

Abstract
Methylmercury (MeHg) has been previously shown to affect neurotransmitter release. Short-term synaptic plasticity (STP) is primarily related to changes in the probability of neurotransmitter release. To determine if MeHg affects STP development, we examined STP forms in the visual cortex of rat following in vivo MeHg exposure. Neonatal rats received 0 (0.9% NaCl), 0.75 or 1.5 mg/kg/day MeHg subcutaneously for 15 or 30 days beginning on postnatal day 5, after which visual cortical slices were prepared for field potential recordings. In slices prepared from rats treated with vehicle, field excitatory postsynaptic potentials (fEPSPs) evoked by paired-pulse stimulation at 20 - 200 ms inter-stimulus intervals showed a depression (PPD) of the second fEPSP (fEPSP2). PPD was also seen in slices prepared from rats after 15 day treatment with 0.75 or 1.5 mg/kg/day MeHg. However, longer duration treatment (30 days) with either dose of MeHg resulted in paired-pulse facilitation (PPF) of fEPSP2 in the majority of slices examined. PPF remained observable in slices prepared from animals in which MeHg exposure had been terminated for 30 days after completion of the initial 30 day MeHg treatment, whereas slices from control animals still showed PPD. MeHg did not cause any frequency- or region-preferential effect on STP. Manipulations of [Ca2+]e or application of the GABAA receptor antagonist bicuculline could alter the strength and polarity of MeHg-induced changes in STP. Thus, these data suggest that low level postnatal MeHg exposure interferes with the developmental transformation of STP in the visual cortex, which is a long-lasting effect.
Keywords: Methylmercury, extracellular microelectrode recording, synaptic transmission, short-term synaptic plasticity, visual cortical slice of rat
Introduction
One of the most conspicuous and consistent neurological signs seen in humans and animals following acute and chronic exposure to methylmercury (MeHg), a prominent environmental neurotoxicant, is visual functional deficits (Hunter and Russell, 1954; Takeuchi et al., 1959; Tokuomi and Okajima, 1961; Bakir et al., 1973; Rustam and Hamdi, 1974; Chang, 1980; Mukuno et al., 1981; Merigan et al., 1983; Rice and Gilbert, 1982, 1990; Eto, 1997; Clarkson et al., 2003; Burbacher et al., 2005; Ekino et al., 2007). These are characterized by concentric constriction of the visual fields and reduced visual acuity. Neuropathological evidence suggests that these disorders are primarily central in origin and caused by the region-selective lesions in the visual cortex of the cerebrum. However, the exact mechanisms underlying the MeHg-induced visual functional deficits remain poorly understood.
Although region-selective neuropathlogical lesions in the visual cortex appear to be responsible for MeHg-induced irreversible concentric constriction of the visual fields, it does not necessarily mean that the pathological changes are the initial causes of MeHg-induced visual functional deficits. In fact, evidence indicates that some of the visual functional disturbances such as reduced visual acuity and impaired spatial contrast sensitivity occurred in the absence of or preceding pathological changes in the visual cortex in animals (Mattson et al. 1981; Rice and Gilbert, 1982, 1990; Burbacher et al., 2005) and humans with MeHg poisoning (Mukuno et al., 1981). Furthermore, some of the visual deficits demonstrated in patients (Bakir et al., 1973; Rustam and Hamdi, 1974, Korogi et al., 1994) and animals with MeHg poisoning (Berlin et al., 1975; Merigan et al., 1983) were reversible. Therefore, it is possible that the visual functional disturbances, particularly at low level MeHg exposure, are initially the result of a temporary disruption of neuronal function in the visual system, i.e. disruption by MeHg of synaptic transmission in the visual cortex. Consistent with this assumption is that children with prenatal and early childhood exposure to low levels of MeHg via consumption of seafood showed delayed visual-evoked potentials (Murata et al., 1999). Similarly, rats (Zenick, 1976; Dyer et al., 1978) and dogs (Mattsson et al., 1981), following exposure to low levels of MeHg, also demonstrated altered visual-evoked potentials prior to appearance of any overt signs of neurotoxicity. Thus, it is conceivable that low level MeHg may affect synaptic transmission in the visual pathway. In other words, effects of MeHg on the visual synaptic function should, at least in part, contribute to MeHg-induced visual functional deficits. To date, however, no direct evidence for effects of MeHg on synaptic transmission in the visual cortex has been demonstrated.
The efficacy of excitatory synaptic transmission between neurons is activity-dependent. When a presynaptic neuron fires repetitively, the strength or amplitude of postsynaptic responses can be enhanced (facilitation, augmentation, post-tetanic potentiation) or decreased (depression). Synaptic facilitation or depression that occurs on a timescale of tens to hundreds of milliseconds is often referred to short-term synaptic plasticity (STP) (for review, see Thomson, 2000; Zucker and Regehr, 2002; Blitz et al., 2004; Xu-Friedman and Regehr, 2004). Visual cortical neurons express both long- and short-term synaptic plasticity (Kirkwood and Bear, 1994; Ramoa and Sur, 1996; Berardi et al., 2003; Yoshimura et al., 2003; Daw et al., 2004; Jia et al., 2004, 2006) which regulate the strength and dynamics of synaptic transmission in the visual cortex. In the developing visual cortex, STP is thought to play a critical role in activity-dependent refinement of synaptic connections, including ocular dominance plasticity during an early critical period (Rumpel et al., 1998; Reyes and Sakmann, 1999; Fagiolini and Hensch, 2000). STP is also believed to play a role in specific temporal filtering of excitatory synaptic inputs (Varela et al., 1997; Abbott et al., 1997; Buonomano, 2000; Fortune and Rose, 2000; Abbott and Regehr, 2004). Thus, STP provides a mechanism to allow neurons to transmit information by responding selectively to changes in the spatial and temporal patterns of presynaptic inputs arriving at synapses.
STP in the visual cortex is regulated developmentally. During early postnatal development, STP shifts gradually from predominant short-term facilitation in juvenile visual cortex to predominant short-term depression in adult visual cortex of rat (Ramoa and Sur, 1996). Maturation of GABAergic inhibition is thought to be involved in the developmental transformation of STP in the visual cortex (Ramoa and Sur, 1996; Jia et al., 2004). Theoretically, any factor that interferes with this developmental transformation of STP could potentially alter the STP forms in the visual cortex and thus affect the visual synaptic function. MeHg has been previously shown to disrupt central synaptic transmission in brain slices in vitro (Yuan and Atchison, 1993, 1995, 1997, 1999, 2003 and 2007). More relevantly, MeHg has also been shown to affect preferentially GABAergic systems in vivo (Shaw et al., 1975; O’Kusky, 1985) and in vitro (Yuan and Atchison, 1997). Therefore, it is possible that early postnatal MeHg exposure interferes with the developmental transformation of STP in the visual cortex. As a first step in testing this, the present study was designed specifically to examine if early postnatal exposure of animals to MeHg in vivo alters STP in the visual cortex. Specifically, we sought to determine: 1) if MeHg exposure in vivo interferes with the maturation of STP in the primary visual cortex; 2) if effects of MeHg on STP demonstrate any region-selectivity (anterior versus posterior visual cortex), and 3) if intracellular Ca2+ homeostasis and GABAergic inhibitory systems are involved in MeHg-induced changes in STP in the visual cortex. Using extracellular microelectrode recording technique in acutely isolated visual cortical slices of rat, we showed that postnatal exposure of animals to low levels of MeHg in vivo reversed the developmental form of STP from prominent paired-pulse depression (PPD) to paired-pulse facilitation (PPF) in layer II/III neurons of the visual cortex of rat, and this change was long-lasting. Our results suggest that early postnatal exposure of animals to low levels of MeHg interferes with developmental changes in STP in the visual cortex of rat.
Materials and Methods
Chemicals and Solutions
Methylmercury chloride (MeHg) was obtained from ICN Biomedical Inc. (Costa Mesa, CA). It was dissolved in physiological saline to the final concentration of 4 mg/ml as a stock solution. Solutions for sc injection were diluted from the stock solution with 0.9% NaCl based on the principle of equal volume injection per kg of body weight. 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX), amino-5-phosphonopentanoic acid (APV), (-)-bicuculline methiodide and tetradotoxin (TTX) were all purchased from Sigma Chemical Co. (St Louis, MO).
Brain “slicing” solution contained (in mM): 125 NaCl; 2.5 KCl; 4 MgCl2; 1.25 KH2PO4; 26 NaHCO3; 1 CaCl2 and 20 D-glucose, pH 7.35-7.4 at room temperature when saturated with 95% O2 -5% CO2.
Artificial cerebrospinal fluid (ACSF) contained (in mM): 125 NaCl; 2.5 KCl; 1 MgCl2; 1.25 KH2PO4; 26 NaHCO3; 2 CaCl2 and 25 D-glucose; pH 7.35-7.4 when saturated with 95% O2 -5% CO2. In experiments involving use of low Ca2+-containing ACSF, the standard 2 mM Ca2+ in ACSF was replaced by equimolar or 4 mM Mg2+, whereas in experiments in which a higher external Ca2+ concentration ([Ca2+]e) was used, extra Ca2+ was added to ACSF to reach a final Ca2+ concentration of 5 mM.
