

Abstract
Fanconi anemia (FA) is a rare autosomal recessive or X-linked disorder characterized by aplastic anemia, cancer susceptibility and cellular sensitivity to DNA crosslinking agents. Eight FA proteins (FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL and FANCM) and three non-FA proteins (FAAP100, FAAP24 and HES1) form an FA nuclear core complex, which is required for monoubiquitination of the FANCD2-FANCI dimer upon DNA damage. FANCL possesses a PHD/RING-finger domain and is a putative E3 ubiquitin ligase subunit of the core complex. In this study, we report an FA patient with an unusual presentation belonging to the FA-L complementation group. The patient lacks an obvious FA phenotype except for the presence of a cafe-au-lait spot, mild hypocellularity and a family history of leukemia. The molecular diagnosis and identification of the FA subgroup was achieved by FA complementation assay. We identified bi-allelic novel mutations in the FANCL gene and functionally characterized them. To the best of our knowledge, this is the second reported case belonging to the FA-L complementation group.
Keywords: FA-L, FANCL, Mutation, Fanconi anemia
INTRODUCTION
Fanconi anemia (FA; MIM# 227650) is a rare autosomal recessive or X-linked disorder (Levitus, et al., 2006; Wang, 2007). The disease has a highly variable clinical presentation and is characterized by congenital abnormalities, predisposition to hematological and non-hematological malignancies and progressive fatal bone marrow failure (Levitus, et al., 2006; Wang, 2007). FA is a genetically heterogeneous disease, and, to date, 13 genes and their corresponding protein products have been identified (FANCA; MIM# 607139; FANCB; MIM# 300515; FANCC; MIM# 227645; FANCD1 (BRCA2); MIM# 605724; FANCD2; MIM# 227646; FANCE; MIM# 600901; FANCF; MIM# 603467; FANCG; MIM# 602956; FANCI; MIM# 611360; FANCJ (BRIP1/BACH1); MIM# 605882; FANCL; MIM# 608111; FANCM; MIM# 609644 and FANCN (PALB2); MIM# 610832) (Wang, 2007). Eight FA proteins (FANCA, -B, -C, -E, -F, -G, -L and -M) and three non-FA proteins (FAAP100, FAAP24 and HES1) form a complex known as the FA nuclear core complex which is required for the monoubiquitination of FANCD2-FANCI dimer upon DNA damage (Wang, 2007; Tremblay, et al., 2008). DNA replication inhibitors, such as hydroxyurea (HU), induce FANCD2 monoubiquitination. FA patients who lack expression of any one of the core complex proteins or FANCI show complete absence of monoubiquitinated FANCD2 (Levitus, et al., 2006; Wang, 2007). Cells derived from FA patients are hypersensitive to DNA cross-linkers, such as mitomycin C (MMC), diepoxybutane (DEB), or cisplatin (Levitus, et al., 2006; Wang, 2007). When compared to wild-type cells, FA cells treated with these DNA cross-linkers show 1) Increased chromosomal breakage, 2) G2/M arrest and 3) reduced survival (Levitus, et al., 2006; Wang, 2007).
The relative prevalence of each FA complementation group is not uniform (Kutler, et al., 2003). Complementation groups were determined either by cell fusion experiments or by correction of G2/M defect or cross-linker hypersensitivity of a patient’s cells by transduction with retroviral vectors containing the normal cDNAs (Kutler, et al., 2003; Levitus, et al., 2006). Most patients belong to the FA-A group (60-80%), followed by groups FA-C (10%) and FA-G (9%) (Kutler, et al., 2003; Wang, 2007). So far, only one known case of FA belonging to the FA-L complementation group has been reported, making it a very rare group (Meetei, et al., 2003a). The gene mutated in the FA-L subgroup, FANCL, contains a PHD (plant homeodomain) or RING-finger domain that exhibits auto-ubiquitination in vitro (Meetei, et al., 2003a; Gurtan, et al., 2006). It associates with UBE2T, an E2, via its PHD/RING-finger domain in yeast two-hybrid assays (Machida, et al., 2006). The interaction of FANCD2, the core complex, and UBE2T has been proposed to occur on chromatin where they are recruited independently (Alpi, et al., 2007). Ubiquitin plays a very important role in the regulation of the FA pathway (Jacquemont and Taniguchi, 2007). The FA core complex has been proposed to act as a multi-subunit E3 ubiquitin ligase complex for FANCD2-FANCI and FANCL is the catalytic subunit that is responsible for ubiquitination (Meetei, et al., 2003a; Wang, 2007).
During this study, we report an FA patient who belongs to the FA-L complementation group, but without obvious hematological abnormalities. To best of our knowledge this is the second reported case belonging to the FA-L complementation group. We have functionally characterized the effect of mutations identified in this patient.
