinases via nongenomic mechanisms that involve a cytoplasmic or membrane-related VDR (10) and that, when sustained, ultimately lead to activation of genes involved in cell cycle arrest, with an overall decrease in cell numbers. To study this hypothesis, we created a stably transfected siRNA VDR SaOS-2 subclone, and discriminated between signaling cascades activated by either sustained (3-5 days) or transient (15 min) 1,25D treatment. The model studied here for antiproliferative 1,25D signaling in human osteosarcoma is presented in Figure 6.
Fig. 6.

Proposed model for the integration of anti-proliferative genomic and nongenomic 1,25D pathways via MAPK cross-talk through a classic VDR in human osteosarcoma cells. Shortly after arrival at the cell surface, 1,25D hormone interacts with a VDR protein localized at the cytoplasm in close proximity to the plasma membrane that triggers nongenomic effects of the steroid. Cytoplasmic 1,25D/VDR complex rapidly (transient pathway) activates a JNK MAP kinase, on one hand, and upregulates c-Fos expression via a MEK1/MEK2/ERK pathway, on the other. Only under sustained presence of hormone, phosphorylated c-Jun, a downstream product of JNK activation, dimerizes with c-Fos at the cell nucleus, and the complex binds to an AP-1 site upstream the p21waf1 gene involved in the control of cell cycle progression. In addition, under continuous presence of hormone and according to traditional genomic pathways, 1,25D/VDR complex translocates to the cell nucleus and binds to a vitamin D response element (VDRE) upstream of the p21 gene. As a result of this genomic and nongenomic cross-talk, the cdk inhibitor p21 is expressed leading to cell cycle arrest and decreased cell proliferation in a time frame of days. As seen from our results, a classic VDR is required for nongenomic activation of MAPK signaling.
We found that sustained but not transient 1,25D treatment caused a significant reduction of human osteosarcoma SaOS-2 cell proliferation only in the presence of a functional VDR. This antiproliferative effect of the hormone correlated with activation of JNK and ERK1/2 MAPK pathways, and upregulation of activator protein 1 (AP-1) and the cell cycle regulator p21waf1 gene. JNK and its downstream target c-Jun have been shown to be involved in the control of cell cycle, proliferation and apoptosis in a variety of cancer cell systems (41). Activated (phosphorylated) c-Jun heterodimerizes with c-Fos, a downstream product of ERK1/2 MAPK activation (21), and constitutes one of multiple forms of the ubiquitous AP-1 transcription factor complex. AP-1 modulates the expression of genes directly involved in the progression of the cell cycle, including the gene that encodes for the cell cycle regulator protein p21, which mediates cell growth arrest by inhibiting the action of G1 cyclin-dependent kinases. It has been previously demonstrated that the p21 gene promoter region has both AP-1 and VDR binding sites (33), and that p21 gene expression is triggered by multiple differentiation-inducing agents including 1,25D by a p53-independent pathway. Our data show for the first time that both transient (15 min) and sustained (3-5 days) treatment with 10 nM 1,25D significantly activated JNK/c-Jun, and MEK1/MEK2/c-Fos in SaOS-2 cells. On the contrary, only sustained 1,25D stimulus upregulated AP-1 and p21. As a result, cell growth was only reduced after a 3 day-long treatment with 1,25D. This coincides with reports in the literature that indicate that short-term activation of MAPK promotes cell growth, whereas long-term accumulation of activated MAPK induces cell death in some cell types (20). Western blot results obtained in rat ROS 17/2.8 osteosarcoma cells, on the other hand, showed increased JNK activation but not c-Fos upregulation upon sustained 1,25D treatment (see Figure 1B). This is may explain at least in part survival effects of the steroid described recently (11). It has been reported, for example, that the duration and magnitude of a MAPK ERK signal is a critical factor in determining the proliferative response of cells to a signal from the environment (42).
Our results are in agreement with a previous report on 1,25D induction of a time-dependent loss of cell viability in a rat (UMR-106) and human (TE 85) osteosarcoma cell lines (4). However, those studies did not clarify the mechanisms. We found that our newly created siRNA VDR SaOS-2 subclone, which expresses significantly reduced levels of VDR protein, is insensitive to 1,25D-induced cell growth inhibition. In addition, we verified in native SaOS-2 cells that 1,25D treatment activates nongenomic MAPK pathways, and a genomic AP-1 cell cycle signaling, which work together towards inhibiting cell growth. We found that a classic VDR appears to be required for cytoplasmic activation of JNK and subsequent phosphorylation of a c-Jun transcription factor that triggers a genomic pathway. Expression of a cytoplasmic or membrane-localized classic VDR has been shown recently in different cell systems including osteoblasts (10). Our study thus provides clear evidence of a cross-talk between nongenomic and genomic actions of 1,25D at the level of MAP kinases and AP-1 via a classic VDR, and the combined regulation of these pathways leading to control of cell proliferation in human osteosarcoma cells. As summarized in Figure 6, our data show that VDR activation leads to p21 expression via direct and indirect mechanisms. In the first case, VDR localized at the nucleus activates p21 via binding to a VDR responsive element (VDRE) that leads to AP-1 promoted upregulation of the gene. In the second case, a classic VDR localized in the cell cytoplasm activates –by means of unknown mechanisms- JNK and ERK1/2 MAP kinases that upregulate a downstream AP-1 complex.