Animals and Treatment
All animal handling procedures complied with the National Institutes of Health of the USA guidelines on animal care and use and were approved by Michigan State University Institutional Animal Use and Care Committee. Male Sprague-Dawley rat pups of postnatal day 4 (PND4) along with mother rats were purchased from Charles River Laboratories (Wilmington, MA). Animals were provided with water and food ad libitum with 12:12 light/dark cycle. 72 rat pups at age PND5 from 12 litters were randomly assigned to three groups: control (0.9% NaCl), 0.75 and 1.5 mg/kg/day MeHg. The MeHg dosages were chosen based on previous literature (Kakita et al., 2000; Shigematsu et al., 2000; Sakamoto et al., 2004) in which the selected doses for the present study did not induce any significant pathological alterations in the visual cortex. Physiological saline or MeHg was injected subcutaneously (sc) beginning at PND5 and continued for 15 (PND20) or 30 consecutive days (PND35), respectively (Figure 1A). During exposure, the body weight, general health and behavior of animals were monitored daily. After completion of 15 or 30 day injections, 8 rats from each group were sacrificed for brain slice preparations. The remaining 8 rats of each group were kept for an additional 30 days without any further treatment. These rats were euthanized at 60 days and brain slices were prepared using identical procedures to those for rats at 15 or 30 days. These subgroups were designed specifically to examine if any effect of MeHg on synaptic transmission is reversible after an extra 30 days of tissue “clearance” of MeHg.
Figure 1.

Schematic illustration of the procedure for in vivo MeHg treatment and representative demonstration of field potentials recorded in layer II/III neurons of visual cortical slices of rat. (A) MeHg exposure procedure: rats began to receive subcutaneous injections (sc) of 0.9% NaCl or 0.75 or 1.5 mg/kg/day MeHg at PND5 and continued for 15 (PND20) or 30 (PND35) consecutive days, respectively. At PND20 or 35, 8 rats from each group were sacrificed for brain slice preparations, respectively. The remaining 8 rats of each group were kept for an extra 30 days without any further treatment after completion of the 30 day injections (Clearance). At PND65, these animals were also sacrificed for brain slice preparations. (B) A representative field excitatory postsynaptic potential (fEPSP) was recorded from layer II/III neurons by a single-pulse stimulation of layer IV neurons of a visual cortical slice of rat. Following the stimulation artifact (S) are two downward components. The first one is a population spike corresponding to compound action potentials activated by antidromic stimulation of axons of layer II/III neurons (Anti-PS), which is TTX-sensitive. The second one is the glutamatergic fEPSP that is mediated by orthodromic stimulation of presynaptic fibers. (C) A representative recording from another visual cortical slice demonstrates sensitivity of fEPSPs to the AMPA receptor antagonist CNQX and NMDA receptor antagonist APV. In this case, the Anti-PS component was smaller relative to the fEPSP component before CNQX and APV treatment (Control, black trace). Application of 10 μM CNQX plus 50 μM APV blocked the fEPSP but not the Anti-PS (Grey trace). (D) Paired-pulse responses in a slice prepared from a control animal with 30 day injection of 0.9% NaCl demonstrated paired-pulse depression (PPD). Note: the second stimulus-evoked fEPSPs (fEPSP2) was smaller than the first stimulus-evoked fEPSP (fEPSP1) although the two Anti-PSs were virtually identical. (E) paired-pulse responses in a slice prepared from a rat receiving injections of 1.5 mg/kg/day MeHg for 30 days demonstrated paired-pulse facilitation (PPF). The fEPSP2 was larger than fEPSP1 although the two Anti-PSs were similar. All recordings were made from the posterior visual cortical slices. The same is true for all the rest of figures unless specified.
Pre- and postnatal stress including postnatal handling, neonatal maternal separation and environmental changes can induce long-term neurochemical and neurobehavioral effects (Vallée et al., 1997, 1999; Huot et al., 2001; Kikusui and Mori, 2009). For the purpose of control, we also included three age-matched control rats, respectively, for each age subgroup. The age-matched animals stayed with their mothers until weaning at PND21 and received no daily handling or injection. At PND20, 35 or 65, these rats were sacrificed and brain slices were prepared and examined using the same procedures as described above.
Neurodevelopment
General postnatal physical development and behavior of animals were examined daily. Neurological reflex development of rats was assessed beginning on PND5 using the method described by Šlamberová et al. (2006), Mesquita et al. (2007) and Sugawara et al. (2008).
Surface righting reflex
An individual rat pup was placed on its back and the time needed to turn over and restore its normal prone position was recorded.
Air righting reflex
An individual rat pup was held on its back in mid-air 30 - 40 cm above a soft surface before being released. The position of rat pup reaching the soft surface was recorded. The reflex was considered to be achieved when the rat pup landed on the surface with all four limbs.
Eye opening
The days on which bilateral eye opened were recorded.
Visual cliff avoidance test
Visual cliff avoidance test was performed beginning on PND14 (after rats opened eye) in a home-made clear Perpex box (80 × 80 cm square × 40 cm high, open-topped) as described by Glynn et al. (2003). Three of the four walls were covered with black and white checkerboard. Half of the base was also covered with the same pattern of checkerboard (bench side). The box was placed on a table with the bench side on the table, while the clear half base (cliff side) was suspended in air over the table edge so that it created a cliff appearance without an actual drop-off. A similar patterned checkerboard was placed on the floor underneath the box (80 cm) to emphasize the cliff drop-off. Individual rats were placed on the bench side in the box and allowed to move freely. The activity of rats in the box was observed and video taped for 5 min. The latency to and number of individual rats that ventured over the cliff side was recorded. Occasionally, visual cliff avoidance tests were simply performed on a table edge as described by Dong et al. (1996).
Tissue Hg content measurement
Total Hg content in visual cortex was analyzed by Chemical Solution LTD (Mechanicsburg, PA). In brief, 500 mg brain tissues was digested in 2.5 ml HNO3 and heated in an open graphite block at 95 ± 5 °C for 30 min. After cooling to room temperature, the sample ash was re-dissolved in 2 ml H2O2 and heated again at 95 ± 5 °C for 15 min. The digested sample was finally dissolved in 25 ml deionized water. Total Hg content in samples was then analyzed using Inductively Coupled Plasma-Mass Spectrometers. The detection limit was set at 0.02 ppm.
Preparation of visual cortical slices
At PND20, 35 or 65, after completion of designed durations for injections or “clearance”, the rat brains were quickly isolated after decapitation and bisected sagittally. One side was frozen immediately for later total Hg measurement or molecular biology experiments. The other side was used for brain slice preparation. Thick coronal visual cortical slices (350 - 400 μm) were prepared in ice-cold, oxygenated “slicing” solution saturated with 95% O2 /5% CO2 using procedures modified from those described by Kirkwood and Bear (1994). A brain tissue block containing the visual cortex portion was glued onto the tissue pedestal of an NVMSL1 motorized advanced Slicer (World Precision Instruments, Inc. Sarasota, FL) with cyanocrylate glue. After removing the first 400 - 600 μm from the occipital pole, the tissue block between atlas 45 and 35 transverse levels (Swanson, 1998) was transected transversely to produce 5 - 8 coronal slices. Slices were then transferred to a home-made holding chamber containing ACSF (see Solutions) aerated continuously with 95% O2 /5% CO2, and incubated at room temperature of 22-25 °C for at least 60 min before electrophysiological recording. The slices in the approximate range of atlas 43 - 45 levels were considered as the posterior visual cortical slices, whereas slices in the approximate range of atlas 35 - 37 levels were considered as the anterior visual cortical slices in the present study.
Extracellular recordings of field potentials
One slice was transferred to a recording chamber and mechanically fixed using a “U” shape anchor made of a platinum wire frame with nylon mesh. The slice was superfused (2 - 4 ml/min) continuously with ACSF by gravity. The laminar structure of the primary visual cortex (V1 region) in slices was visually identified under low power (4 x) magnification of an upright microscope. The field potentials were recorded from layer II/III neurons using a glass electrode with resistance of 2 - 7 MΩ when filled with ACSF and evoked by a broken-tip glass stimulating electrode in layer IV. Isolated stimuli were generated from a Grass S88 stimulator (Grass, Inc, Quincy, MA) at a frequency of 0.2 Hz, 0.1 ms duration and varied voltages that caused 50 - 60% of maximum amplitude of responses. The stimulating electrode had an impedance of 1 - 4 MΩ when filled with ACSF. Consistent with previous reports (Varela et al., 1997), the field potentials of layer II/III neurons in the primary visual cortex evoked by single pulse stimulation of layer IV neurons usually consist of two components as shown in Figure 1B-C. Short-term synaptic plasticity (STP) was examined by using a paired-pulse stimulation paradigm with pulse-pulse or inter-stimulus intervals (ISIs) varying from 20 to 200 ms without changing stimulation intensity. Figure 1D and E show two representative recordings of paired-pulse evoked responses in layer II/III neurons. Each recording consists of two fEPSPs (fEPSP1 and fEPSP2) that were activated by consecutive and identical stimulus pulses at 50 ms ISI. If the ratio of fEPSP2/fEPSP1 was less than 1 (Figure 1D), the paired-pulse evoked response was defined as paired-pulse depression (PPD). In contrast, if the ratio of fEPSP2/fEPSP1 was larger than 1 (Figure 1E), then the paired-pulse evoked response would be defined as paired-pulse facilitation (PPF). Recordings were made primarily in the posterior visual cortical slices. However, for examining any potential regional-specific differential effect of MeHg on visual synaptic function, similar recordings were made in the anterior slices. The shapes of field potentials, particularly the antidromic component, recorded from layer II/III neurons by stimulating layer IV neurons usually vary somewhat depending on the ages of animals, the region of the visual cortex and the relative positions of stimulating and recording electrode. In general, the antidromically-evoked responses (Anti-PS) appear larger in slices prepared from younger animals such as PND10-20 and from the anterior regions of the visual cortex compared with those of adults and posterior visual cortical slices. Data acquisition was as described in Yuan and Atchison (1993). Field potential signals from recording electrodes were amplified using a Model 3000 AC/DC differential amplifier (A-M Systems, Inc., Carlsborg, WA) and filtered at 5 kHz with a 4-pole low-pass Bessel filter and digitized at 10 - 20 kHz for later off-line analysis using pClamp 9.0 program (Axon Instruments, Union City, CA). All experiments were carried out at room temperature of 22 - 25 °C.
