


Abstract
Many enzymes acting on DNA require Mg2+ ions not only for catalysis but also to bind DNA. Binding studies often employ Ca2+ as a substitute for Mg2+, to promote DNA binding whilst disallowing catalysis. The SfiI endonuclease requires divalent metal ions to bind DNA but, in contrast to many systems where Ca2+ mimics Mg2+, Ca2+ causes SfiI to bind DNA almost irreversibly. Equilibrium binding by wild-type SfiI cannot be conducted with Mg2+ present as the DNA is cleaved so, to study the effect of Mg2+ on DNA binding, two catalytically-inactive mutants were constructed. The mutants bound DNA in the presence of either Ca2+ or Mg2+ but, unlike wild-type SfiI with Ca2+, the binding was reversible. With both mutants, dissociation was slow with Ca2+ but was in one case much faster with Mg2+. Hence, Ca2+ can affect DNA binding differently from Mg2+. Moreover, SfiI is an archetypal system for DNA looping; on DNA with two recognition sites, it binds to both sites and loops out the intervening DNA. While the dynamics of looping cannot be measured with wild-type SfiI and Ca2+, it becomes accessible with the mutant and Mg2+.
INTRODUCTION
Most enzymes that act at phosphodiester bonds in DNA require divalent metal ions as catalytic cofactors, usually Mg2+ (1–3). These include, with just one independent exception (4), all of the several thousand restriction endonucleases identified to date (5). The majority of the restriction enzymes discovered by biochemical methods fall into the Type II category (6). The Type II enzymes recognize specific sequences of 4–8 bp, though these are sometimes interrupted by a non-specific sequence of specified length, and cleave the DNA at fixed locations within or close to their recognition sites (7–9). Most show their maximal activity with Mg2+, have lower activities with some metal ions such as Mn2+ but have no activity with Ca2+ (7,9), though some Type II enzymes deviate from the majority in this respect (10).
In the absence of divalent ions, certain restriction enzymes bind DNA specifically at their recognition sites even though they cannot cut the DNA (11–13). Others bind DNA non-specifically under these conditions, without significant selectivity for their recognition sites (14–17), while further systems show no detectable affinity for any DNA sequence without metal ions (18–20). However, the enzymes that have either no detectable affinity for DNA in the absence of divalent metal, or no significant selectivity for their recognition sites, almost invariably show strongly enhanced affinities for their target sites in the presence of Ca2+ (18,20–23). Since first being applied to the EcoRV endonuclease (21), Ca2+ has been used extensively in both equilibrium binding and structural studies on enzyme–DNA complexes, not only with restriction nucleases but also with many other Mg2+-dependent enzymes acting on DNA (1–3). Moreover, with EcoRV, Ca2+ was shown to be a ‘near-perfect analogue’ for Mg2+ in DNA binding (24).
Restriction enzymes often contain at their active sites a partially conserved sequence motif, (E/D) … PD … (D/E)XK, containing three carboxylates and a lysine (2,25). The carboxylates usually bind two metal ions: the metal used in the crystal structures of the enzyme–substrate complexes is normally Ca2+. Replacement of any one of these carboxylates with an alanine obliterates catalytic activity, but the resultant proteins often bind DNA more avidly than the wt (wild-type) enzyme, sometimes specifically at their recognition sites even in the absence of divalent metal ions (13,16,26,27). The enhancement in binding is most likely due to the removal of charge repulsion between the active-site carboxylates and the DNA phosphate at the scissile bond, as a similar effect can be obtained by lowering the pH to protonate the carboxylates (13,22,28). Moreover, DNA binding by these catalytically inactive proteins can also be studied in the presence of the natural cofactor Mg2+ (23,24,29): like Ca2+ with the wt enzymes, Mg2+ can induce the mutants to bind specifically at their recognition sites.
Many Type II restriction endonucleases are dimeric proteins that interact symmetrically with palindromic recognition sequences (5–7), so that one active site is positioned to cleave one strand of the DNA and the second the complementary strand (2,8,9). Such enzymes bind to individual copies of their recognition site and cleave each site in a separate reaction. However, a large number of the Type II enzymes are fully active only after binding two copies of the recognition sequence and are virtually inactive when bound to a single copy (30–34). Some such enzymes, the Type IIE systems (6), use one copy of the site as an allosteric ligand to activate the catalytic reaction at the other copy (30,33). A Type IIE enzyme thus cleaves DNA with two sites by binding to both sites and looping out the intervening DNA (34,35) before cutting just one site (36). Other systems, the Type IIF enzymes, form tetramers with two identical DNA-binding clefts, but these become catalytically competent only when both clefts are filled with cognate DNA (20,37–39). Hence, the Type IIF enzymes also act on DNA with two sites by binding to both sites and trapping the intervening DNA in a loop, but they then usually cut both sites before dissociating from the DNA, without liberating intermediates cut at one site (36–41).