MATERIAL AND METHODS
Constructs, retrovirus, cell lines and cell culture
The retroviral vector pMIEG3 and the His-FLAG tagged wild-type FANCL (pMIEG3-HF-FANCL) construct has been described previously (Ling, et al., 2007). This construct has been shown to functionally correct FA-L lymphoblast (EUFA868) (Ling, et al., 2007). The His-FLAG-tagged mutant 3-bp deletion and 4-bp duplication FANCL (HF-FANCL-delTAT and HF-FANCL-dupAATT, respectively) constructs were made by site-directed mutagenesis of the pMIEG3-HF-FANCL construct. Retroviral particles were generated at the Viral Vector Core facility at Cincinnati Children’s Hospital Medical Center. EUFA868 cells and EUFA868 cells stably expressing HF-FANCL have been described previously (Meetei, et al., 2003a; Ling, et al., 2007). RA3056 cells are Epstein-Barr virus transformed lymphoblast cells (LCLs) derived from an FA patient (IFAR1088/1) reported in this study. RA3056 cells stably expressing HF-FANCL and EUFA868 cells stably expressing HF-FANCL-delTAT and HF-FANCL-dupAATT were obtained by infecting these cells with retroviral supernatant and sorting the cells according to their EGFP levels using a FACS Vantage cell sorter (BD Biosciences). All the lymphoblast cell lines were maintained at 37°C and 5% CO2 in RPMI medium (Invitrogen) with 15% fetal bovine serum (Atlanta Biologicals). RA3100 cells are primary fibroblasts derived from the same patient presenting with RA3056 LCLs. HeLa S3 cells stably expressing HF-FANCL, HF-FANCL-delTAT and HF-FANCL-dupAATT were obtained by infecting these cells with retroviral supernatant and sorting cells as described above. HeLa S3 and RA3100 cells and the corresponding stable cells were maintained at 37°C and 5% CO2 in DMEM medium (Invitrogen) with 10% fetal bovine serum.
Mutational analysis
FANCL mutational analysis was done by direct sequencing of the PCR products from genomic DNA and cDNAs. For genomic DNA mutational analysis, genomic DNA was isolated from the RA3056 cell line using the PUREGENE DNA Extraction kit (Gentra Systems). All 14 exons and 300 bp of the upstream promoter region were tested for variations from the reference sequence (NM_018062.3). For PCR amplification, pairs of primers were designed for the intronic regions flanking individual exons to include splice sites. Each primer was tailed 5’ with the universal M13 forward (5’ primer) or M13 reverse (3’ primer) primer sequence to allow for bi-directional direct sequencing of the PCR products. Primer sequences are available upon request. Product size was restricted to no more than 900 bp so that amplification of exon 12 of FANCL was divided into two overlapping fragments. Prior to capillary sequencing (Applied Biosystems 3730), PCR products were further purified using an ExoSAP-IT (United States Biochemical Corp.). Comparison of the sequencing results to the wild-type reference sequence was performed using the MutationSurveyor software (SoftGenetics, LLC). For cDNA mutational analysis, total RNA was isolated from RA3056 cells and reverse transcribed to cDNA using the One-Step RT-PCR Kit (Qiagen) according to the manufacturer’s instructions. Specific amplification of the FANCL cDNA was performed, and the product was purified as described above and subsequently cloned in TOPO T/A cloning vector (Invitrogen). Twelve cDNA clones were sequenced on both strands using M13 forward and reverse primers.
Mutation nomenclature was generated using the Mutalyzer program (http://www.LOVD.nl/mutalyzer/) (Wildeman et al., 2008). The reference nucleotide sequence NM_018062.3 was used for both cDNA and gDNA nomenclature. For cDNA nomenclature, the +1 corresponds to the A of the translation initiation codon ATG. For gDNA nomenclature, the +1 corresponds to the first nucleotide of NM_018062.3. For mutation nomenclature at protein level, NP_060532.2 was used as reference sequence.
Immunoprecipitation and Immunoblotting
One 150-mm plate of HeLa S3 cells stably expressing HF-FANCL was grown up to 85–95% confluence. Cells were washed with phosphate-buffered saline (Invitrogen) and collected as a pellet. Total cell lysate was made by lysing the cells in 1.5 ml of 300-lysis buffer (30mM Hepes pH 7.9, 300mM NaCl, 1mM EDTA, 1% Triton and 0.1% NP-40) supplemented with complete protease inhibitors (Roche). Total cell lysate was incubated with 20 μl anti-FLAG M2 agarose beads (Sigma-Aldrich) overnight, washed four times with 300-lysis buffer and eluted with 3X FLAG peptide (Sigma-Aldrich) for 1-2 h. For immunoprecipitation from LCLs, the cells were grown in T75 flasks, and approximately 100 μL cells were collected and processed as described above for HeLa S3 cells.