Statistical analysis
Data were analyzed statistically using Student’s t test or one-way analysis of variance (ANOVA) for MeHg dose- or time-dependent measures, unless otherwise specified. Dunnett’s procedure was used for post hoc comparisons (Tukey-Kramer multiple comparison test). In the case of testing statistical significance of MeHg-induced PPF change, Fisher’s exact test was used. Values were considered statistically significant at p ≤ 0.05. The data are presented as mean ± standard deviation (SD). Multiple recordings were routinely made from one or several slices from any given animal, but the values were then averaged and counted as sample # (n) = 1. Each experiment was replicated in a minimum of five animals; the actual number of replicates for each experiment is listed in the corresponding figure legend.
Results
Low level MeHg exposure did not significantly affect the rate of body weight gain and general behaviors
There was no significant difference in the rate of body weight gain of rats among the three groups during MeHg treatments and “clearance”. The average body weights for control, 0.75 and 1.5 mg/kg/day MeHg groups were 50.2 ± 6.6, 50.5 ± 5.5 and 48.4 ± 5.4 g at PND20 (p > 0.05, n = 24), 145.4 ± 11.9, 144.0 ± 11.4 and 135.8 ± 11.7 g at PND35 (p > 0.05, n = 16), and 373.2 ± 31.9, 378.2 ± 18.14 and 347.9 ± 14.9 g at PND65 (p > 0.05, n = 8), respectively.
MeHg did not appear to cause any overt abnormal physical and behavioral changes. Surface- and air-righting reflex tests revealed no difference among control and MeHg-treated animals. All animals opened both eyes around PND14 and no difference could be identified between control and MeHg-treated animals. Hair growth and gloss showed no difference between control and MeHg-treated animals. Cliff avoidance tests also revealed no significant difference between control and MeHg-exposed animals. No apparent MeHg exposure-related hindlimb crossing was observed during MeHg injections and the designed extra 30 days of MeHg “clearance”. Thus, MeHg exposure did not appear to cause any significant effect on the rate of body weight gain, general physical development and behavior under the present experimental conditions.
Total Hg contents in the visual cortex were MeHg exposure level-dependent
At the time of completion of 15 or 30 days of injections or 30 day injection plus an extra 30 days of MeHg “clearance”, animals were sacrificed and total Hg content in brain tissues (primarily the visual cortex) was analyzed. The data are summarized in Table 1. As shown in table 1, animals with exposure to 0.75 or 1.5 mg/kg/day MeHg for 15 or 30 days showed a higher total Hg content in the visual cortex in a dose- dependent manner compared with the MeHg-free controls. After 30 days of MeHg “clearance”, however, the tissue total Hg content decreased dramatically. The differences among the three groups at the designed time points are statistically significant (p < 0.05). Thus, the total Hg contents in the visual cortex increased in a MeHg exposure dose-dependent manner.
Table 1.
Total Hg contents in rat visual cortex following MeHg treatment
| MeHg (mg/kg/day) | 15 Days (μg/g) | 30 Days (μg/g) | 60 Days (μg/g)* |
|---|---|---|---|
| Control | <0.02*** | <0.02 | <0.02 |
| 0.75 | 0.70 ± 0.31** | 0.86 ± 0.46** | 0.08 ± 0.03** |
| 1.5 | 1.19 ± 0.79** | 1.87 ± 0.87** | 0.18 ± 0.06** |
Rats were injected sc with 0.9% (Control) or 0.75 or 1.5 mg/kg/day MeHg for 15 or 30 consecutive days or 30 days plus another 30 days clearance (60 days) at the time when the tissue samples were collected.
Differences are statistically significant among three groups (p < 0.05), but no statistical difference was seen between 0.75 and 1.5 mg/kg/day groups at 15 days. Values are Mean ± SD (n = 4 - 8).
The samples were measured at a detection limit of 0.02 μg/g.
MeHg reversed the developmental course of STP in the visual cortical slices of rat
One previous study has shown that STP in the visual cortex of rat underwent a transformational change from predominant PPF to predominant PPD during the first few postnatal weeks (Ramoa and Sur, 1996). To test the possibility that MeHg interferes with this developmental transition of STP forms, we examined changes in paired-pulse evoked synaptic responses in layer II/III neurons of V1 region in the posterior visual cortical slices following early postnatal MeHg exposure. In agreement with previous findings (Ramoa and Sur, 1996; Jia et al., 2004, 2006), all recordings in slices prepared from control animals that were given 0.9% NaCl for 15 (PND20) or 30 days (PND35) showed PPD (Figure 2). The mean ratios of fEPSP2/fEPSP1 at 50 ms ISI were 0.75 ± 0.11 and 0.75 ± 0.09 (n = 6 - 8), respectively. Consistently, recordings in slices prepared from age-matched rats at PND20, 35 and 65 all showed PPD responses (data not shown), suggesting that daily handling of animals under our experimental conditions did not induce any developmental change in STP in the visual cortex. All recordings in slices prepared from rats exposed to 0.75 mg/kg/day MeHg for 15 days also showed PPD. Whereas most recordings in slices prepared from rats exposed to 1.5 mg/kg/day MeHg for 15 days showed PPD, recordings from one animal showed PPF. The mean fEPSP2/fEPSP1 ratios were 0.73 ± 0.12 and 0.79 ± 0.34 (n = 5 - 6), respectively, for 15 day treatments with 0.75 and 1.5 mg/kg/day. However, all recordings in slices prepared from rats that were exposed either to 0.75 or 1.5 mg/kg/day MeHg for 30 days showed PPF. The average fEPSP2/fEPSP1 ratios were 2.19 ± 1.07 and 1.32 ± 0.09 (n = 6 - 8), respectively. Qualitatively, the changes in STP forms (PPD vs PPF) between control and MeHg-treated animals are statistically significant (p < 0.01, Fisher’s exact test). Quantitatively, however, the statistically significant differences in the fEPSP2/fEPSP1 ratios are seen only between control and low MeHg dose groups (p < 0.05, ANOVA), not between control and high MeHg dose groups (Figure 3). Interestingly, PPF remained observable in slices prepared from rats even after termination of MeHg exposure for 30 days following completion of the 30 days of consecutive injections of 0.75 or 1.5 mg/kg/day MeHg, whereas slices prepared from control animals still showed PPD. The average fEPSP2/fEPSP1 ratios were 0.86 ± 0.11, 1.70 ± 0.54 and 1.47 ± 0.26 (n = 5 - 6), respectively, for control, 0.75 and 1.5 mg/kg/day groups at PND65. Thus, these data suggest that in vivo MeHg exposure interfered with the developmental transition of STP forms in the visual cortex and this effect is long-lasting.
Figure 2.

MeHg altered the developmental form of short-term synaptic plasticity (STP) in the visual cortex of rat in a MeHg exposure time-dependent manner. Representative recordings of paired-pulse responses in slices prepared from rats treated with 0.9% NaCl (Top), 0.75 (Middle) and 1.5 mg/kg/day MeHg (Bottom), respectively, for 15 or 30 days or 30 days injections plus extra 30 days of “clearance”(total 60 days). Each trace is a representative example of 6 - 8 individual experiments.
Figure 3.

Comparison of effects of MeHg on fEPSP2/fEPSP1 ratios. Data shown are the ratios of fEPSP2/fEPSP1 evoked by paired-pulse stimulation at 50 ms inter-pulse interval. Each value is the mean ± SD of 5 - 8 animals. The asterisk indicates a value significantly different than control (p < 0.05).
Both PPD and PPF are frequency- or inter-pulse interval-dependent. To determine if MeHg-induced changes in STP are also affected by ISI, we next examined paired-pulse synaptic responses evoked at varied ISIs. As shown in Figure 4 are three representative recordings in slices prepared from animals that were injected with 0.9% NaCl, 0.75 or 1.5 mg/kg/day MeHg for 30 consecutive days, respectively. As expected, in the range of 20 - 200 ms ISIs, all paired-pulse responses in slices prepared from control animals were PPD in nature and reducing the ISI from 200 to 20 ms tended to strength the PPD interactions (Figure 4. Note: for simplicity, the responses evoked by 200 ms ISI are not shown in Figure 4). In contrast, recordings in slices prepared from rats that were exposed to 0.75 or 1.5 mg/kg/day MeHg for 30 days demonstrated increased PPF interactions (Figure 4). A similar pattern was seen in PND65 rats (data not shown). These data suggest that the MeHg-induced PPF remained frequency-dependent. Interestingly, the mean fEPSP2/fEPSP1 ratios at different ISIs were all greater at 0.75 mg/kg/day MeHg than those at 1.5 mg/kg/day MeHg at both PND35 and 65 although the differences between the two doses are not statistically significant (p > 0.05, data not shown).
Figure 4.

MeHg-induced PPF in visual cortical slices remained frequency-dependent. Representative recordings of paired-pulse responses in slices prepared from rats with sc injection of 0.9% NaCl (A), 0.75 (B) and 1.5 mg/kg/day MeHg (C), respectively, for 30 days. Paired-pulse responses were evoked by varying stimulus inter-pulse intervals (ISIs) from 100 - 20 ms (from top to bottom in each panel). Each trace is representative of 4 - 5 individual experiments.