The archetype of the Type IIF systems, the first to be identified as tetrameric enzyme acting at two DNA sites, is the SfiI endonuclease (37). In the presence of Mg2+, SfiI cleaves DNA at the sequence GGCCNNNN↓NGGCC (where N indicates any base and ↓ the point of cleavage) (42). Without any divalent metal ion, it shows no detectable binding to DNA with this or any other sequence but, in the presence of Ca2+, it binds specific duplexes much more readily than non-specific DNA, forming complexes containing the tetramer bound to two duplexes (18). The binding is highly cooperative: the complex with one duplex is not formed to a significant extent during the association and has virtually no activity (18,39). Even though SfiI cannot bind two duplexes in trans in the absence of metal ions, it can under these conditions trap loops on plasmids with two SfiI sites in cis, albeit only transiently as the loops dissociate rapidly (43,44). Conversely, in the presence of Ca2+, SfiI forms extremely stable loops on two-site plasmids that last for >> 7 h, much longer than the loops formed with other Type IIE and IIF restriction enzymes (35,44).
The complex of the SfiI nuclease bound to two DNA duplexes was crystallized in the presence of Ca2+ (45). Two subunits bind one duplex on one side of the tetramer and the other two the second duplex on the opposite side of the protein: each subunit contacts the specified base pair in one half of the recognition sequence. However, their active sites contain only one Ca2+ ion and this is located too far away from the scissile phosphodiester to interact with it directly (Figure 1a). This situation contrasts with that in a related restriction enzyme, BglI (Figure 1b), which was also crystallized as a DNA–protein complex with Ca2+ (46). BglI is a dimeric enzyme that recognizes a truncated SfiI site, GCCNNNN↓NGGC, but despite their dissimilar amino acid sequences, the monomers of BglI and SfiI have similar structures (47). The active site in BglI has the same arrangement of three carboxylates and a lysine as SfiI but contains two Ca2+ ions coordinated by the carboxylates and by the phosphate at the scissile bond. The two Ca2+ ions in the active site of BglI lie close to the positions from which Mg2+ ions could catalyse phosphodiester hydrolysis by a two-metal mechanism (1–3,25), while a Mg2+ at the site of the single Ca2+ in SfiI cannot function catalytically. It seems likely that the crystal structure of the SfiI–Ca2+–DNA complex differs from the SfiI–Mg2+–DNA complex that carries out the catalytic reaction.
Figure 1.

Active sites of (a) SfiI and (b) BglI. The crystal structures of SfiI and BglI, in their complexes with cognate DNA and Ca2+, both contain three carboxylates and a lysine: in SfiI (a), E55, D79, D100 and K102; in BglI (b), E87, D116, D142 and K144. Also shown in both (a) and (b) is the tri-nucleotide sequence centred on the scissile phosphate (marked with an arrow indicating the direction of the attacking nucleophile). The nucleotides and the side chains of the key amino acids are in ‘stick’ format (the DNA in green; the amino acids in yellow), with functional groups coloured as follows: red, oxygen; blue, nitrogen; purple, phosphorous. The sections of peptide main-chain included here are in ‘ribbon’ format: in brown for the residues noted above; in green for the remainder. The Ca2+ ions are shown as non-bonded grey spheres: water molecules have been omitted. Data from the RCSB Protein Data Bank; accession codes 2EZV for SfiI and 1DMU for BglI.
The aim of this study was to determine the role of the metal ion in DNA binding by SfiI and the extent to which Ca2+ can mimic Mg2+ in this respect. This study also yielded an experimental system that is exploited in the following paper (48) to observe directly loop capture and release by SfiI on single DNA molecules.
MATERIALS AND METHODS
Mutagenesis
Plasmids carrying the genes for the SfiI modification and restriction enzymes, pSYX33-SfiIM+ and pRRS-SfiIR+ respectively, were gifts from I. Schildkraut and S-Y Xu (New England Biolabs). These were used to transform Escherichia coli ER2353, first with the modification plasmid to give E. coli ER2353[pSYX33-SfiIM+] and then with the nuclease plasmid to give ER2353[pSYX33-SfiIM+, pRRS-SfiIR+]. The mixture of plasmids from this strain was subjected to site-directed mutagenesis of the gene for the SfiI nuclease by the QuikChange method (Stratagene), using primers that specified either the D79A or the D100A substitution. The resultant products were used to transform ER2353[pSYX33-SfiIM+]. Plasmids were isolated from the transformants and the derivatives of pRRS-SfiIR+ sequenced across the entire gene for the SfiI nuclease (University of Dundee Sequencing Service): no mutations other than those targeted were introduced.
