

Abstract
A systematic stereocontrolled synthesis of benzannulated spiroketals has been developed, using kinetic spirocyclization reactions of glycal epoxides, leading to a new AcOH-induced cyclization and valuable insights into the reactivity and conformations of these systems. One stereochemical series accommodates axial positioning of the aromatic ring while another adopts an alternative 1C4 chair conformation to avoid it. Equatorial aromatic rings also participate in non-obvious steric interactions that impact thermodynamic stability. A discovery library of 68 benzannulated spiroketals with systematic variations in stereochemistry, ring size, and positioning of the aromatic substituent has been synthesized for broad biological evaluation.
Benzannulated spiroketals are found in a variety of natural products with diverse, compelling biological activities. Key examples include the rubromycin family of telomerase inhibitors, the related DNA helicase inhibitor heliquinomycin, the papulacandin family of fungal cell wall glucan synthase inhibitors, the griseusin class of antibacterial agents, the antimitotic paecilospirone, and the novel matrix metalloproteinase inhibitor berkelic acid.1 As the benzannulated spiroketal motif has been directly implicated in the biological activity of several of these compounds, this class is an attractive target for the diversity-oriented synthesis of natural product-based libraries for use in discovery screening.2 In this vein, the aromatic ring can be expected to influence the reactivity, three-dimensional conformation, and physicochemical properties of these molecules relative to aliphatic spiroketals. To evaluate the impact of the aromatic ring on the chemistry and biology of benzannulated spiroketals, we have carried out a systematic study of their stereocontrolled synthesis using kinetic spirocyclization reactions. This work has revealed unexpected reactivity patterns and conformational preferences and paves the way to comprehensive biological evaluation of this class.
The synthesis of benzannulated spiroketals has depended largely upon thermodynamically controlled transketalization reactions of glycoside or ketoalcohol precursors.3,4 Several kinetically controlled approaches have also been developed, but still generally do not provide selective access to contrathermodynamic products. Fully stereocontrolled access to either diastereomer at the anomeric carbon is preferable both for total synthesis applications and to leverage stereochemical diversity in spiroketal libraries.5 Early work by Wallace suggested the feasibility of overcoming inherent thermodynamic preferences in kinetic chromone epoxide spirocyclizations, albeit with limited stereoselectivity.6 Recently, Pettus has also advanced elegant cycloaddition-based approaches to the stereoselective synthesis of rubromycin family members.7 We have previously developed a stereocontrolled approach to aliphatic spiroketals using two complementary kinetic spirocyclization reactions of glycal epoxides that proceed with either inversion or retention of configuration at the anomeric carbon, independently of thermodynamic considerations.8 We envisioned that these reactions might also be useful for the synthesis of benzannulated spiroketals.
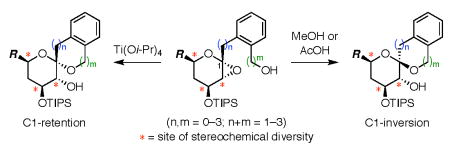
Our overall synthetic strategy is outlined in Figure 1.9 The threo- and erythro-glycal stannanes 18a,10 were functionalized at C1, either by direct Stille cross couplings of aryl and benzyl bromides,11 or via conversion to the corresponding glycal iodides followed by B-alkyl Suzuki–Miyaura cross couplings with styrenes and allylbenzenes.12 Stille couplings of benzyl bromide substrates required copper iodide to suppress glycal dimer formation and yields were further increased by protection from light.13 Deacetylation then provided all nine precursors to 5-, 6-, and 7-membered rings with the aromatic ring systematically positioned along the sidechain (2a–r).
Figure 1.

Overall approach to stereocontrolled synthesis of benzannulated spiroketals using kinetic spirocyclization reactions. (*) = site of stereochemical diversity.
Stereoselective anti-epoxidation at low temperature afforded glycal epoxide intermediates 3a–r, which were subjected in situ to various spirocyclization conditions (Figures 2 and 3). Warming the glycal epoxides (−78 °C → rt) resulted in spontaneous cyclization with variable selectivity for retention of configuration (4a–r). Acid-catalyzed spirocyclization with TsOH (−78 °C → rt) led to 4a–r exclusively, although these reactions were often compromised by side reactions.14 In contrast, our Ti(Oi-Pr)4-mediated spirocyclization (−78 °C → 0 °C)8b afforded 4a–r in high yields and with complete stereocontrol for all substrates.9 Notably, the conformational constraint provided by the aromatic ring allowed highly efficient formation of 7-membered rings (4c,e,f,i,l,n,o,r), which was not the case for the corresponding aliphatic systems.8b
Figure 2.