MeHg did not appear to affect STP in the anterior and posterior visual cortical slices differentially
Pathological studies have previously demonstrated that MeHg-induced neuropathological lesions in the visual cortex are region-selective (Hunter and Russell, 1954, Takeuchi et al., 1959, 1962; Eto, 1997; Eto et al., 2001, 2002). To test whether the effect of MeHg on synaptic function such as STP is also region-preferential, we next examined changes in paired-pulse evoked synaptic responses in the anterior visual cortical slices. In general, paired-pulse evoked synaptic responses recorded in the anterior slices were similar to those observed in the posterior slices prepared from control and MeHg-treated animals i.e., PPD in control slices and PPF in MeHg-treated slices (data not shown). However, recordings from the anterior slices showed some variability and inconsistency. Although the majority of recordings from the anterior slices prepared from control animals demonstrated PPD, occasionally, a few recordings (1/12 and 2/17 recordings in slices prepared from 7 rats at PND20 or PND35, respectively) actually showed PPF. On the other hand, PPD were also exhibited in 5/17 and 1/12 recordings in the anterior slices prepared from 0.75 mg/kg/day MeHg-treated rats (8 rats) at both PND35 and PND65 and in 4/14 recordings in slices prepared from 1.5 mg/kg/day MeHg-treated rats (6 rats) at PND65, respectively. No PPD was seen in slices prepared from rats treated with 1.5 mg/kg/day at PND35. Compared with those values obtained from the posterior visual cortical slices shown in Figure 2, the mean fEPS2/fEPS1 ratios obtained from anterior slices are not statistically significantly different from those in posterior slices (p > 0.05). Thus, these data suggest that MeHg does not appear to cause a region-preferential effect on STP in the primary visual cortex of rat
MeHg-induced change in STP in the visual cortex is extracellular Ca2+ concentration-dependent
STP is generally attributed to alterations of release of transmitter presynaptically. Since this process depends on presynaptic Ca2+ entry, an alteration of extracellular Ca2+ concentration ([Ca2+]e) will be expected to result in a change in the polarity and strength of STP. Thus, the next series of experiments was designed specifically to determine the sensitivity of MeHg-induced PPF to changes in [Ca2+]e. Figure 5 shows two representative recordings in slices prepared from control and MeHg-treated rats, respectively. In controls, reducing the external solution from the standard 2 mM Ca2+-containing ACSF to a low Ca2+-containing ACSF (see Solutions) not only decreased the amplitudes of both fEPSP1 and fEPSP2, but also converted STP from PPD into a PPF-like response, due to a more significant reduction of amplitude of fEPSP1 than that of fEPSP2 (Figure 5A). Eventually, both fEPSP1 and fEPSP2 disappeared completely (data not shown in Figure 5A). However, these changes were reversible because re-incubation of slices with standard ACSF completely restored the PPD. In contrast, in slices prepared from MeHg-treated animals, reducing [Ca2+]e only decreased the amplitudes of paired-pulse evoked synaptic responses, but did not change the PPF form (Figure 5B). On the contrary, increasing [Ca2+]e from 2 mM to 5 mM not only strengthened PPD in slices prepared from control animals (Figure 6A), but also switched PPF to PPD in slices prepared from MeHg-treated animals (Figure 6B). Thus, these data suggest that MeHg-induced PPF was possibly a result of presynaptic functional plasticity that is extracellular Ca2+ entry-dependent.
Figure 5.

Reducing extracellular Ca2+ concentrations changed the STP form in control slices but not MeHg-exposed slices. (A) A slice prepared from a rat with injections of 0.9% NaCl for 30 days showed a typical PPD response. Data are shown at selected time points when the slice was incubated in the standard 2 mM Ca2+-containing ACSF (ACSF), in a low Ca2+-containing solution for 1 or 2 min (Low Ca2+ 1 or 2 min), and in standard ACSF again for 10 min (ACSF 10 min). (B) Under the similar recording conditions as described for A, the slice prepared from a rat with injections of 0.75 mg/kg/day MeHg for 30 days demonstrated a typical PPF when incubated in the standard ACSF. Similarly, data are shown at selected time points when the slice was incubated in the standard ACSF (ACSF), in a low Ca2+-containing solution for 2 or 4 min (Low Ca2+ 2 or 4 min), and in standard ACSF again for 10 min (ACSF 10 min). Each trace is representative of 6 - 7 individual experiments.
Figure 6.

Elevating extracellular Ca2+ concentrations changed the STP forms in both control and MeHg-exposed slices. (A) In the standard ACSF, the slice prepared from a rat with injections of 0.9% NaCl for 60 days showed a typical PPD response. Data are shown at selected time points when the slice was incubated in the standard ACSF (ACSF), in a 5 mM Ca2+-containing solution for 1 or 2 min (5 mM Ca2+ 1 or 2 min), and in standard ACSF again for 10 min (ACSF 10 min). (B) A representative slice prepared from a rat with injections of 0.75 mg/kg/day MeHg for 60 days demonstrated a typical PPF when incubated in the standard ACSF. Data are shown at selected time points when the slice was incubated in the standard ACSF (ACSF), in a 5 mM Ca2+-containing solution for 5 or 10 min (5 mM Ca2+ 5 or 10 min), and finally in standard ACSF again for 10 min (ACSF 10 min). Each trace is representative of 6 - 7 individual experiments.
MeHg-induced changes in STP could be associated with changes in GABAergic function
MeHg affects both glutamatergic and GABAergic synaptic transmission (Yuan and Atchison, 1997, 2007), but, GABAergic systems appear to be more sensitive to MeHg than are glutamatergic neurons (Shaw et al., 1975; O’Kusky, 1985; Yuan and Atchison, 1997). Therefore, it is possible that MeHg-induced PPF could be associated with effects of MeHg on GABAergic system because GABA is involved in developmental changes in STP in the visual cortex (Ramao and Sur, 1996; Jia et al., 2004). Thus, the goal of next experiment was to determine if GABAA receptor-mediated synaptic inhibition is involved in MeHg-induced PPF. Theoretically, if MeHg-induced PPF is due to suppression by MeHg of GABAergic inhibitory responses, application of the GABAA receptor antagonist bicuculline should further enhance MeHg-induced PPF or eliminate PPF depending on whether or not bicuculline preferentially affects the second stimulus pulse-evoked GABAergic inhibitory responses. First, we examined effects of 10 μM bicuculline on STP in slices prepared from control animals. Figure 7 demonstrates two representative recordings in slices prepared from control and MeHg-treated rats, respectively. In the control (Left), PPD was clearly seen at 50 ms ISI when this slice was incubated in standard ACSF. Application of 10 μM bicuculline for 1 - 3 min initially increased amplitude of both fEPSP1 and fEPS2, but the increase was more prominent in fEPSP2 than fEPSP1. Therefore, the STP form was switched from PPD to PPF (Bic 1 - 3 min). Prolonged bicuculline treatment over 5 min usually induced a giant and long depolarization response following the first stimulus-evoked response. This giant response always masked the second stimulus-evoked responses - fEPSP2 (data not shown). All these changes were fully recovered after washing the slice with standard ACSF for 10 - 30 min as shown in Figure 7. This pattern of change was consistently seen in all 9 slices prepared from 6 control animals. In contrast, the representative slice prepared from a rat treated with 0.75 mg/kg/day for 30 days plus 30 days of MeHg “clearance” showed a clear PPF response (Right, ACSF), bicuculline also initially enhanced amplitudes of all components of paired-pulse responses including anti-PS and both fEPSP1 and fEPSP2 (Right, Bic 1 min). However, compared with the response prior to bicuculline treatment, the percentage of increase in fEPSP2 was relatively smaller than that in fEPSP1, which thus actually decreased the fEPSP2/fEPSP1 ratio from 4.1 prior to bicuculline treatment to 1.4 after the first min of bicuculline application in this case. This result suggests that a portion of GABAergic inhibition must have been lost in MeHg-treated slices prior to bicuclline treatment. Continuous bicuculline treatment also caused a prolonged, giant depolarization with multiple spikes superimposing on it during the first stimulus pulse (Right, Bic 3 min), which can be clear seen with an ISI at 100 ms (100 ms ISI). At the same time, the MeHg-induced PPF became a PPD-like response, which is possibly due to falling of the second stimulus into the giant, prolonged depolarization (the absolute or relative action potential refractory period). Again, this change has been consistently seen in all slices tested (n = 4). Similarly, all changes were reversible with washout of bicuculline (data not shown). Thus, these data suggest that GABAA receptor-mediated inhibitory synaptic responses are involved in STP in the visual cortex of rat and effects of MeHg on GABAA receptor-mediated synaptic response might partially contribute to MeHg-induced changes in STP.
Figure 7.
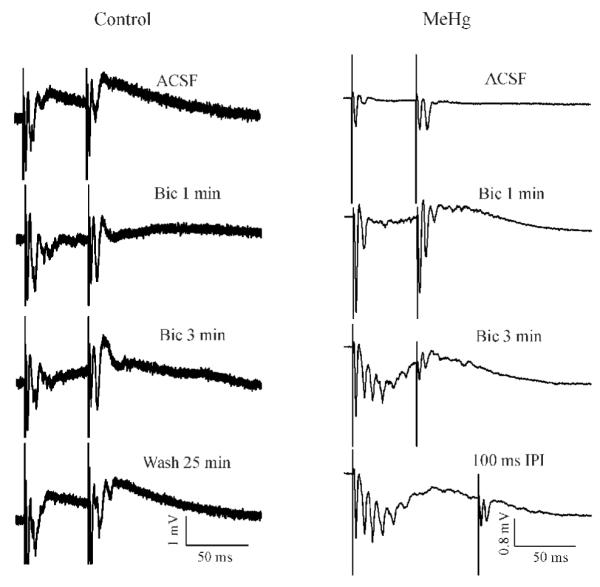
The GABAA receptor antagonist bicuculline altered the pattern of paired-pulse interactions in control and MeHg-treated slices. (Left) Time course of effect of bicuculline on PPD response in a representative slice prepared from a PND35 control rat. Data are shown before (ACSF) and at selected time points after 10 μM bicuclline (Bic) treatment or washing with bicuculline-free ACSF. In this case, all paired-pulse responses were evoked at 50 ms IPI. (Right) Paired-pulse responses were recorded in a representative slice prepared from a PND65 rat after completion of 30 day injections of 0.75 mg/kg/day MeHg from PND5 to PND35. Data are shown before (ACSF) and at selected time points after bicuculline treatment. All responses shown were evoked by paired-pulse stimulation at 50 ms ISI, except at 3 min bicuclline treatment, response is evoked by 100 ms ISIs were also shown. Each trace is a representative of 5 - 8 individual experiments.