Osteosarcoma is the most common type of bone cancer. It is the cancer type with highest incidence in children and adolescents. Each year in the United States, osteosarcoma is diagnosed in approximately 400 children and adolescents younger than 20 years. Osteoblastic osteosarcoma develops from osteoblasts, the bone-forming cells. In the present study, we used the human osteosarcoma SaOS-2 cell line, which is derived from a patient's tumor, and created a stably transfected subclone in which the VDR is knocked-down. We verified that JNK and ERK MAP kinases are a point of cross-talk between cytoplasmic (nongenomic) and nuclear (genomic) functions of the VDR, and that only sustained but not transient 1,25D-induced JNK activation leads to antiproliferative effects by the hormone.
Our results contribute to current understanding on the molecular mechanisms underlying antiproliferative effects of the natural steroid 1,25D. Increasing knowledge in this field is highly required for the proper design and management of therapeutic uses of the steroid hormone and analogs in the treatment of bone tumors.
Acknowledgments
We thank Dr. A. Walker, University of California-Riverside, for kindly providing the pRSV-β-galactosidase plasmid, and p21-luc promoter reporter construct.
Financial support: NIH grant (DK071115) to L.P. Zanello
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Butterwo CE. Vitamin deficiency and cancer. Med Oncol. 1985:T165–174. [Google Scholar]
- 2.Binderup L. Vitamin D analogues: New regulators of cancer cell growth and differentiation. Bioorg Med Chem Lett. 1993;3:1891–1896. [Google Scholar]
- 3.Matsumoto T, Sowa Y, Ohtani-Fujita N, et al. p53-independent induction of WAF1/Cip1 is correlated with osteoblastic differentiation by vitamin D3. Cancer Lett. 1998;129:61–68. doi: 10.1016/s0304-3835(98)00080-9. [DOI] [PubMed] [Google Scholar]
- 4.Witasp E, Gustafsson AC, Cotgreave I, Lind M, Fadeel B. Vitamin D fails to prevent serum starvation- or staurosporine-induced apoptosis in human and rat osteosarcoma-derived cell lines. Biochem Biophys Res Comm. 2005;330:891–897. doi: 10.1016/j.bbrc.2005.03.061. [DOI] [PubMed] [Google Scholar]
- 5.Barroga EF, Kadosawa T, Okumura M, Fujinaga T. Effects of vitamin D and retinoids on the differentiation and growth in vitro of canine osteosarcoma and its clonal cell lines. Res Vet Sci. 1999;66:231–236. doi: 10.1053/rvsc.1998.0265. [DOI] [PubMed] [Google Scholar]
- 6.Fuchs B, Pritchard DJ. Etiology of osteosarcoma. Clin Orthop Relat Res. 2002;397:40–52. doi: 10.1097/00003086-200204000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Ryhanen S, Jaaskelainen T, Saarela JT, Maenpaa PH. Inhibition of proliferation and induction of differentiation of osteoblastic cells by a novel 1α,25(OH)2-vitamin D3 analog with an extensively modified side chain (CB1093) J Cell Biochem. 1998;70:414–424. doi: 10.1002/(sici)1097-4644(19980901)70:3<414::aid-jcb14>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 8.Barroga EF, Kadosawa T, Asano K, Okumura M, Fujinaga T. Apoptosis induction of POS canine osteosarcoma cells by vitamin D and retinoids. J Vet Med Sci. 1998;60:1269–1272. doi: 10.1292/jvms.60.1269. [DOI] [PubMed] [Google Scholar]
- 9.Bouillon R, Okamura WH, Norman AW. Structure-function relationships in the vitamin D endocrine system. Endocr Rev. 1995;16:200–257. doi: 10.1210/edrv-16-2-200. [DOI] [PubMed] [Google Scholar]
- 10.Huhtakangas JA, Olivera CJ, Bishop JE, Zanello LP, Norman AW. The vitamin D receptor is present in caveolae-enriched plasma membranes and binds 1α,25(OH)2-vitamin D3 in vivo and in vitro. Molec Endocrinol. 2004;18:2660–71. doi: 10.1210/me.2004-0116. [DOI] [PubMed] [Google Scholar]
