

Abstract
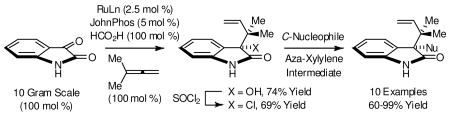
Ruthenium catalyzed tert-prenylation of isatin 1 occurs efficiently in the absence of N-protecting groups under the conditions of C-C bond forming transfer hydrogenation employing 1,1-dimethylallene as the prenyl donor. The prenylated adduct, 3-hydroxy-3-tert-prenyl-oxindole 2, is converted to the tertiary neopentyl chloride 3, which participates in nucleophilic substitution by way of an aza-ortho-xylylene intermediate to furnish adducts 4a-4i. Through tertiary neopentyl substitution, two contiguous all carbon quaternary centers are established.
Prenylated indole alkaloids have attracted attention due to their remarkable biological effects and challenging structural features. 1 Those incorporating tert-prenyl moieties at the 2- or 3-position include the brevianamides, austamides, paraherquamides, marcfortines, echinulins, aspergamides, norgeamides, avrainvillamides, stephacidins, notamides, roquefortines, as well as the amauromine, ardeemin, and flustramine families of natural products. The construction of indole alkaloids that incorporate a 3-tert-prenyl moiety requires construction of two contiguous all-carbon quaternary centers. Typically, this substructure is installed through the reaction of bis-N-protected tryptophan derivatives with N-(phenylseleno)phthalimide to form 3-phenylselenio-pyrroloindoline adducts, which are ionized with methyl triflate in the presence of prenyl tributylstannane.2 Considerable pre-activation attends this method, which requires stoichiometric use of both tin and selenium reagents, as well as protection of the indolic nitrogen.
In the course of studies aimed at the development of C-C bond forming hydrogenations beyond hydroformylation,3 we recently developed a suite of catalytic methods for carbonyl allylation, 4 b,d,e,f,i,j,k crotylation4b,c,g,k and reverse prenylation4a,b,h,k in the absence of stoichiometric allylmetal reagents. In the specific case of reverse prenylation,4a,b,h,k it was found that reductive C-C bond formation is achieved simply upon hydrogenation4a or transfer hydrogenation4b,h,k of 1,1-dimethylallene in the presence of carbonyl partners, including isatins.4a,k Although the synthesis of 3-tert-prenylated oxindoles can be achieved through the addition of n-prenylindium reagents to isatins 5 or through enolate-Claisen rearrangement, 6 N-protected isatins are generally required.7,8e
Here, we report that under the mild conditions of transfer hydrogenation, direct tert-prenylation of isatin occurs in the absence of N-protecting groups. Furthermore, the resulting adduct, 3-hydroxy-3-tert-prenyl-oxindole 2, is readily converted to the chloride 3, which engages in tertiary neopentyl substitution with C-nucleophiles to furnish adducts possessing two contiguous all-carbon quaternary centers, presumably by way of an aza-ortho-xylylene intermediate.8,9 To our knowledge, these studies represent the first general protocol for intermolecular substitution in a tertiary neopentyl system.10,11
Our initial studies focused on the reaction of isatin 1 with 1,1-dimethylallene under the conditions of ruthenium catalyzed transfer hydrogenation. Our prior work on allene couplings of this type took advantage of a catalyst derived from RuBr(CO)3(η3-C3H5) and t-BuPPh2 in combination with isopropanol as terminal reductant.12 For the present study, a process better suited to gram scale synthesis was sought. Hence, our optimization focused on the use of commercially available RuHCl(CO)(PPh3)3 as pre-catalyst. For this catalyst precursor, formic acid was found to be superior to isopropanol as terminal reductant. Additionally, the choice of ligand proved to be crucial. Use of RuHCl(CO)(PPh3)3 in the absence of added ligand did not result in product formation. However, use of RuHCl(CO)(PPh3)3 in the presence of more electron rich phosphines, such as tris(4-methoxyphenyl)phosphine or JohnPhos, provided the desired product of tert-prenylation 2 in isolated yields of 61% and 74%, respectively, at catalyst loadings of 2.5 mol % employing equimolar quantities of isatin 1, 1,1-dimethylallene and formic acid at 65°C. The latter conditions employing JohnPhos as ligand were employed on 10 gram scale with isolation of the product via crystallization from ethyl acetate-hexane (Scheme 1).
Scheme 1.