Discussion
The main finding of the present study is that MeHg reversed the course of developmental transition of STP form in layer II/III neurons of the primary visual cortex of rat. Unlike the normal postnatal developmental course in which the STP form in the visual cortex of rat is switched gradually from PPF in juvenile to PPD in young adult rats, the STP forms in the visual cortex of young adult rats were reversed to PPF instead of PPD following MeHg exposure. This change remained observable even after MeHg exposure had been terminated for 30 days. In addition, the MeHg-induced PPF appeared to be sensitive to changes in [Ca2+]e: reducing or increasing [Ca2+]e could reverse the polarity and strength of MeHg-induced changes in STP. In addition, block of GABAA receptor by its antagonist bicuculline also changed the polarity of STP. To the best of our knowledge, this is the first evidence that early postnatal MeHg exposure interferes with the developmental changes in STP in the visual cortex.
In the present study, we did not see a significant change in body weight gain or overt signs of neurological disorders in rats following MeHg exposure. However, because only insensitive methods were used for examining potential effects of MeHg on general physical development and behavior of animals in this study, the current results do not rule out the possibility that MeHg may induce subtle abnormal changes in neurodevelopment and neurobehaviors at these exposure levels. In fact, studies conducted by Newland’s group (Rasmussen and Newland, 2001; Day et al., 2005; Reed et al., 2006; Paletz et al., 2007) have consistently shown that exposure of rats to low levels of MeHg during gestation and lactation induced neurobehavioral changes although MeHg did not affect food or water consumption, litter size and body weight gain of rats at those doses (Newland and Reile, 1999). Comparing with the rat brain Hg contents measured in those studies, which are 0.5 - 10 ppm at birth (PND0) and 0.04 - 0.5 ppm at weaning (PND21), respectively (Newland and Reile, 1999), the brain Hg contents in the present study are within the range of brain Hg contents that induced behavioral changes. Therefore, more sensitive methods should be used in the future study in order to monitor the effects of low level MeHg exposure on neurodevelopment.
Ramoa and Sur (1996) have previously shown that STP development in the visual cortex of rat was age-dependent. Using intracellular recordings in neurons in visual cortical slices of rat, they showed that PPF was present in about half of the neurons tested in PND5 - 10 rats, whereas the remaining neurons studied at these ages did not show any paired-pulse interactions. However, the proportion of neurons displaying PPF decreased progressively in an age-dependent manner, whereas the proportion of neurons displaying PPD increased gradually as animals became more mature. After the first postnatal month, about 50% of the neurons tested demonstrated PPD, whereas PPF was rarely seen. Consistently, using field potential recording technique in vivo, Jia et al. (2004, 2006) also showed that PPD was prominent in the visual cortex of rats at PND20, 30 and >60 and the strength of PPD increased in an age-dependent manner. In agreement with those findings, our recordings from control rats including the age-matched controls at PND20, 35 and 65 almost all expressed PPD, whereas PPF was rarely seen. Thus, our data further confirm that the primary form of STP, at least at layer IV-II/III synapses, in the primary visual cortex of normal mature rats was PPD.
However, the STP form in the visual cortex of rat was altered following MeHg exposure in vivo during the early postnatal development. Although almost all recordings in slices prepared from MeHg-treated rats at either dose for 15 days showed PPD, all recordings in slices prepared from rats receiving MeHg treatment for 30 days demonstrated PPF. These results suggest that postnatal MeHg exposure affects STP in the primary visual cortex and this effect is MeHg exposure time-dependent. Surprisingly, the fEPSP2/fEPSP1 ratios evoked by 50 ms and all other ISIs in slices prepared from rats receiving injection of 0.75 mg/kg/day MeHg for 30 days were slightly larger than those from rats receiving injections of 1.5 mg/kg/day MeHg for the same duration of exposure. But, the differences between the two doses are not statistically significant (p > 0.05). Therefore, no clear dose-dependent effect of MeHg on STP in the visual cortex was observed in the present study. However, further confirmation with a wider range of MeHg exposure doses, more time points and lager sample size may be needed in order to determine the dose-dependent effect of MeHg on STP in the visual cortex.
One interesting observation in the present study is that MeHg-induced PPF in the visual cortex remained observable even after MeHg exposure had been terminated for 30 days and total Hg contents in tissue had decreased significantly. Clearly, these results suggest that the effect of MeHg on STP in the primary visual cortex was long-lasting. The mechanisms responsible for this long-lasting effect remain to be determined. The exchangeable nature of MeHg-induced PPF by manipulation of [Ca2+]e (Figure 5 and 6) suggests that the MeHg-induced PPF should be a result of functional change, perhaps a long-term change in the process of transmitter release. However, possible involvement of postsynaptic factors could not be ruled out completely because postsynaptic factors are known to contribute to STP (Rumpel et al., 1998; Xu-Friedman and Regehr, 2004). It has been previously shown that prenatal exposure of animals to 5 mg/kg/day during days 6 to 9 of gestation caused striking dendritic spine abnormalities in the offspring brains, which consisted of a reduction of stubby and mushroom-shaped spines and a predominance of long, thin and tortuous spines. In addition, the number of spines was greater and the diameter of the main apical dendrites was reduced (Stoltenburg-Didinger and Markwort, 1990). Similarly, neonatal exposure of rats to 5 mg/kg/day MeHg for 17 - 20 days induced significant changes in synaptic connections and number and morphology of dendritic spines of neurons in the visual cortex (O’Kusky, 1985). Such severe alterations of the postsynaptic microstructures might not occur under our experimental conditions, but subtle changes in synaptic circuits, dendritic spines, postsynaptic receptor density or sensitivity could still be possible. If it occurs, it may explain why the MeHg-induced change in STP is long-lasting. Clearly, further investigation with longer durations for MeHg “clearance” may be necessary to determine if this effect is reversible.
Frequency- or use-dependence is a general property of STP including both PPD and PPF. Consistent with this property, both PPD from the control rats and PPF from MeHg-treated rats in our study remained frequency-dependent. Moreover, MeHg did not appear to cause any special effect on PPF at a preferential frequency or ISI. Thus, these data suggest that MeHg does not alter the synaptic functional property that is responsible for generation of frequency-dependent STP in the visual cortex of rat.
One of the most striking features of MeHg poisoning in humans and animals is the region- or layer-selective neuropathological lesions in the CNS (Hunter and Russell, 1954, Takeuchi et al., 1959, 1962; Shaw et al., 1975; Merigan et al., 1983; O’Kusky, 1985; Wakabayashi et al., 1995; Eto et al., 2001, 2002). In the present study, our results did not suggest any region-selective effect of MeHg on STP. The lack of region-selective effects of MeHg on STP could be due to differences in brain anatomy between rodents and human or nonhuman primates or age-dependent differential responses of animals to MeHg exposure. Region-specific pathological changes are usually seen in adult brains, whereas a more diffuse pathological change is often seen in young brains following MeHg exposure (Takeuchi et al., 1959; Chang, 1977; Eto, 1997).
STP is generally considered to be due to changes in the probability for transmitter release from presynaptic terminals (for review, see Thomson, 2000; Zucker and Regehr, 2002; Xu-Friedman and Regehr, 2004). One important theory about PPF is the “residual Ca2+ hypothesis”: facilitation is caused by residual Ca2+ remaining in the presynaptic terminals after the conditioning stimulus. In the case of paired-pulse stimulation, extracellular Ca2+ enters nerve terminals through voltage-gated Ca2+ channels during the first action potential. At shorter ISIs, some residual Ca2+ will remain in the nerve terminals when the second action potential arrives. This will result in the summation of the “residual Ca2+” and the Ca2+ that entered during the second impulse leading to enhanced probability for transmitter release and facilitation of the second synaptic response. Thus, STP is [Ca2+]e-dependent because altering [Ca2+]e affects Ca2+ influx and the probability of transmitter release (for review see Thomson, 2000; Zucker and Regehr, 2002; Xu-Friedman and Regehr, 2004). The question is how MeHg exposure induces PPF in the visual cortex. One of the most consistent effects of MeHg in vitro is disruption of Ca2+ homeostasis regulation leading to an increase in [Ca2+]i in cells (Hare et al., 1993; Hare and Atchison, 1995a, b; Marty and Atchison, 1997, 1998; Limke and Atchison, 2002; Limke et al., 2003; Edwards et al., 2005; Yuan and Atchison, 2007). MeHg does this by inducing release of Ca2+ from intracellular stores and influx of extracellular Ca2+ 1(Hare et al., 1993; Hare and Atchison, 1995a, b; Marty and Atchison, 1997, 1998; Limke and Atchison, 2002; Limke et al., 2003, 2004; Edwards et al., 2005). In addition, MeHg also appears to inhibit Ca2+ uptake by mitochondria (Levesque and Atchison, 1991), which could presumably slow down Ca2+ removal from nerve terminals. Therefore, one possible mechanism underlying MeHg-induced PPF is that MeHg-induced increase in [Ca2+]i plus decrease in Ca2+ removal somehow facilitates accumulation of more residual Ca2+ in nerve terminals, which leads to increased transmitter release and subsequent PPF.