Diastereomeric ratios of benzannulated spiroketals formed from d-threo-glycal epoxides 3a–i. Isolated yields of 4 (Ti[Oi-Pr]4) and 5 (MeOH or AcOH) shown in parentheses. a Spontaneous spirocyclization (−78 °C → rt). b Remainder methyl glycoside 6. c Inseparable mixture of 4 and 5 (separable after desilylation).
Figure 3.

Diastereomeric ratios of benzannulated spiroketals formed from d-erythro-glycal epoxides 3j–r. Isolated yields of 4 (Ti[Oi-Pr]4) and 5 (MeOH or AcOH) shown in parentheses. a Spontaneous spirocyclization (−78 °C → rt). b Remainder methyl glycoside 6. c Inseparable mixture of 4 and 5 (separable after desilylation).
Conversely, our MeOH-induced spirocyclization (−63 °C)8a provided stereocontrol for the inherently thermodynamically and kinetically disfavored inversion products in several cases (5a,b,d–f,j,m,n). The aromatic ring constraint provided reduced competing intermolecular formation of methyl glycoside side products (6) compared to the corresponding aliphatic systems,8a and again allowed several 7-membered rings to be formed (5e,f,n). Two additional reactivity trends were noted in these MeOH-induced spirocyclizations. C1-Aryl substrates yielded decreased selectivity for inversion of configuration (5b vs. 5d; 5c vs. 5e,f; 5k vs. 5m; 5l vs. 5n), which we attributed to stabilization of a cyclic oxocarbenium intermediate leading to the retention products. Phenolic substrates also afforded decreased stereoselectivity (5g–i,p–r) and increased methyl glycoside formation, presumably due to the lower nucleophilicity of the phenol sidechains.
To achieve more efficient access to the inversion products, we explored alternative Brønsted acids and were gratified to find that AcOH (10 equiv., −63 °C → −44 °C) provided increased yields for both the problematic 7-membered ring and phenolic systems (5e–i,n–r).15,16 Treatment of the isolated products with AcOH confirmed that this reaction remains under kinetic control. The increased yields could be attributed largely to the absence of competing intermolecular glycosylation. AcOH may also provide increased epoxide reactivity for less-reactive phenol nucleophiles, consistent with our previous proposal that the MeOH-induced spirocyclization proceeds via activation of the epoxide by MeOH hydrogen bonding.8a,17
To evaluate the impact of the aromatic ring upon the conformation of benzannulated spiroketals, we carried out detailed structural analyses of several products (Figure 4).9 Analyses of NOESY spectra and J values18 indicated that both threo series products 4a and 5a adopt standard 4C1 chair conformations. Notably, this is despite the axial orientation of the aromatic ring in 5a, due to unfavorable 1,3,5-triaxial interactions in the alternative 1C4 conformation (5a′). In contrast, the corresponding erythro series product 4j adopts the alternative 1C4 conformation, due to the additional steric impact of the C3-OTIPS group (4j′). While both 4j and 5j have anomeric stabilization and comparable 1,3-diaxial interactions involving C1-O, the thermodynamic preference for 4j can be rationalized by an additional, non-obvious steric interaction between the ortho-proton of the aromatic ring and C2-OH in 5j. Strikingly, after desilylation (R′ = H), the thermodynamic preference is reversed, with 8 stabilized by an intramolecular hydrogen bond. These results highlight the profound impact of the aromatic ring upon both the conformational and thermodynamic preferences of benzannulated spiroketals and the increased conformational diversity in this class compared to the corresponding aliphatic spiroketals.
Figure 4.

Alternative conformations of benzannulated spiroketals 4a, 5a, 4j, 5j and desilylated congeners 7, 8. NOESY interactions are indicated in blue; steric interactions are indicated in red. A non-obvious steric interaction in 5j between the ortho-proton of the aromatic ring and C2-OH is highlighted in bold red.
Desilylation of all products provided a discovery library of 68 out of the 72 possible benzannulated spiroketals (both enantiomeric series) having all combinations of five-, six-, and seven-membered rings and aromatic ring positions, with nearly comprehensive stereochemical diversity. These compounds have been deposited in the NIH Molecular Libraries Small Molecule Repository19 and are undergoing biological evaluation to assess the effectiveness of this structural class against a wide range of targets. Analysis of those results will be reported in due course.