Untreated or HU (Fluka)-treated cells were lysed with 2X SDS buffer and boiled for 8-10 min. The cell lysate, or the FLAG-immunoprecipitated samples, were separated on 8-16% gradient gel (Invitrogen) and transferred onto a nitrocellulose membrane (Bio-Rad) and probed with anti-FANCD2 (Novus Biologicals), anti-FANCM (Meetei, et al., 2005), anti-FANCL (Meetei, et al., 2003a), anti-FANCA (Meetei, et al., 2003b), anti-FANCG (Fanconi Anemia Research Fund) and anti-FAAP100 (Ling, et al., 2007) antibodies at a dilution of 1:3000, 1:2000, 1:1000, 1:2000, 1:500 and 1:3000, respectively. The excess antibody was washed with PBST-NaCl and probed with HRP-conjugated anti-rabbit secondary antibody at a dilution of 1:5000. Detection was performed by the chemiluminescence technique with the use of enhanced chemiluminescence reagent (GE Healthcare). Multiple exposures were recorded on an X-ray film (ISC Bioexpress) and developed in an automatic developer (Kodak).
Assays for functional effects
Cell cycle analysis was done as described previously (Chandra, et al., 2005). Briefly, the untreated or melphalan-treated cells were fixed in 1% paraformaldehyde (in PBS) for 20 min and then washed in PBS and resuspended for 10 min at room temperature in 0.1% solution of Triton X-100 (Sigma-Aldrich) in PBS. After being washed with PBS, cells were stained for 1h at 4°C with a solution of PBS containing 2 mg/ml RNase A (Qiagen) and 50 μg/ml propidium iodide. The stained cells were subjected to flow cytometric analysis using the FACScan Analytic Flow Cytometer (BD Biosciences). The sensitivity of cells to MMC (Sigma-Aldrich) was evaluated using CellTiter 96 AQueous One cell proliferation Assay kit (Promega). Trypan blue (Mediatech) stained cells were counted using a hematocytometer, and 5,000 cells were seeded into 96-well plates with 100 μl of RPMI culture medium, incubated for 16 h, and exposed to various concentrations of MMC in 10 μL RPMI. After 72 h, 20 μL CellTiter 96 AQueous One Solution was added to each well and incubated for 2 h. The absorbance of each well was measured in a microplate reader at 490 nm. DEB sensitivity assay was performed to assess chromosomal breaks using a standard protocol (Auerbach, 2003).
RESULTS
Patient
The male patient (IFAR 1088/1) was diagnosed at the age of 15 months after presenting with slow growth, poor feeding and irritability. During an evaluation for developmental delay in fine motor skills and speech, a DEB-induced chromosomal instability test was performed at a reference laboratory, and breakage at 10-fold higher than control was reported. The karyotype was normal. A brain MRI at 15 months showed mildly delayed myelination. He was diagnosed with attention deficit hyperactivity disorder. This patient was first seen in the Cincinnati Children’s Hospital Fanconi Anemia Comprehensive Care Center at age 8 years and was noted to have normal stature (15% for height) and weight (50%) with no obvious physical stigmata of Fanconi anemia. He did have one small cafe-au-lait spot on the abdomen. Family history was only remarkable for a paternal first cousin with childhood leukemia. Extensive multi-system analysis revealed no cardiac, hearing or urogenital abnormalities, although there was a mild glucose intolerance detected by glucose/insulin levels in an oral glucose tolerance test. Blood counts were entirely normal with a normal erythrocyte mean cell volume. Bone marrow aspirate/biopsy showed trilineage hematopoiesis with mild hypocellularity. The cytogenetics study was normal. FISH analysis showed 1p25, 1p36, monosomy 7 and 3q27 within normal ranges. Repeat DEB studies performed with PHA-stimulated peripheral blood on LCLs on two separate occasions revealed both spontaneous and diepoxybutane (DEB)-induced mean chromosomal breaks above the control range, but not in the typical FA range (Auerbach, 2003). In both cases, multiple cells exhibited chromosomal aberrations, including chromatid breaks, but no cells demonstrated chromatid exchanges. DEB studies on fibroblasts showed normal breakage at 0.01 μg/ml of DEB. However, metaphases could not be obtained at 0.1 μg/ml DEB, a result suggestive of a possible chromosomal breakage syndrome, although not highly suggestive of FA. Detailed complementation studies were initiated after this patient’s LCLs (RA3056) revealed substantial G2 arrest following melphalan treatment (Chandra, et al., 2005).