Reverse prenylation of isatin 1 and conversion to 3-chloro-3-tert-prenyl oxindole 3.a
aCompound 2 was isolated by crystallization from ethyl acetate-hexane. Compound 3 was isolated by silica gel chromatography. See Supporting Information for experimental details.
With gram quantities of alcohol 2 in hand, methods for the synthesis of tertiary neopentyl chloride 3 were explored. Standard conditions employing thionyl chloride and a tertiary amine base led to isolated yields ranging between 30%-90%. However, the desired chloride 3 was contaminated with substantial quantities of Wagner-Meerwein product. It was postulated that Wagner-Meerwein rearrangement occurs upon ionization of the transient chlorosulfite to form the protonated aza-ortho-xylylene. Based on this interpretation, irreversible dianion formation followed by the addition of thionyl chloride should generate a transient chlorosulfite that should eliminate to furnish a neutral aza-ortho-xylylene, which should be less susceptible to Wagner-Meerwein shift. Indeed, treatment of alcohol 2 with 2.2 equivalents of LHMDS followed by thionyl chloride provided the tertiary neopentyl chloride 3 in 69% yield as a single constitutional isomer (Scheme 1).
Acquisition of chloride 3 set the stage for tertiary neopentyl substitution. Upon exposure of chloride 3 to dimethyl malonate in the presence of potassium carbonate in dichloromethane solvent,8 the desired product of tertiary neopentyl substitution 4a was obtained in 84% isolated yield. These conditions were applied to a range of C-nucleophiles. As demonstrated by the formation of adducts 4a-4i, active methylene compounds, cyanide and electron rich arenes engage in efficient tertiary neopentyl substitution with chloride 3 (Scheme 2). Finally, borohydride reduction of chloride 3 also is possible, as demonstrated by the formation of 4j (Scheme 3).
Scheme 2.

Tertiary neopentyl substitution of chloride 3 to furnish adducts 4a-4i possessing contiguous all-carbon quaternary centers.a
aFor adducts 4a-4f, 300 mol % of NuH was employed. For adducts 4g-4i, 150 mol % of NuH was employed. Cited yields are of material isolated by silica gel chromatography. See Supporting Information for experimental details. bObtained as a mixture of diastereomers and keto-enol tautomers. cFor the formation of 4d, 10 mol % Bu4NI, THF-H2O (1:3) was used as solvent and the reaction was run for 24 h.
Scheme 3.

Dehalogenation of chloride 3 mediated by NaBH4.
aAs described in Scheme 2.
In summary, we report a protecting group-free method for the gram-scale synthesis of 3-hydroxy-3-tert-prenyl-oxindole 2 via ruthenium catalyzed C-C bond forming transfer hydrogenation. Conditions were identified for the conversion of tertiary neopentyl alcohol 2 to the corresponding chloride 3 in the absence of Wagner-Meerwein shift. Finally, chloride 3 engages in tertiary neopentyl substitution by way of an aza-ortho-xylylene intermediate to furnish the tert-prenylated oxindoles 4a-4i. Future studies will focus on the development of related asymmetric neopentyl substitutions and application of these methods toward the synthesis of naturally occurring 3-tert-prenylated indoles.
Supplementary Material
Figure 1.