In conclusion, we have carried out a systematic analysis of kinetic and thermodynamic spirocyclization reactions to form benzannulated spiroketals, leading to the development of a new AcOH-induced kinetic spirocyclization. Notably, the systematic nature of this study, mandated by our interest in diversity-oriented synthesis applications, has revealed significant reactivity changes and conformational effects imparted by the aromatic ring, which may be useful in the design and synthesis of a broad range of molecules with related structural motifs. Finally, this work sets the stage for further biological investigation of this compelling class of molecules.
Supplementary Material
Acknowledgments
We thank Prof. Ian Fairlamb (University of York) for helpful discussions and Dr. George Sukenick, Dr. Hui Liu, Hui Fang, and Dr. Sylvia Rusli for expert mass spectral analyses. D.S.T. is an Alfred P. Sloan Research Fellow. Financial support from the NIH (P41 GM076267), NYSTAR Watson Investigator Program, William H. Goodwin and Alice Goodwin and the Commonwealth Foundation for Cancer Research, and MSKCC Experimental Therapeutics Center is gratefully acknowledged.
Footnotes
Supporting Information Available: Detailed experimental procedures and analytical data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Rubromycins/heliquinomycin: Brasholz M, Sörgel S, Azap C, Reißig HU. Eur J Org Chem. 2007:3801–3814.. Papulacandins: Traxler P, Fritz H, Fuhrer H, Richter WJ. J Antibiot. 1980;33:967–978. doi: 10.7164/antibiotics.33.967.Varona R, Perez P, Duran A. FEMS Microbiol Lett. 1983;20:243–247.. Griseusins: Tsuji N, Kobayashi M, Wakisaka Y, Kawamura Y, Mayama M, Matsumoto K. J Antibiot. 1976;29:7–9. doi: 10.7164/antibiotics.29.7.. Paecilospirone: Namikoshi M, Kobayashi H, Yoshimoto T, Meguro S. Chem Lett. 2000:308–309. doi: 10.1248/cpb.48.1452.. Berkelic acid: Stierle AA, Stierle DB, Kelly K. J Org Chem. 2006;71:5357–5360. doi: 10.1021/jo060018d.
- 2.Tan DS. Nat Chem Biol. 2005;1:74–84. doi: 10.1038/nchembio0705-74. [DOI] [PubMed] [Google Scholar]
- 3.For examples from natural product total synthesis, see: Rubromycins: (a) Reviewed in ref. 1a. Akai S, Kakiguchi K, Nakamura Y, Kuriwaki I, Dohi T, Harada S, Kubo O, Morita N, Kita Y. Angew Chem, Int Ed. 2007;46:7458–7461. doi: 10.1002/anie.200702382.. Heliquinomycinone: Siu T, Qin D, Danishefsky SJ. Angew Chem, Int Ed. 2001;40:4713–4716. doi: 10.1002/1521-3773(20011217)40:24<4713::aid-anie4713>3.0.co;2-n.. Papulacandins: Denmark SE, Regens CS, Kobayashi T. J Am Chem Soc. 2007;129:2774–2776. doi: 10.1021/ja070071z.. Griseusins: Kometani T, Takeuchi Y, Yoshii E. J Org Chem. 1983;48:2311–2314.. Berkelic acid: Wu X, Zhou J, Snider BB. Angew Chem, Int Ed. 2009;48:1283–1286. doi: 10.1002/anie.200805488.Buchgraber P, Snaddon TN, Wirtz C, Mynott R, Goddard R, Fürstner A. Angew Chem, Int Ed. 2008;47:8450–8454. doi: 10.1002/anie.200803339.