- 11.Vertino AM, Bula CM, Chen JR, et al. Nongenotropic, anti-apoptotic signaling of 1α,25(OH)2-vitamin D3 and analogs through the ligand binding domain of the vitamin D receptor in osteoblasts and osteocytes. Mediation by Src, phosphatidylinositol 3-, and JNK kinases. J Biol Chem. 2005;280:14130–14137. doi: 10.1074/jbc.M410720200. [DOI] [PubMed] [Google Scholar]
- 12.Zanello LP, Norman AW. Stimulation by 1α,25(OH)2-vitamin D3 of whole cell chloride currents in osteoblastic ROS 17/2.8 cells: A structure-function study. J Biol Chem. 1997;272:22617–22622. doi: 10.1074/jbc.272.36.22617. [DOI] [PubMed] [Google Scholar]
- 13.Zanello LP, Norman AW. Multiple molecular mechanisms of 1α,25(OH)2-vitamin D3 rapid modulation of three ion channel activities in osteoblasts. Bone. 2003;33:71–79. doi: 10.1016/s8756-3282(03)00162-5. [DOI] [PubMed] [Google Scholar]
- 14.Zanello LP, Norman AW. Rapid modulation of osteoblast ion channel responses by 1α,25(OH)2-vitamin D3 requires the presence of a functional vitamin D nuclear receptor. Proc Natl Acad Sci USA. 2004;101:1589–1594. doi: 10.1073/pnas.0305802101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lieberherr M. Effects of vitamin-D3 metabolites on cytosolic free calcium in confluent mouse osteoblasts. J Biol Chem. 1987;262:13168–13173. doi: 10.1515/9783110846713.769. [DOI] [PubMed] [Google Scholar]
- 16.Ulsperger E, Hamilton G, Olszewski U, et al. Effects of 1α,25(OH)2-vitamin D3 pretreatment and MAP kinase inhibitor PD 98059 on response of osteoblasts to prostate-derived osteoblastic factors. Oncol Rep. 2003;10:1529–15234. [PubMed] [Google Scholar]
- 17.Wang X, Rao J, Studzinski GP. Inhibition of p38 MAP kinase activity upregulates multiple MAP kinase pathways and potentiates 1α,25-(OH)2-vitamin D3-induced differentiation of human leukemia HL60 cells. Exp Cell Res. 2000;258:425–437. doi: 10.1006/excr.2000.4939. [DOI] [PubMed] [Google Scholar]
- 18.Liu M, Lee MH, Cohen M, Bommakanti M, Freedman LP. Transcriptional activation of the Cdk inhibitor p21 by vitamin D3 leads to the induced differentiation of the myelomonocytic cell line U937. Genes Dev. 1996;10:142–153. doi: 10.1101/gad.10.2.142. [DOI] [PubMed] [Google Scholar]
- 19.Saramaki A, Banwell CM, Campbell MJ, Carlberg C. Regulation of the human p21waf1/cip1 gene promoter via multiple binding sites for p53 and the vitamin D3 receptor. Nucleic Acids Res. 2006;34:543–554. doi: 10.1093/nar/gkj460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Martindale JL, Gorospe M, Holbrook NJ. Regulation of p21waf1/cip1 expression through mitogen-activated protein kinase signaling pathway. Cancer Res. 1996;56:31–35. [PubMed] [Google Scholar]
- 21.Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 22.Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 23.Binetruy B, Smeal T, Karin M. Ha-Ras augments c-Jun activity and stimulates phosphorylation of its activation domain. Nature. 1991;351:122–127. doi: 10.1038/351122a0. [DOI] [PubMed] [Google Scholar]
- 24.Chen A, Davis BH, Bissonnette M, Scaglione-Sewell B, Brasitus TA. 1α,25-(OH)2-vitamin D3 stimulates activator protein-1-dependent caco-2 cell differentiation. J Biol Chem. 1999;274:35505–35513. doi: 10.1074/jbc.274.50.35505. [DOI] [PubMed] [Google Scholar]
- 25.Wang Q, Salman H, Danilenko M, Studzinski GP. Cooperation between antioxidants and 1α,25(OH)2 vitamin D3 in induction of leukemia HL60 cell differentiation through the JNK/AP-1/Egr-1 pathway. J Cell Physiol. 2005;204:964–974. doi: 10.1002/jcp.20355. [DOI] [PubMed] [Google Scholar]
- 26.Ryhanen S, Jaaskelainen T, Mahonen A, Maenpaa PH. Inhibition of MG-63 cell cycle progression by synthetic vitamin D3 analogs mediated by p27, Cdk2, cyclin E, and the retinoblastoma protein. Biochem Pharmacol. 2003;66:495–504. doi: 10.1016/s0006-2952(03)00283-1. [DOI] [PubMed] [Google Scholar]
- 27.Hodge C, Liao J, Stofega M, et al. Growth hormone stimulates phosphorylation and activation of elk-1 and expression of c-fos, egr-1, and junB through activation of extracellular signal-regulated kinases 1 and 2. J Biol Chem. 1998;273:31327–31336. doi: 10.1074/jbc.273.47.31327. [DOI] [PubMed] [Google Scholar]