The probability for neurotransmitter release is a key factor in determining the polarity of STP (Thomson, 2000; Zucker and Regehr, 2002; Xu-Friedman and Regehr, 2004). Synapses with high release probability usually display PPD because the readily releasable pool of vesicles is significantly depleted after the first action potential, whereas synapses with low release probability usually demonstrate PPF because depletion of readily releasable pool of vesicles is minimal after the first action potential. However, the release probability itself is in turn affected by [Ca2+]i in nerve terminals. Any change in [Ca2+]i or [Ca2+]e is expected to change the probability for transmitter release and thus STP polarity. Consistently, incubation of control slices in low [Ca2+]e-containing ACSF not only reduced the baseline transmission (amplitudes of fEPSP1 and fEPSP2), but also relieved the depression and converted the paired pulse responses from PPD to PPF. This conversion was possibly due to decreased probability for neurotransmitter release at synapses in a low [Ca2+]e environment. As described above, synapses with low release probability then displayed PPF. Similarly, lowering [Ca2+]e reduced amplitudes of both fEPSP1 and fEPSP2 recorded in MeHg-treated slices and led to decreased probability for transmitter release. However, the reduced release probability would not further change the STP that was already in PPF form in MeHg-treated slices. In contrast, elevation of [Ca2+]e not only enhanced the strength of PPD in control slices, but also converted PPF into PPD in MeHg-treated slices. These changes could be due to increased extracellular Ca2+ entry by elevation of [Ca2+]e leading to higher release probability, which in turn caused rapid depletion of readily releasable pool of vesicles after the first stimulus and thus PPD. Therefore, these data suggest that MeHg-induced PPF in the visual cortex should still be attributed to a functional change in the process of presynaptic transmitter release.
The maturation of GABAergic inhibition has been implicated to play a role in the developmental switch of STP from PPF to PPD in the visual cortex during early postnatal life, because development of PPD was temporally correlated closely with the maturation of GABAergic inhibition (Ramoa and Sur, 1996). Furthermore, application of GABAB receptor antagonist 2-hydroxy-saclofen partially and reversibly reduced PPD in mature cortical neurons (Ramoa and Sur, 1996). Application of the GABAA receptor antagonist bicuculline also significantly reduced PPD in an age-dependent manner in rat visual cortex (Jia et al., 2004). These results suggest that both GABAA and GABAB receptors are involved in PPD development in the visual cortex of rat. Interestingly, application of bicuculline not only reduced PPD, but also sometimes transformed PPD into PPF in brain slices isolated from temporal lobe epileptic patients (Uruno et al., 1995). Consistent with those observations, our data showed that bicuculline almost always initially switched the STP from PPD to PPF in control slices. Thus, these data again confirm that GABAA receptor plays a role in PPD generation in the visual cortex.
The question is how effect of MeHg on GABAergic inhibition could be linked to MeHg-induced PPF. Since GABAergic neurons, particular the aspinous or sparsely-spinous GABAergic interneurons in the visual cortex, are highly sensitive to MeHg-induced degeneration (Shaw et al., 1975; O’Kusky, 1985 O’Kusky and Mcgeer, 1985; O’Kusky et al., 1988), it is possible that a preferential effect of low level MeHg on GABAergic neuronal function, causes a reduction of GABA release and disinhibition of glutamatergic excitatory synaptic responses pre- and postsynaptically, may contribute to MeHg-induced PPF. Presynaptic disinhibition of GABAB receptor inhibition would cause membrane depolarization and increased Ca2+ entry and glutamate release from nerve terminals; postsynaptic disinhibition of inhibitory actions of GABAA and/or GABAB receptors would further increase postsynaptic responses. Both actions plus MeHg-induced increase in [Ca2+]i could thus result in PPF. However, further investigation of the effect of MeHg on GABAergic interneurons and its role in MeHg-induced changes in STP is needed.
STP has been proposed to be involved in a number of important functions including specific temporal filtering of synaptic inputs and transforming temporal information into spatial code during early postnatal development (Varela et al., 1997; Abbott et al., 1997; Rumpel et al., 1998; Reyes and Sakmann, 1999; Buonomano, 2000; Fagiolini and Hensch, 2000; Fortune and Rose, 2000; Abbott and Regehr, 2004). In the primary visual cortex, temporal filter properties are believed to play a critical role in adaptation and gain control, velocity tuning and direction selectivity (for review, see Abbott and Regehr, 2004). Therefore, it has been suggested that short-term synaptic depression may underlie the mechanism of contrast adaption (Chance et al., 1998). Theoretically, any manipulation that affects STP in the visual cortex may potentially cause deficits in temporal processing in the visual cortical synapses. Thus, it is possible that effects of MeHg on developmental maturation of STP in the visual cortex interfere with temporal and spatial visual functions. Consistent with this assumption, it has been previously shown that pre- and postnatal exposure of monkeys to low levels of MeHg induced deficits in spatial and temporal visual function (Rice and Gilbert, 1982, 1990; Burbacher et al., 2005). In addition, patients with adult or prenatal MeHg exposure also showed impaired spatial contrast sensitivity (Mukuno et al., 1981). However, whether or not MeHg-induced deficits in temporal and spatial contrast visual function are directly associated with the effects of MeHg on STP in the visual cortex remains to be determined.
In conclusion, this study provides the first evidence that early postnatal exposure to low levels of MeHg altered the developmental form of STP in the primary visual cortex, which was a long-lasting effect. No region-preferential effect of MeHg on STP was observed in the present study. Multiple mechanisms including effects of MeHg on [Ca2+]i homeostasis and GABAergic systems are likely involved in the effects of MeHg on STP. Given the importance of STP in the establishment and modulation of synaptic connections and function of neurons in the visual cortex during the early postnatal development, the effects of MeHg on STP in the visual cortex might potentially be associated with MeHg-induced visual functional deficits following pre- or early postnatal exposure to low levels of MeHg.
Acknowledgements
We thank Dr. William D. Atchison for his constructive suggestions and generous laboratory equipment supports during preparation of this manuscript. We also thank Drs, Atchison and Ravindra Hajela for critical reviewing this manuscript. This work is supported by NIEHS grants R21ES013767 and Michigan State University funding 06-HBRI-II-616.
Footnotes
A preliminary report of part of these results was presented in an abstract form at the 25th International Neurotoxicity Conference, October 12-16, 2008, Rochester, NY.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
References
- Abbott LF, Regehr WG. Synaptic computation. Nature. 2004;431:796–803. doi: 10.1038/nature03010. [DOI] [PubMed] [Google Scholar]
- Abbott LF, Varela JA, Sen K, Nelson SB. Synaptic depression and cortical gain control. Science. 1997;275:220–224. doi: 10.1126/science.275.5297.221. [DOI] [PubMed] [Google Scholar]
- Bakir F, Damluji SF, Amin-Zaki L, Murtadha M, Khalidi A, al-Rawi NY, Tikriti S, Dahahir HI, Clarkson TW, Smith JC, Doherty RA. Methylmercury poisoning in Iraq. Science. 1973;181:230–241. doi: 10.1126/science.181.4096.230. [DOI] [PubMed] [Google Scholar]
- Berardi N, Pizzorusso T, Ratto GM, Maffei L. Molecular basis of plasticity in the visual cortex. Trends Neurosci. 2003;26:369–378. doi: 10.1016/S0166-2236(03)00168-1. [DOI] [PubMed] [Google Scholar]
- Berlin M, Grant CA, Hellberg J, Hellström J, Schültz A. Neurotoxicity of methylmercury in squirrel monkeys. Cerebral cortical pathology, interference with scotopic vision, and changes in operant behavior. Arch. Environ. Health. 1975;30:340–348. doi: 10.1080/00039896.1975.10666717. [DOI] [PubMed] [Google Scholar]
- Blitz DM, Foster KA, Regehr WG. Short-term synaptic plasticity: a comparison of two synapses. Nat. Rev. Neurosci. 2004;5:630–640. doi: 10.1038/nrn1475. [DOI] [PubMed] [Google Scholar]
- Buonomano DV. Decoding temporal information: A model based on short-term synaptic plasticity. J Neurosci. 2000;20:1129–1141. doi: 10.1523/JNEUROSCI.20-03-01129.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbacher TM, Grant KS, Mayfield DB, Gilbert SG, Rice DC. Prenatal methylmercury exposure affects spatial vision in adult monkeys. Toxicol. Appl. Pharmacol. 2005;208:21–28. doi: 10.1016/j.taap.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Chance FS, Nelson SB, Abbott LF. Synaptic depression and the temporal response characteristics of V1 cells. J Neurosci. 1998;18:4785–4799. doi: 10.1523/JNEUROSCI.18-12-04785.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LW. Neurotoxic effects of mercury--a review. Environ. Res. 1977;14:329–373. doi: 10.1016/0013-9351(77)90044-5. [DOI] [PubMed] [Google Scholar]