Cellular defects observed in patient-derived cells were complemented by FANCL gene
Patient-derived LCL (RA3056) and fibroblast cells (RA3100) were tested for cellular features characteristic of FA cells (Fig 1). RA3056 cells showed substantial melphalan-induced G2/M arrest which was higher than wild-type cells and lower than the previously reported LCLs from an FA-L patient (EUFA868) (Fig 1A). In addition, treatment with HU was associated with reduced FANCD2 monoubiquitination compared to wild-type cells, although some FANCD2-L form representing apparent monoubiquitination was still observed (Fig 1B). No spontaneous FANCD2 monoubiquitination was observed in the RA3056 cells. The FANCD2-L form was observed only in cells treated with HU. To determine the potential relevance of these findings with respect to cell growth, LCLs were exposed to increasing concentrations of MMC. RA3056 cells demonstrated higher MMC sensitivity than HCKE wt cells, but the reduction in survival was less severe than EUFA868 cells (Fig 1C). Finally, the level of chromosomal breakage was almost 50% lower than that of EUFA868 cells (Fig 1D). The RA3100 fibroblast cells also showed substantial melphalan-induced G2/M arrest (data not shown).
Figure 1. Cellular defects observed in RA3056 cells were complemented by FANCL.
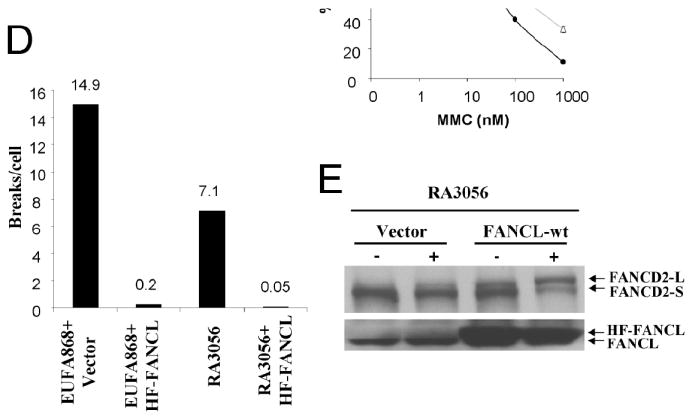
A. Cell cycle analysis of wild-type cells (HCKE), FA-L (EUFA868), patient-derived LCLs (RA3056) and RA3056 cells expressing functional FANCL. The RA3056 cells showed a higher number of G2/M arrested cells, and this defect was corrected by expressing functional FANCL. The number of cells arrested in G2 is denoted as a percentage above the graphs. B. Immunoblot analysis showing reduced FANCD2 monoubiquitination in RA3056 cells compared to that of wild-type (HCKE) cells. C. Survival curve showing MMC sensitivity of wild-type (HCKE), FA-L (EUFA868), RA3056 and RA3056 stably expressing functional FANCL (RA3056+HF-FANCL). Note that the RA3056 cells are more sensitive compared to wild-type cells, but they show more resistance compared to FA-L cells, a defect which is corrected by expressing functional FANCL. D. Bar diagram showing chromosome breakage analysis data of EUFA868 and RA3056 cells with or without functional FANCL correction. While the RA3056 cells showed higher chromosome breakage, this was corrected by FANCL.
In order to find the defective complementation group, RA3056/RA3100 cells were transduced with retroviral vectors, each carrying FANCA, FANCC, FANCE, FANCF, FANCG or FANCL. The transduced cells were exposed to melphalan, and cell cycle analysis was carried out using flow cytometry. A reduction of the G2/M fraction was observed only in the FANCL-transduced cells (Fig. 1A). Transduction of FANCA, FANCC, FANCE, FANCF and FANCG genes could not complement cell cycle defect of RA3056 and RA3100 cells (data not shown). RA3056 cells expressing functional FANCL were assayed for FANCD2 monoubiquitination, MMC sensitivity and chromosomal breakage (Fig. 1). Ectopic expression of FANCL results in correction of defects in basal and HU-induced monoubiquitination of FANCD2 (Fig. 1E). The cells became resistant to MMC (Fig. 1C), and there was a substantial reduction in chromosomal breakage (Fig. 1D).
Mutations in FANCL
Sequencing of RA3056 genomic DNA and cDNA of FANCL revealed bi-allelic mutations (Fig. 2). An in-frame 3-bp deletion mutation, c.1007_1009delTAT (p.Ile336_Cys337delinsSer), was observed in exon 12 involving two adjacent codons, 336 and 337. The deletion was in the PHD/RING-finger domain and resulted in the loss of one of two amino acids, isoleucine-336, and conversion of cysteine-337 to serine. The other allele carried a 4-bp duplication, c.1095_1098dupAATT (p.Thr367AsnfsX13), located in exon 14. The duplication was just outside the RING-finger domain and resulted in a frameshift. The new protein formed as a result would be predicted i) to carry a frameshift with a series of 12 missense amino acids and ii) to be three amino acids longer than the wild-type FANCL (Fig. 2). Sequencing of parental DNA revealed that the c.1007_1009delTAT was present in the mother’s genomic DNA and that the c.1095_1098dupAATT was present in the father’s genomic DNA.