- 28.Fleet JC. Rapid, membrane-initiated actions of 1α,25(OH)2-vitamin D3: What are they and what do they mean? J Nutr. 2004;134:3215–3218. doi: 10.1093/jn/134.12.3215. [DOI] [PubMed] [Google Scholar]
- 29.Nemere I, Norman AW. Steroid hormone actions at the plasma membrane: Induced calcium uptake and exocytotic events. Mol Cell Endocrinol. 1991;80:C165–169. doi: 10.1016/0303-7207(91)90132-c. [DOI] [PubMed] [Google Scholar]
- 30.Boyan BD, Schwartz Z, Bonewald L, Swain L. Localization of 1α,25(OH)2-vitamin D3 responsive alkaline phosphatase in osteoblast-like cells (ROS 17/2.8, MG 63, and MC 3T3) and growth cartilage cells in culture. J Biol Chem. 1989;264:11879–11886. [PubMed] [Google Scholar]
- 31.Ho AK, Price DM, Terriff D, Chik CL. Timing of mitogen-activated protein kinase (MAPK) activation in the rat pineal gland. Mol Cell Endocrinol. 2006;252:34–39. doi: 10.1016/j.mce.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 32.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 33.Zenmyo M, Komiya S, Hamada T, et al. Transcriptional activation of p21 by vitamin D3 or vitamin K2 leads to differentiation of p53-deficient MG-63 osteosarcoma cells. Hum Pathol. 2001;32:410–416. doi: 10.1053/hupa.2001.23524. [DOI] [PubMed] [Google Scholar]
- 34.Blutt SE, Allegretto EA, Pike JW, Weigel NL. 1α,25-(OH)2-vitamin D3 and 9-cis-retinoic acid act synergistically to inhibit the growth of LNCaP prostate cells and cause accumulation of cells in G1. Endocrinology. 1997;138:1491–1497. doi: 10.1210/endo.138.4.5063. [DOI] [PubMed] [Google Scholar]
- 35.Wang XN, Ponzio NM, Studzinski GP. Long-term exposure of HL60 cells to 1α,25-(OH)2-vitamin D3 reduces their tumorigenicity: A model for cancer chemoprevention. Proc Soc Exp Biol Med. 1997;215:399–404. doi: 10.3181/00379727-215-44150. [DOI] [PubMed] [Google Scholar]
- 36.Witasp E, Gustafsson AC, Cotgreave I, Lind M, Fadeel B. Vitamin D fails to prevent serum starvation- or staurosporine-induced apoptosis in human and rat osteosarcoma-derived cell lines. Biochem Biophys Res Comm. 2005;330:891–897. doi: 10.1016/j.bbrc.2005.03.061. [DOI] [PubMed] [Google Scholar]
- 37.Abe J, Nakano T, Nishii Y, et al. A novel vitamin D3 analog, 22-oxa-1,25-dihydroxyvitamin D3, inhibits the growth of human breast cancer in vitro and in vivo without causing hypercalcemia. Endocrinology. 1991;129:832–837. doi: 10.1210/endo-129-2-832. [DOI] [PubMed] [Google Scholar]
- 38.Song RX, Santen R. Membrane initiated estrogen signaling in breast cancer. Biol Reprod. 2006;75:9–16. doi: 10.1095/biolreprod.105.050070. [DOI] [PubMed] [Google Scholar]
- 39.Gatson JW, Kaur P, Singh M. Dihydrotestosterone differentially modulates the mitogen-activated protein kinase and the phosphoinositide 3-kinase/Akt pathways through the nuclear and novel membrane androgen receptor in C6 cells. Endocrinology. 2006;147:2028–2034. doi: 10.1210/en.2005-1395. [DOI] [PubMed] [Google Scholar]
- 40.Wessler S, Otto C, Wilck N, Stangl V, Fritzemeier KH. Identification of estrogen receptor ligands leading to activation of non-genomic signaling pathways while exhibiting only weak transcriptional activity. J Steroid Biochem Mol Biol. 2006;98:25–35. doi: 10.1016/j.jsbmb.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 41.Johnson GL, Vaillancourt R. Sequential protein kinase reactions controlling cell growth and differentiation. Curr Opin Cell Biol. 1994;6:230–238. doi: 10.1016/0955-0674(94)90141-4. [DOI] [PubMed] [Google Scholar]
- 42.Roovers K, Assoian R. Integrating the MAP kinase signal into the G1 phase cell cycle machinery. Bioessays. 2000;22:818–826. doi: 10.1002/1521-1878(200009)22:9<818::AID-BIES7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]