- Chang LW. Mercury. In: Spencer PS, Schaumburg HH, editors. Experimental and Clinical Neurotoxicology. The Williams and Wilkins Co.; New York: 1980. pp. 508–526. [Google Scholar]
- Clarkson TW, Magod l., Myers G. The toxicology of mercury-current exposure and clinical manifestations. N. Engl. .J Med. 2003;349:1731–1737. doi: 10.1056/NEJMra022471. [DOI] [PubMed] [Google Scholar]
- Daw N, Rao Y, Wang XF, Fischer Q, Yang Y. LTP and LTD vary with layer in rodent visual cortex. Vision Res. 2004;44:3377–3380. doi: 10.1016/j.visres.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Day JJ, Reed MN, Newland MC. Neuromotor deficits and mercury concentrations in rats exposed to methyl mercury and fish oil. Neurotoxicol teratol. 2005;27:629–641. doi: 10.1016/j.ntt.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Dong Z, Chen P, Li X-Q. Neurobehavioral study of prenatal exposure to hyperthermia combined with irradiation in mice. Neuroltoxicol Teratol. 1996;18:703–709. doi: 10.1016/s0892-0362(96)00127-4. [DOI] [PubMed] [Google Scholar]
- Dyer RS, Eccles CU, Annau Z. Evoked potential alterations following prenatal methyl mercury exposure. Pharmacol. Biochem. Behav. 1978;8:137–141. doi: 10.1016/0091-3057(78)90330-1. [DOI] [PubMed] [Google Scholar]
- Edwards JR, Marty MS, Atchison WD. Comparative sensitivity of rat cerebellar neurons to dysregulation of divalent cation homeostasis and cytotoxicity caused by methylmercury. Toxicol. Appl. Pharmacol. 2005;208:222–232. doi: 10.1016/j.taap.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Ekino S, Susa M, Ninomiya T, Imamura K, Kitamura T. Minamata disease revisited: an update on the acute and chronic manifestations of methyl mercury poisoning. J Neurol. Sci. 2007;262:131–144. doi: 10.1016/j.jns.2007.06.036. [DOI] [PubMed] [Google Scholar]
- Eto K. Pathology of Minamata disease. Toxicol. Pathol. 1997;25:614–623. doi: 10.1177/019262339702500612. [DOI] [PubMed] [Google Scholar]
- Eto K, Yasutake A, Kuwana T, Korogi Y, Akima M, Shimozeki T, Tokunaga H, Kaneko Y. Methylmercury poisoning in common marmosets-a study of selective vulnerability within the cerebral cortex. Toxicol. Pathol. 2001;29:565–573. doi: 10.1080/019262301317226375. [DOI] [PubMed] [Google Scholar]
- Eto K, Yasutake A, Korogi Y, Akima M, Shimozeki T, Tokunaga H, Juwana T, Kaneko Y. Methylmercuty poisoning in common marmosets-MRI findings and peripheral nerve leisions. Toxicol. Pathol. 2002;30:723–734. doi: 10.1080/01926230290166814. [DOI] [PubMed] [Google Scholar]
- Fagiolini M, Hensch TK. Inhibitory threshold for critical-period activation in primary visual cortex. Nature. 2000;404:183–186. doi: 10.1038/35004582. [DOI] [PubMed] [Google Scholar]
- Fortune ES, Rose GJ. Short-term synaptic plasticity contributes to the temporal filtering of electrosensory information. J Neurosci. 2000;20:7122–7130. doi: 10.1523/JNEUROSCI.20-18-07122.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn D, Bortnick RA, Morton AJ. Complexin II is essential for normal neurological function in mice. Human Mol. Genet. 2003;12:2431–1448. doi: 10.1093/hmg/ddg249. [DOI] [PubMed] [Google Scholar]
- Hare MF, Atchison WD. Methylmercury mobilizes Ca++ from intracellular stores sensitive to inositol 1,4,5-trisphosphate in NG108-15 cells. J Pharmacol. Exp. Ther. 1995a;272:1016–1023. [PubMed] [Google Scholar]
- Hare MF, Atchison WD. Nifedipine and tetrodotoxin delay the onset of methylmercury-induced increase in [Ca2+]i in NG108-15 cells. Toxicol. Appl. Pharmacol. 1995b;135:299–307. doi: 10.1006/taap.1995.1236. [DOI] [PubMed] [Google Scholar]
- Hare MF, McGinnis KM, Atchison WD. Methylmercury increases intracellular concentrations of Ca++ and heavy metals in NG108-15 cells. J Pharmacol. Exp. Ther. 1993;266:1626–1635. [PubMed] [Google Scholar]
- Hunter D, Russell DS. Focal cerebellar and cerebellar atrophy in a human subject due to organic mercury compounds. J Neurol. Neurosurg. Psychiatry. 1954;17:235–241. doi: 10.1136/jnnp.17.4.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huot RL, Thrivikraman KV, Meaney MJ, Plotsky PM. Development of adult ethanol preference and anxiety as a consequence of neonatal maternal separation in Long Evens rats and reversal with antidepressant treatment. Psychopharmacology. 2001;158:366–373. doi: 10.1007/s002130100701. [DOI] [PubMed] [Google Scholar]
- Jia F, Xie X, Zhou Y. Short-term depression of synaptic transmission from rat lateral geniculate nucleus to primary visual cortex in vivo. Brain Res. 2004;1002:158–161. doi: 10.1016/j.brainres.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Jia F, Wei H, Li X, Xie X, Zhou Y. Short-term synaptic plasticity in the rat geniculo-cortical pathway during development in vivo. Neurosci. Lett. 2006;398:73–77. doi: 10.1016/j.neulet.2005.12.054. [DOI] [PubMed] [Google Scholar]
- Kakita A, Wakabayshi K, Su M, Sakamoto M, Ikuta F, Takahashi H. Distinct pattern of neuronal degeneration in the fetal rat brain induced by consecutive transplacental administration of methylmercury. Brain Res. 2000;859:233–239. doi: 10.1016/s0006-8993(00)01964-8. [DOI] [PubMed] [Google Scholar]
- Kikusui T, Mori Y. Behavioural and neurochemical consequences of early weaning in rodents. J Neuroendocrinol. 2009;21:427–431. doi: 10.1111/j.1365-2826.2009.01837.x. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Bear MF. Homosynaptic long-term depression in the visual cortex. J Neurosci. 1994;14:3404–3412. doi: 10.1523/JNEUROSCI.14-05-03404.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korogi Y, Takahashi M, Shinzato J, Okajima T. MR findings in seven patients with organic mercury poisoning (Minamata disease) AJNR Am. J Neuroradiol. 1994;15:1575–1578. [PMC free article] [PubMed] [Google Scholar]
- Levesque PC, Atchison WD. Disruption of brain mitochondrial calcium sequestration by methylmercury. J Pharmacol. Exp. Ther. 1991;256:236–242. [PubMed] [Google Scholar]
- Limke TL, Atchison WD. Acute exposure to methylmercury opens the mitochondrial permeability transition pore in rat cerebellar granule cells. Toxicol. Appl. Pharmacol. 2002;178:52–61. doi: 10.1006/taap.2001.9327. [DOI] [PubMed] [Google Scholar]
- Limke TL, Otero-Montañez JK, Atchison WD. Evidence for interactions between intracellular calcium stores during methylmercury-induced intracellular calcium dysregulation in rat cerebellar granule neurons. J Pharmacol. Exp. Ther. 2003;304:949–958. doi: 10.1124/jpet.102.042457. [DOI] [PubMed] [Google Scholar]
- Limke TL, Bearss JJ, Atchison WD. Acute exposure to methylmercury causes Ca2+ dysregulation and neuronal death in rat cerebellar granule cells through an M3 muscarinic receptor-linked pathway. Toxicol. Sci. 2004;80:60–68. doi: 10.1093/toxsci/kfh131. [DOI] [PubMed] [Google Scholar]
- Marty MS, Atchison WD. Pathways mediating Ca2+ entry in rat cerebellar granule cells following in vitro exposure to methyl mercury. Toxicol. Appl. Pharmacol. 1997;147:319–330. doi: 10.1006/taap.1997.8262. [DOI] [PubMed] [Google Scholar]
- Marty MS, Atchison WD. Elevations of intracellular Ca2+ as a probable contributor to decreased viability in cerebellar granule cells following acute exposure to methylmercury. Toxicol. Appl. Pharmacol. 1998;150:98–105. doi: 10.1006/taap.1998.8383. [DOI] [PubMed] [Google Scholar]
- Mattsson JL, Miller E, Alligood JP, Koering JE, Levin SG. Early effects of methylmercury on the visual evoked response of the dog. Neurotoxicol. 1981;2:499–514. [PubMed] [Google Scholar]
- Merigan WH, Maurissen JP, Weiss B, Eskin T, Lapham LW. Neurotoxic actions of methylmercury on the primate visual system. Neurobehav. Toxicol. Teratol. 1983;5:649–658. [PubMed] [Google Scholar]
- Mesquita AR, PêGo JM, Summavielle T, Maciel P, Almeida OFX, Sousa N. Neurodevelopment Milestone Abnormalities In Rats Exposed To Stress In Early Life. Neuroscience. 2007;147:1022–1033. doi: 10.1016/j.neuroscience.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Mukuno K, Ishikawa S, Okamura R. Grating test of contrast sensitivity in patients with Minamata disease. Br. J Ophthalmol. 1981;65:284–290. doi: 10.1136/bjo.65.4.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata K, Weihe P, Renzoni A, Debes F, Vasconcelos R, Zino F, Araki S, Jørgensen PJ, White RF, Grandjean P. Delayed evoked potentials in children exposed to methylmercury from seafood. Neurotoxicol. Teratol. 1999;21:343–348. doi: 10.1016/s0892-0362(99)00011-2. [DOI] [PubMed] [Google Scholar]
- Newland MC, Reile PA. Blood and brain mercury levels after chronic gestational exposure to methylmercury in rats. Tox. Sci. 1999;50:106–116. doi: 10.1093/toxsci/50.1.106. [DOI] [PubMed] [Google Scholar]