Figure 2. Mutations observed in FANCL gene.
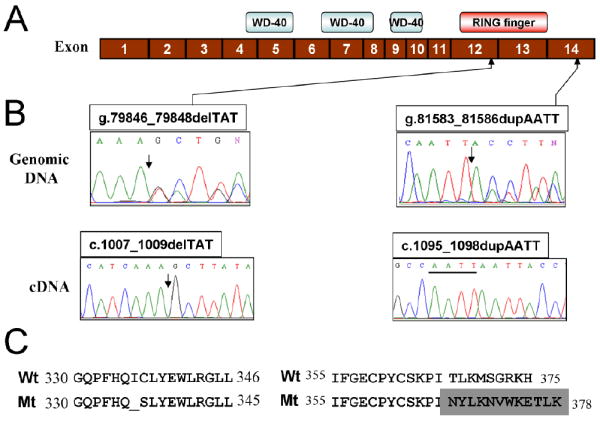
A. Schematic diagram of FANCL cDNA showing WD-40 repeats and PHD/RING-finger domains. The positions of the mutations are marked by upward arrows. The numbers in the boxes represent exon numbers. B. Sequence chromatogram showing the altered region in genomic DNA and cloned cDNA obtained from RA3056 cells. The site of mutations is indicated by a downward arrow. The duplication is underlined. C. The altered sequence of FANCL protein caused by mutation.
3-bp deletion mutation is a null mutation
The 3-bp deletion (c.1007_1009delTAT) was tested for its functional effects on cell cycle, FANCD2 monoubiquitination, MMC sensitivity and chromosomal breakage (Fig. 3). Ectopic expression of HF-FANCL-delTAT in EUFA868 cells, which lack FANCL protein, could not correct any of these cellular defects (Fig. 3). The cells showed substantial G2/M arrest, (Fig. 3A), no FANCD2 monoubiquitination (Fig. 3B), increased sensitivity to MMC (Fig. 3C), and a high rate of chromosome breakage (Fig. 3D). Because the 3-bp deletion mutation is in the RING-finger domain, and not in the region that is required for interaction with other core complex proteins, we hypothesize that this mutation may behave as a dominant negative. In order to confirm this, we overexpressed HF-FANCL-delTAT in HeLa S3 cells and assessed the effect on FANCD2 monoubiquitination (Fig. 4A). Overexpression of this mutant in HeLa S3 did not alter the FANCD2 monoubiquitination levels suggesting that this mutation is not a dominant negative. That the nature of this mutation apparently lacks a dominant negative characteristic is a consequence of reduced binding with FANCA, FANCG, FAAP100 and FANCM in both HeLa S3 cells and EUFA868 cells (Fig 4B). This lack of binding can be correlated with complete lack of FANCD2 monoubiquitination in EUFA868 cells overexpressed with HF-FANCLdelTAT (Fig 4B).
Figure 3. Effect of mutations on the cellular phenotype.
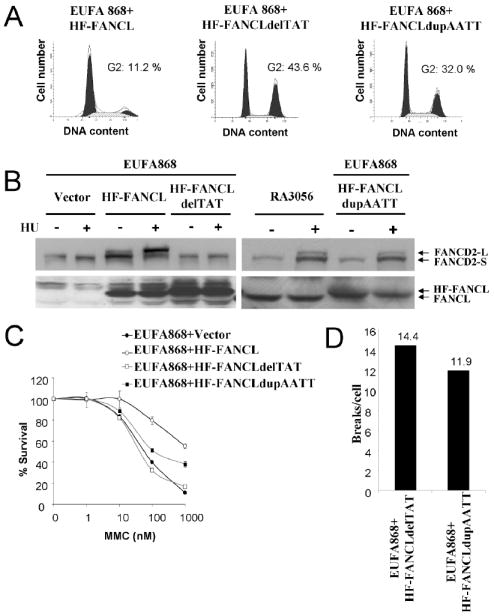
EUFA868 cells were transduced with virus carrying functional FANCL (HF-FANCL), FANCL with 3-bp deletion (HF-FANCLdelTAT) or 4-bp insertion (HF-FANCLdupAATT). A. Cell cycle analysis shows that the 3-bp deletion failed to complement these cells and that the 4-bp duplication showed partial restoration of this defect. B. Immunoblot analysis showing FANCD2 monoubiquitination. The FANCD2-L form was completely absent in cells transduced with 3-bp deletion, but the 4-bp duplication showed FANCD2-L form that was similar in intensity toRA3056 cells. C. Survival curve showing MMC sensitivity. The cells expressing 3-bp del mutant show similar sensitivity to EUFA868 cells, and the cells expressing 4-bp duplication show partial sensitivity. D. Bar diagram showing chromosome breakage analysis data.