Examples of indole alkaloids that incorporate a tert-prenyl moiety at the indole 3-position.
Acknowledgments
The Acknowledgment is made to the Robert A. Welch Foundation and the NIH-NIGMS (R01-GM069445-S109) for partial support of this research.
Footnotes
Supporting Information Available. Spectral data for all new compounds (1H NMR, 13C NMR, IR, HRMS). Single crystal X-ray diffraction data for 4e. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For selected reviews encompassing the synthesis and biosynthesis of prenylated indole alkaloids, see: Williams RM, Stocking EM, Sanz-Cervera JF. Top Cur Chem. 2000;209:97.Williams RM. Chem Pharm Bull. 2002;50:711. doi: 10.1248/cpb.50.711.von Nussbaum F. Angew Chem Int Ed. 2003;42:3068. doi: 10.1002/anie.200301646.Williams RM, Cox RJ. Acc Chem Res. 2003;36:127. doi: 10.1021/ar020229e.Steffan N, Grundmann A, Yin WB, Kremer A, Li SM. Curr Med Chem. 2009;16:218. doi: 10.2174/092986709787002772.
- 2.For application of this strategy toward the synthesis of 3-tert-prenylated indoles, see: Marsden SP, Depew KM, Danishefsky SJ. J Am Chem Soc. 1994;116:11143. Amauramine and ardeemin families of natural products:Depew KM, Marsden SP, Zatorska D, Zatorski A, Bornmann WG, Danishefsky SJ. J Am Chem Soc. 1999;121:11953.Chen WC, Joullié MM. Tetrahedron Lett. 1989;39:8401. Roquefortine family of natural products:Schiavi B, Richard DJ, Joullié MM. J Org Chem. 2002;67:620. doi: 10.1021/jo0106593.Richard DJ, Schiavi B, Joullié MM. Proc Nat Acad Sci. 2004;101:11971. doi: 10.1073/pnas.0401407101.Shangguan N, Hehre WJ, Ohlinger WS, Beavers MP, Joullié MM. J Am Chem Soc. 2008;130:6281. doi: 10.1021/ja800067q.Sunazuka T, Shirahata T, Tsuchiya S, Hirose T, Mori R, Harigaya Y, Kuwajima I, Ohmura S. Org Lett. 2005;7:941. doi: 10.1021/ol050077y. The oxaline and neoxaline core, see:
- 3.For selected reviews on C-C bond forming hydrogenation and transfer hydrogenation, see: Ngai MY, Kong JR, Krische MJ. J Org Chem. 2007;72:1063. doi: 10.1021/jo061895m.Skucas E, Ngai MY, Komanduri V, Krische MJ. Acc Chem Res. 2007;40:1394. doi: 10.1021/ar7001123.Shibahara F, Krische MJ. Chem Lett. 2008;37:1102. doi: 10.1246/cl.2008.1102.Bower JF, Kim IS, Patman RL, Krische MJ. Angew Chem Int Ed. 2009;48:34. doi: 10.1002/anie.200802938.
- 4.(a) Skucas E, Bower JF, Krische MJ. J Am Chem Soc. 2007;129:12678. doi: 10.1021/ja075971u. [DOI] [PubMed] [Google Scholar]; (b) Bower JF, Skucas E, Patman RL, Krische MJ. J Am Chem Soc. 2007;129:15134. doi: 10.1021/ja077389b. [DOI] [PubMed] [Google Scholar]; (c) Shibahara F, Bower JF, Krische MJ. J Am Chem Soc. 2008;130:6338. doi: 10.1021/ja801213x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:6340. doi: 10.1021/ja802001b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:14891. doi: 10.1021/ja805722e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Lu Y, Kim IS, Hassan A, Del Valle DJ, Krische MJ. Angew Chem Int Ed. 2009;48:5018. doi: 10.1002/anie.200901648. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Kim IS, Han SB, Krische MJ. J Am Chem Soc. 2009;131:2514. doi: 10.1021/ja808857w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Han SB, Kim IS, Han H, Krische MJ. J Am Chem Soc. 2009;131:6916. doi: 10.1021/ja902437k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Lu Y, Krische MJ. Org Lett. 2009;11:3108. doi: 10.1021/ol901096d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Hassan A, Lu Y, Krische MJ. Org Lett. 2009;11:3112. doi: 10.1021/ol901136w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Itoh J, Han SB, Krische MJ. Angew Chem Int Ed. 2009;48:6316. doi: 10.1002/anie.200902328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nair V, Ros S, Jayan CN, Viji S. Synthesis. 2003:2542. [Google Scholar]
- 6.(a) Malapel-Andrieu B, Piroëlle S, Mérour JY. J Chem Res. 1998:594. [Google Scholar]; (b) Kawasaki T, Nagaoka M, Satoh T, Okamoto A, Ukon R, Ogawa A. Tetrahedron. 2004;60:3493. [Google Scholar]
- 7.As described in reference 8e, treatment of 3-methyl-3-bromooxindole with n-prenyl tributylstannane delivers the 3-methyl-3-tert-prenyl-oxindole in the absence of an N-protecting group. However, stoichiometric quantities of tin byproducts are generated.
- 8.While many substitution reactions involving C-nucleophiles and 3-substituted-3-halo-oxindoles are reported, examples of tertiary neopentyl substitution are absent. While in earlier literature aza-xylylene intermediates are not proposed as intermediates (e.g. refs. a-c), their intervention is possible and highly likely: Labroo RB, Labroo VM, King MM, Cohen LA. J Org Chem. 1991;56:3637.Kobayashi M, Aoki S, Gato K, Matsunami K, Kurosu M, Kitagawa I. Chem Pharm Bull. 1994;42:2449. doi: 10.1248/cpb.42.2449.Rajeswaran WG, Labroo RB, Cohen LA. J Org Chem. 1999;64:1369.Fuchs JR, Funk RL. J Am Chem Soc. 2004;126:5068. doi: 10.1021/ja049569g.Fuchs JR, Funk RL. Org Lett. 2005;7:677. doi: 10.1021/ol047532v.England DB, Merey G, Padwa A. Org Lett. 2007;9:3805. doi: 10.1021/ol7016438.England DB, Merey G, Padwa A. Heterocycles. 2007;74:491.Krishnan S, Stoltz BM. Tetrahedron Lett. 2007;48:7571.
- 9.For a seminal observation of substitution reactions involving heteroatom nucleophiles and 3-halo-oxindoles, see: Hinman RL, Bauman CP. J Org Chem. 1964;29:2431. Intervention of aza-xylylene intermediates is not proposed, yet is highly probable.
- 10.For selected examples of primary neopentyl substitution reactions, see: Lewis RG, Gustafson DH, Erman WF. Tetrahedron Lett. 1967;8:401.Paquette LA, Philips JC. Tetrahedron Lett. 1967;8:4645.Weiss RG, Snyder EI. J Org Chem. 1971;36:403.Stephenson B, Solladie G, Mosher HS. J Am Chem Soc. 1972;94:4184.Anderson PH, Stephenson B, Mosher HS. J Am Chem Soc. 1974;96:3171.
- 11.For reviews encompassing neopentyl substitution, see: Mosher HS. Tetrahedron. 1974;30:1733.Rossi RA, Postigo AI. Curr Org Chem. 2003;7:747.
- 12.For ruthenium catalyzed reductive coupling of 1,1-disubstituted allenes to paraformaldehyde and higher aldehydes, see: Ngai MY, Skucas E, Krische MJ. Org Lett. 2008;10:2705. doi: 10.1021/ol800836v.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.