- Nierenberg DW, Nordgren RE, Chang MB, Siegler RW, Blayney MB, Hochberg F, Toribara TY, Cernichiari E, Clarkson T. Delayed cerebellar disease and death after accidental exposure to dimethylmercury. New. Eng. J Med. 1998;338:1672–1676. doi: 10.1056/NEJM199806043382305. [DOI] [PubMed] [Google Scholar]
- O’Kusky JR. Synaptic degeneration in rat visual cortex after neonatal administration of methylmercury. Exp. Neurol. 1985;89:32–47. doi: 10.1016/0014-4886(85)90263-8. [DOI] [PubMed] [Google Scholar]
- O’Ksky JR, McGeer EG. Methylmercury poisoning of the developing nervous system in the rat; decreased activity of glutamic acid decarboxylase in cerebral cortex and neostriatum. Develop. Brain. Res. 1985;21:299–306. doi: 10.1016/0165-3806(85)90219-6. [DOI] [PubMed] [Google Scholar]
- O’Kusky JR, Radke JM, Vincent SR. Methylmercury-induced movement and postural disorders in developing rat: loss of somatostatin-immunoreactive interneurons in the striatum. Develop. Brain. Res. 1988;40:11–23. doi: 10.1016/0165-3806(88)90003-x. [DOI] [PubMed] [Google Scholar]
- Paletz EM, Day JJ, Craig-Schmidt MC, Newland MC. Spatial and visual discrimination reversals in adult and geriatric rats exposed during gestation to methylmercury and n - 3 polyunsaturated fatty acids. NeuroToxicology. 2007;28:707–719. doi: 10.1016/j.neuro.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramoa AS, Sur M. Short-term synaptic plasticity in the visual cortex during development. Cereb. Cortex. 1996;6:640–646. doi: 10.1093/cercor/6.4.640. [DOI] [PubMed] [Google Scholar]
- Rasmussen EB, Newland MC. Developmental exposure to methylmercury alters behavioral sensitivity to d-amphetamine and pentobarbital in the adult rats. Neurotoxicol Teratol. 2001;23:45–55. doi: 10.1016/s0892-0362(00)00112-4. [DOI] [PubMed] [Google Scholar]
- Reed MN, Paletz EM, Newland MC. Gestational exposure to methylmercury and selenium: Effects on a spatial discrimination reversal in adulthood. NeuroToxicology. 2006;27:721–723. doi: 10.1016/j.neuro.2006.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, Sakmann B. Developmental switch in the short-term modification of unitary EPSPs evoked in layer 2/3 and layer 5 pyramidal neurons of rat neocortex. J Neurosci. 1999;19:3827–3835. doi: 10.1523/JNEUROSCI.19-10-03827.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DC, Gilbert SG. Early chronic low-level methylmercury poisoning in monkeys impairs spatial vision. Science. 1982;216:759–761. doi: 10.1126/science.7079739. [DOI] [PubMed] [Google Scholar]
- Rice DC, Gilbert SG. Effects of developmental exposure to methylmercury on spatial and temporal visual function in monkeys. Toxicol. Appl. Pharmacol. 1990;102:151–163. doi: 10.1016/0041-008x(90)90092-9. [DOI] [PubMed] [Google Scholar]
- Rumpel S, Hatt H, Gottmann K. Silent synapses in the developing rat visual cortex: evidence for postsynaptic expression of synaptic plasticity. J neurosci. 1998;18:8863–8874. doi: 10.1523/JNEUROSCI.18-21-08863.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustam H, Hamdi T. Methyl mercury poisoning in Iraq: a neurological study. Brain. 1974;97:499–510. [PubMed] [Google Scholar]
- Sakamoto M, Kakita A, de Oliveira RB, Pan HS, Takahashi H. Dose-dependent effects of methylmercury administered during neonatal brain spurt in rats. Dev. Brain Res. 2004;152:171–176. doi: 10.1016/j.devbrainres.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Shaw CM, Mottet NK, Body RL, Luschei ES. Variability of neuropathologic lesions in experimental methylmercurial encephalopathy in primates. Am. J Pathol. 1975;80:451–470. [PMC free article] [PubMed] [Google Scholar]
- Shigematsu J, Yasuda T, Goto Y, Tanaka K, Tobimatsu S. Chronic effects of methylmercury on the cerebral function in rats. J Neurol. Sci. 2000;182:69–75. doi: 10.1016/s0022-510x(00)00454-8. [DOI] [PubMed] [Google Scholar]
- Šlamberová R, Pometlová M, Charousová P. Postnatal development of rat pups is altered by prenatal methamphetamine exposure. Prog Neuro-Psychopharmacol. Biol. Psych. 2006;30:82–88. doi: 10.1016/j.pnpbp.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Stoltenburg-Didinger G, Markwort S. Prenatal methylmercury exposure results in dendritic spine dysgenesis in rats. Neurotoxicol. Teratol. 1990;12:573–576. doi: 10.1016/0892-0362(90)90064-j. [DOI] [PubMed] [Google Scholar]
- Sugawara N, Ohha T, Nakai K, Kakita A, Nakamura T, Suzuki K, Kameo S, Shimada M, Kurokawa N, Satoh C, Satoh H. Effects of perinatal coexposure to methylmercury and polychlorinated biphenyls on neurobehavioral development in mice. Arch Toxicol. 2008;82:387–397. doi: 10.1007/s00204-007-0254-x. [DOI] [PubMed] [Google Scholar]
- Swanson LW. Brain Maps: Structure of the Rat Brain. 2nd rev. (ed). Elsevier; Amsterdam: 1998. [Google Scholar]
- Takeuchi T, Morikawa N, Matsumoto H, Shiraishi Y. A patholgical study of Minamata Disearse in Japan. Acta Neuropathol. 1962;2:40–57. [Google Scholar]
- Takeuchi T, Kambara T, Morikawa N, Matsumoto H, Shiraishi Y, Ito H. Pathologic observations of the Minamata disease. Acta. Pathol. 1959;9:769–783. doi: 10.1111/j.1440-1827.1959.tb02965.x. [DOI] [PubMed] [Google Scholar]
- Thomson AM. Facilitation, augmentation and potentiation at central synapses. Trends Neurosci. 2000;23:305–312. doi: 10.1016/s0166-2236(00)01580-0. [DOI] [PubMed] [Google Scholar]
- Tokuomi H, Okajima T. Minamata Disease. World Neurol. 1961;2:536–545. [PubMed] [Google Scholar]
- Uruno K, O’Connor MJ, Masukawa LM. Effects of bicuculline and baclofen on paired-pulse depression in the dentate gyrus of epileptic patients. Brain Res. 1995;695:163–172. doi: 10.1016/0006-8993(95)00652-7. [DOI] [PubMed] [Google Scholar]
- Vallée M, Mayo W, Dellu F, Le Moal M, Simon H, Maccari S. Prenatal stress induces high anxiety and postnatal handling induces low anxiety in adult offspring: correlation with stress induced-corticosterone secretion. J. Neurosci. 1997;17:2626–2636. doi: 10.1523/JNEUROSCI.17-07-02626.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallée M, Maccari S, Dellu F, Simon H, Le Moal M, Mayo W. Long-term effects of prenatal stress and postnatal handling on age-related glucocorticoid secretion and cognitive performance: a longitudinal study in the rat. Eur. J. Neurosci. 1999;11:2906–2916. doi: 10.1046/j.1460-9568.1999.00705.x. [DOI] [PubMed] [Google Scholar]
- Varela JA, Sen K, Gibson J, Fost J, Abbott LF, Nelson SB. A quantitative description of short-term plasticity at excitatory synapses in layer 2/3 of rat primary visual cortex. J Neurosci. 1997;17:7926–7940. doi: 10.1523/JNEUROSCI.17-20-07926.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayshi K, Kakita A, Sakamoto M, Su M, Iwanaga K, Ikuta F. Variability of brain lesions in rats administered methylmercury at various postnatal development phases. Brain Res. 1995;705:267–272. doi: 10.1016/0006-8993(95)01208-7. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG. Structural contributions to short-term synaptic plasticity. Physiol. Rev. 2004;84:69–85. doi: 10.1152/physrev.00016.2003. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y, Ohmura T, Komatsu Y. Two forms of synaptic plasticity with distinct dependence on age, experience, and NMDA receptor subtype in rat visual cortex. J Neurosci. 2003;23:6557–6566. doi: 10.1523/JNEUROSCI.23-16-06557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Atchison WD. Disruption by methylmercury of membrane excitability and synaptic transmission in hippocampal slices of the rat. Toxicol. Appl. Pharmacol. 1993;120:203–215. doi: 10.1006/taap.1993.1104. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Atchison WD. Methylmercury acts at multiple sites to block hippocampal synaptic transmission. J. Pharmacol. Exp. Ther. 1995;275:1308–1316. [PubMed] [Google Scholar]
- Yuan Y, Atchison WD. Action of methylmercury on GABAA receptor-mediated inhibitory synaptic transmission is primarily responsible for its early stimulatory effects on hippocampal CA1 excitatory synaptic transmission. J Pharmacol. Exp. Ther. 1997;282:64–73. [PubMed] [Google Scholar]
- Yuan Y, Atchison WD. Comparative effects of methylmercury on parallel fiber and climbing fiber responses in rat cerebellar slices. J Pharmacol. Exp. Ther. 1999;288:1015–1025. [PubMed] [Google Scholar]
- Yuan Y, Atchison WD. Methylmercury differentially affects GABAA receptor-mediated spontaneous IPSCs in Purkinje and granule cells of rat cerebellar slices. J. Physiol. (Lond) 2003;550:191–204. doi: 10.1113/jphysiol.2003.040543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Atchison WD. Methylmercury-induced increase of intracellular Ca2+ increases spontaneous synaptic current frequency in rat cerebellar slices. Mol. Pharmacol. 2007;71:1109–1121. doi: 10.1124/mol.106.031286. [DOI] [PubMed] [Google Scholar]
- Zenick H. Evoked potential alterations in methylmercury chloride toxicity. Pharmacol. Biochem. & Behav. 1976;5:253–255. doi: 10.1016/0091-3057(76)90075-7. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu. Rev. Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]