Figure 4.
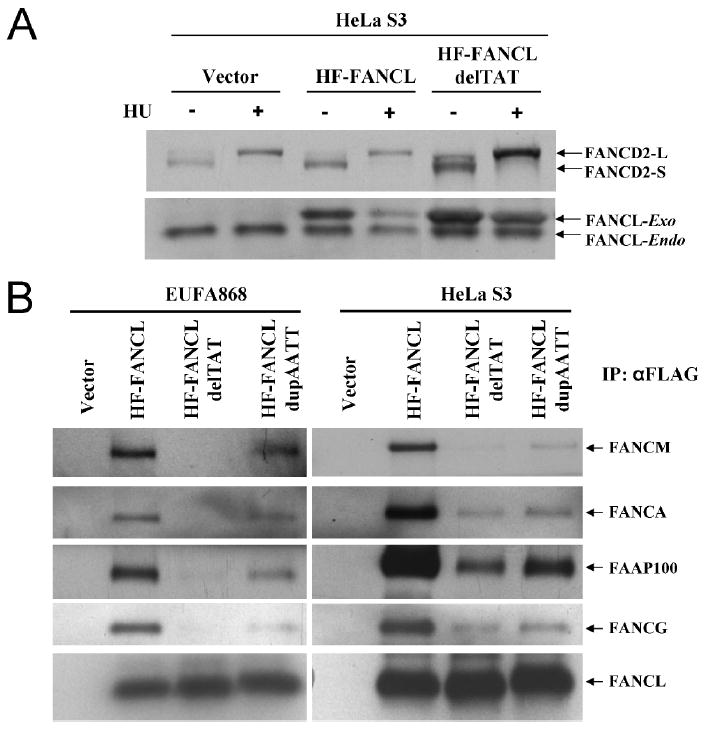
A. Immunoblot analysis showing FANCD2 monoubiquitination in HeLa S3 cells transduced with vector alone, HF-FANCL or HF-FANCLdelTAT. Over expression of 3-bp deletion does not abolish FANCD2-L band. B. Immunoblot showing immunoprecipitated complex from FA-L or HeLa S3 cells transduced with vector alone, HF-FANCL, HF-FANCLdelTAT or HF-FANCLdupAATT and probed with FANCA, FANCG, FANCL, FANCM and FAAP100 antibodies. The 3-bp del shows negligible or weak interaction, and the 4 bp duplication shows weak to moderate interaction compared to wild-type FANCL with core complex proteins.
4-bp duplication mutation is a hypomorphic mutation
Similarly, the 4-bp duplication (c.1095_1098dupAATT) was tested for its functional effects on cellular phenotypes. In contrast to HF-FANCLdelTAT, ectopic expression of HF-FANCL-dupAATT in EUFA868 cells resulted in partial correction of G2/M arrest (Fig. 3A). This partial phenotype appeared similar to RA3056 cells (Fig 1A). Also, FANCD2 monoubiquitination was similar to RA3056 cells (Fig. 3B). MMC sensitivity and chromosomal defects also showed partial correction (Fig. 3C and D). In summary, the phenotype of the cells was intermediate when compared to EUFA868 cells transduced with vector alone or with HF-FANCL and was similar to RA3056 cells (Fig. 1). We also checked the ability of the HF-FANCL-dupAATT to interact with other FA proteins. The HF-FANCL-dupAATT mutant was able to bind weakly, but with higher affinity compared with the 3bp-del, with FANCA, FANCG, FAAP100 and FANCM (Fig. 4B). The weak binding can be correlated with weak FANCD2 monoubiquitination and reduced radial chromosomes (Fig. 3).
DISCUSSION
FA patients display a wide range of clinical variability. This clinical variability not only arises from genotypic difference, but also from difference in ethnic and individual genetic background, as well as environmental factors (Adachi, et al., 2002). There are several clinical phenotypes associated with FA, including developmental delay, microcephaly, cafe-au-lait spots and cancer predisposition, which overlap with another chromosome instability disorder, Nijmegen breakage Syndrome (NBS; MIM# 251260) (Gennery, et al., 2004). Not only the clinical phenotypes, but some NBS cells also show an overlap of the biological phenotypes such as sensitivity to DNA crosslinker agents MMC and DEB (Gennery, et al., 2004). This overlap in clinical and biological phenotypes makes diagnosis challenging in some cases (Gennery, et al., 2004). An early accurate diagnosis of the disorder results in the proper management of the disease (Chandra, et al., 2005). The patient described in this study shows mild clinical features that include developmental delay, cafe-au-lait spot and mild hypocellularity which are common features to both FA and NBS. Accurate diagnosis in such cases is challenging, and, in the present case, it is much more challenging because the standard chromosome breakage test yielded results that were lower than the typical FA range (Table 1). In such cases, accurate diagnosis can only be achieved by using an “FA Complementation assay”. The FA Complementation assay is an efficient method for the identification of specific complementation groups by exploiting a key characteristic of FA cells which undergo arrest in the G2/M phase of cell cycle in response to DNA damaging agents (Shimamura, et al., 2002; Chandra, et al., 2005). By using this assay, not only can an accurate diagnosis can be made, but the patient can also be classified to a particular FA subtype thus confirming the validity of FA complementation assay for accurate diagnosis and sub-typing in all FA cases. The severity of clinical and biological phenotype associated with the FA-L complementation group could not be assessed as a result of small number of subjects.