- 4.For other approaches to benzannulated spiroketals, see: Alkoxyselenation: Elsley DA, MacLeod D, Miller JA, Quayle P, Davies GM. Tetrahedron Lett. 1992;33:409–412.. Alkyne cyclotrimerization: McDonald FE, Zhu HYH, Holmquist CR. J Am Chem Soc. 1995;117:6605–6606.. Glycoside rearrangement: Kumazawa T, Asahi N, Matsuba S, Sato S, Furuhata K, Onodera JI. Carbohydr Res. 1998;308:213–216.. Michael addition: Carretero JC, De Diego JE, Hamdouchi C. Tetrahedron. 1999;55:15159–15166.Choi PJ, Rathwell DCK, Brimble MA. Tetrahedron Lett. 2009;50:3245–3248.. Ring-closing metathesis: Van Hooft PAV, Van Swieten PF, Van der Marel GA, Van Boeckel CAA, Van Boom JH. Synlett. 2001:269–271.. o-Quinone methide hetero-Diels–Alder: Zhou G, Zheng D, Da S, Xie Z, Li Y. Tetrahedron Lett. 2006;47:3349–3352.Lindsey CC, Wu KL, Pettus TRR. Org Lett. 2006;8:2365–2367. doi: 10.1021/ol0606886.. Alkyne hydroalkoxylation: Messerle BA, Vuong KQ. Organometallics. 2007;26:3031–3040.Zhang Y, Xue J, Xin Z, Xie Z, Li Y. Synlett. 2008:940–944.. Radical cyclization: Liu YC, Sperry J, Rathwell DCK, Brimble MA. Synlett. 2009:793–797.
- 5.For a recent review of non-anomeric spiroketal synthesis, see: Aho JE, Pihko PM, Rissa TK. Chem Rev. 2005;105:4406–4440. doi: 10.1021/cr050559n.
- 6.Cremins PJ, Wallace TW. J Chem Soc, Chem Commun. 1986:1602–1603. [Google Scholar]
- 7.(a) Wu KL, Wilkinson S, Reich NO, Pettus TRR. Org Lett. 2007;9:5537–5540. doi: 10.1021/ol702450d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Marsini MA, Huang Y, Lindsey CC, Wu KL, Pettus TRR. Org Lett. 2008;10:1477–1480. doi: 10.1021/ol8003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Potuzak JS, Moilanen SB, Tan DS. J Am Chem Soc. 2005;127:13796–13797. doi: 10.1021/ja055033z. [DOI] [PubMed] [Google Scholar]; (b) Moilanen SB, Potuzak JS, Tan DS. J Am Chem Soc. 2006;128:1792–1793. doi: 10.1021/ja057908f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.See Supporting Information for full details.
- 10.Moilanen SB, Tan DS. Org Biomol Chem. 2005;3:798–803. doi: 10.1039/b417429a. [DOI] [PubMed] [Google Scholar]
- 11.(a) Friesen RW, Sturino CF. J Org Chem. 1990;55:2572–2574. [Google Scholar]; (b) Milstein D, Stille JK. J Am Chem Soc. 1979;101:4992–4998. [Google Scholar]; (c) Farina V, Kapadia S, Krishnan B, Wang C, Liebeskind LS. J Org Chem. 1994;59:5905–5911. [Google Scholar]
- 12.Potuzak JS, Tan DS. Tetrahedron Lett. 2004;45:1797–1801. [Google Scholar]
- 13.Crawforth CM, Burling S, Fairlamb IJS, Kapdi AR, Taylor RJK, Whitwood AC. Tetrahedron. 2005;61:9736–9751. [Google Scholar]
- 14.In particular, for erythro series substrates (3j–r), loss of the C3-OTIPS group with concomitant oxidation of the C2-OH group was observed. This may occur via enolization (C2-deprotonation) of the intermediate cyclic oxocarbenium species, followed by Ferrier type elimination of the C3-substituent, tautomerization of the resulting C2 enol to a ketone, and ring reclosure at C1.
- 15.p-NO2PhOH provided comparable 4i:5i selectivity, while weaker (PhOH) and stronger acids (HCO2H, p-NO2PhCO2H, TFA) led to decreased amounts of the desired inversion product 5i. Larger amounts of AcOH (100–1000 equiv.) led to increased formation of the retention products 4, presumably due to increased competing oxocarbenium formation.
- 16.For related reactions of glycals and glycosides, see: Pothier N, Goldstein S, Deslongchamps P. Helv Chim Acta. 1992;75:604–620.Castagnolo D, Breuer I, Pihko PM. J Org Chem. 2007;72:10081–10087. doi: 10.1021/jo702022u.
- 17.Related effects have been proposed in epoxide-opening cascades leading to ladder polyethers: Vilotijevic I, Jamison TF. Science. 2007;317:1189–1192. doi: 10.1126/science.1146421.Byers JA, Jamison TF. J Am Chem Soc. 2009;131:6383–6385. doi: 10.1021/ja9004909.
- 18.Karplus M. J Chem Phys. 1959;30:11–15. [Google Scholar]
- 19.PubChem Substance Identifiers (SID): “DST_SK2_*”; http://pubchem.ncbi.nlm.nih.gov/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.