Table 1.
Chromosomal breakage data of patient’s DEB test performed on peripheral blood lymphocytes compared to FA and control rangea
| Spontaneous | DEB-treated | |
|---|---|---|
| Patient | 0.06 | 0.14 |
| FA | 0.02-0.85 | 1.10-23.9 |
| Non-FA | 0.00-0.12 | 0.00-0.36 |
Patient-derived mutations of several FA genes have been instrumental in delineating the functional domains and residues important for interaction with other members of the FA pathway. As a consequence of small patient number and, hence, only limited samples of patient-derived mutations in the FANCL gene, mutations were generated in vitro in residues that were predicted to be important for FANCL function or interaction or both (Meetei, et al., 2003a; Gurtan, et al., 2006; Medhurst, et al., 2006). Previous studies suggest that the PHD/RING-finger domain is not required for FA core complex assembly and have further suggested a role of PHD/RING-finger domain distinct from FA complex stabilization (Gurtan, et al., 2006). Co-immunoprecipitation experiments using point mutation W341A in FANCL PHD/RING-finger domain show that although this tryptophan is important for ubiquitin ligase activity in vitro it was not important for complex assembly (Gurtan, et al., 2006). Similarly, Medhrust et al. concluded that this residue, Trp-341, is not important for the binding of FANCL to FANCB and was able to correct the MMC sensitivity defect of EUFA868 cells (Medhurst, et al., 2006). In addition, the point mutations E340A and C362A in PHD/RING-finger domain have no effect on either FANCB interaction or MMC sensitivity. However, there are some residues in the PHD/RING-finger domain that are essential for both ubiquitin ligase activity and interaction with FANCL. The Cys-307 mutation not only abolishes ubiquitin ligase activity in vitro, but it also shows a very weak interaction with other core complex proteins (Meetei, et al., 2003a). Studies done with a large number of FANCA mutants suggest that the FA pathway is activated differentially and that the differential activation of FA pathway may partly account for the phenotypic variation in FA patients (Adachi, et al., 2002). The mutation identified in the PHD/RING-finger domain during this study of delTAT, which results in the loss of Isoleucine-336 and conversion of Cysteine-337 to serine also results in complete loss of FANCD2 monoubiquitination in vivo and shows a negligible interaction with core complex (Fig 4B), thus behaving as a null mutation. The observed loss of FANCD2 monoubiquitination could result from the loss of interaction with the core complex or it could also result from loss of structural integrity of the PHD/RING-finger domain. The dupAATT mutation, which occurs just outside the PHD/RING-finger domain, behaves like a hypomorphic mutation in that it result in a frameshift and shows only partial activity. The loss of correct amino acid sequence at the C-terminal region may result in reduced binding of the FANCL with core complex and a decrease in FANCD2 monoubiquitination in RA3056 cells. The mild FA phenotype observed in the patient seems to result from the partially active FANCL allele (FANCLdupAATT) and may not be associated with the FA-L complementation group. A high G2/M arrest and sensitivity to MMC and DEB suggest that the patient’s cells are at high risk of developing cancers due to spontaneous or induced DNA damage.
Acknowledgments
We thank the FA patient and his family for the donation of the samples. We also thank the Translational Trails Development and Support Laboratory (TTDSL), Viral Vector Core and Fluorescent Activated Cell Analyzing and Sorting facility of Cincinnati Children’s Research Foundation.
Contract grant sponsor: National Institutes of Health Research Grants; Contract grant numbers: R01 HL084082 (to A.R.M.), R37HL32987 (to A.D.A.) and HL081499 (to D.A.W and TTDSL). Fanconi Anemia Research Fund (FARF) grant and American Society of Hematology (ASH) grant (to A.R.M.).
Footnotes
Communicated by Dominique Stoppa-Lyonnet
References
- Adachi D, Oda T, Yagasaki H, Nakasato K, Taniguchi T, D’Andrea AD, Asano S, Yamashita T. Heterogeneous activation of the Fanconi anemia pathway by patient-derived FANCA mutants. Hum Mol Genet. 2002;11(25):3125–34. doi: 10.1093/hmg/11.25.3125. [DOI] [PubMed] [Google Scholar]
- Alpi A, Langevin F, Mosedale G, Machida YJ, Dutta A, Patel KJ. UBE2T, the Fanconi anemia core complex, and FANCD2 are recruited independently to chromatin: a basis for the regulation of FANCD2 monoubiquitination. Mol Cell Biol. 2007;27(24):8421–30. doi: 10.1128/MCB.00504-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach AD. Diagnosis of Fanconi anemia by Diepoxybutane Analysis. In: Dracopoli NC, Haines JL, Korf BR, Moir DT, Morton CC, Seidman CE, Seidman JG, Smith DR, editors. Current Protocols in Human Genetics. Hoboken, NJ: John Wiley & Sons, Inc; 2003. pp. 8.7.1–8.7.15. [Google Scholar]
- Chandra S, Levran O, Jurickova I, Maas C, Kapur R, Schindler D, Henry R, Milton K, Batish SD, Cancelas JA, et al. A rapid method for retrovirus-mediated identification of complementation groups in Fanconi anemia patients. Mol Ther. 2005;12(5):976–84. doi: 10.1016/j.ymthe.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Gennery AR, Slatter MA, Bhattacharya A, Barge D, Haigh S, O’Driscoll M, Coleman R, Abinun M, Flood TJ, Cant AJ, et al. The clinical and biological overlap between Nijmegen Breakage Syndrome and Fanconi anemia. Clin Immunol. 2004;113(2):214–9. doi: 10.1016/j.clim.2004.03.024. [DOI] [PubMed] [Google Scholar]
- Gurtan AM, Stuckert P, D’Andrea AD. The WD40 repeats of FANCL are required for Fanconi anemia core complex assembly. J Biol Chem. 2006;281(16):10896–905. doi: 10.1074/jbc.M511411200. [DOI] [PubMed] [Google Scholar]
- Jacquemont C, Taniguchi T. The Fanconi anemia pathway and ubiquitin. BMC Biochem. 2007;8(Suppl 1):S10. doi: 10.1186/1471-2091-8-S1-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutler DI, Singh B, Satagopan J, Batish SD, Berwick M, Giampietro PF, Hanenberg H, Auerbach AD. A 20-year perspective on the International Fanconi Anemia Registry (IFAR) Blood. 2003;101(4):1249–56. doi: 10.1182/blood-2002-07-2170. [DOI] [PubMed] [Google Scholar]
- Levitus M, Joenje H, de Winter JP. The Fanconi anemia pathway of genomic maintenance. Cell Oncol. 2006;28(1-2):3–29. doi: 10.1155/2006/974975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling C, Ishiai M, Ali AM, Medhurst AL, Neveling K, Kalb R, Yan Z, Xue Y, Oostra AB, Auerbach AD, et al. FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. Embo J. 2007;26(8):2104–14. doi: 10.1038/sj.emboj.7601666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida YJ, Machida Y, Chen Y, Gurtan AM, Kupfer GM, D’Andrea AD, Dutta A. UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol Cell. 2006;23(4):589–96. doi: 10.1016/j.molcel.2006.06.024. [DOI] [PubMed] [Google Scholar]
- Medhurst AL, Laghmani el H, Steltenpool J, Ferrer M, Fontaine C, de Groot J, Rooimans MA, Scheper RJ, Meetei AR, Wang W, et al. Evidence for subcomplexes in the Fanconi anemia pathway. Blood. 2006;108(6):2072–80. doi: 10.1182/blood-2005-11-008151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet. 2003a;35(2):165–70. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, Steltenpool J, Stone S, Dokal I, Mathew CG, et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet. 2005;37(9):958–63. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, Hoatlin ME, Wang W. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol. 2003b;23(10):3417–26. doi: 10.1128/MCB.23.10.3417-3426.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura A, Montes de Oca R, Svenson JL, Haining N, Moreau LA, Nathan DG, D’Andrea AD. A novel diagnostic screen for defects in the Fanconi anemia pathway. Blood. 2002;100(13):4649–54. doi: 10.1182/blood-2002-05-1399. [DOI] [PubMed] [Google Scholar]
- Tremblay CS, Huang FF, Habi O, Huard CC, Godin C, Levesque G, Carreau M. HES1 is a novel interactor of the Fanconi anemia core complex. Blood. 2008;112(5):2062–70. doi: 10.1182/blood-2008-04-152710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8(10):735–48. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PEM. Improving Sequence Variant Descriptions in Mutation Databases and Literature Using the Mutalyzer Sequence Variation Nomenclature Checker. Hum Mutat. 2008;29(1):6–13. doi: 10.1002/humu.20654. [DOI] [PubMed] [Google Scholar]