Proteins and DNA
Wt SfiI was purified from E. coli ER2238[pSYX33-SfiIM+, pRRS-SfiIR+] as described previously (37), as were also the D79A and the D100A proteins from strains carrying these mutations. SfiI concentrations were assessed from A280 readings using the extinction coefficient for the tetrameric form of Mr 124 176 (41). Sedimentation equilibrium studies used a Beckman XLA ultracentrifuge as before (20). Protein structures were analysed in INSIGHT II v2005 (Accelrys, San Diego).
The supercoiled form of the plasmid pGB1 (37) was purified as before (39) from E. coli ER2267[pGB1] that had been grown in media containing 37 MBq/l [methyl-3H] thymidine (GE Healthcare). The linear form of pGB1 was generated by cutting the plasmid at its single NdeI site.
Oligodeoxyribonucleotides were obtained HPLC-purified from Sigma Genosys. They were annealed to give the duplexes in Table 1 by mixing equal concentrations of two oligonucleotides with complementary sequences in 20 mM Tris–HCl (pH 8.0), 100 mM NaCl, heating to 95°C and then cooling overnight to room temperature.
Table 1.
Oligoduplexes
| Duplex | Sequence |
|---|---|
| HEX-35 | 5′-HEX-TCGATCCATGTGGCCAACAAGGCCTATTTGTCGAT-3′ |
| 3′-AGCTAGGTACACCGGTTGTTCCGGATAAACAGCTA-5′ | |
| C-21 | 5′-ATGTGGCCAACAAGGCCTATT-3′ |
| 3′-TACACCGGTTGTTCCGGATAA–5′ |
HEX-35 is a 35 bp duplex that carries the recognition sequence for SfiI (underlined) and a hexachlorofluorescein (HEX) moiety attached through a C6 linker to the 5′ end of the top strand. C-21 is a 21 bp duplex that has the same sequence as HEX-35 over the innermost 21 bp of HEX-35, which includes the SfiI recognition sequence. HEX-21 and Alexa-21 (not shown) are derivatives of C-21 that carry at the 5′ end of the top strand either HEX or Alexa Fluor 350, respectively.
Enzyme assays
Activities of wt and mutant SfiI proteins were assessed by adding 10 µl enzyme—diluted to the requisite concentration in SfiI dilution buffer (37)—to 190 µl 3H-labelled DNA in reaction buffer [10 mM Tris–HCl (pH 7.9), 50 mM NaCl, 10 mM MgCl2, 1 mM DTT, 100 μg/ml BSA] at 50°C. Aliquots (15 µl) were removed from the reactions at various times after adding the enzyme (one was removed before, the zero time-point) and mixed immediately with 10 µl Stop Mix (37,41). The samples were analysed by electrophoresis through agarose under conditions that separated the DNA substrate from the cleaved products and the concentration of each form at each time point determined by scintillation counting (36,40).
Gel retardation
Aliquots of SfiI protein (wt, D79A or D100A) in SfiI dilution buffer were added to HEX-35 (Table 1), to HEX-21 or to a mixture of HEX-35 and HEX-21, to give 20 µl solutions that contained 5 nM SfiI tetramer and 10 nM duplex in Ca2+ binding buffer [10 mM Tris–HCl (pH 7.5), 25 mM NaCl, 2 mM CaCl2, 5 mM βME, 100 µg/ml BSA]. After 30 min at room temperature, the samples were mixed with 10 µl 6.6% (w/v) Ficoll 400 in Ca2+ binding buffer and applied to 8% polyacrylamide gels in 45 mM Tris–borate (pH 8.3), 2 mM CaCl2, as described previously (14,39). After electrophoresis, the gels were scanned in a Molecular Dynamics PhosphorImager with illumination at 532 nm: the emission from each HEX-labelled species was recorded though a 555 nm filter and analysed in IMAGEQUANT (Molecular Dynamics).
The displacement of labelled DNA from the SfiI protein (wt or D100A) was examined by first incubating for 30 min at room temperature 10 nM HEX-35 and 7.5 nM SfiI tetramer in Ca2+-binding buffer, before adding C-21 (Table 1) to a final concentration of 100 nM. The additions of C-21 to each sample of HEX-35 and SfiI protein were made at specified times before applying the samples to a polyacrylamide gel: the time intervals between the addition and gel-loading varied from 0.5 to 24 h. The amounts of bound and free HEX-labelled DNA were quantified as above. The changes in the concentration of each form of HEX-35 were analysed as a function of time by fitting to exponential functions in GRAFIT (Erithacus Software). Reaction schemes were modelled in BERKELEY MADONNA (http://www.berkeleymadonna.com).
Fluorescence methods
Fluorescence measurements over extended time scales were recorded at 25°C in a Fluorolog Tau-3 spectrofluorimeter (Horiba Jobin Yvon). Complexes between the SfiI protein (D79A or D100A) and oligoduplexes labelled with Alexa Fluor 350 (Molecular Probes) were detected by fluorescence energy resonance transfer (FRET) between the Trp residues in the protein and the Alexa Fluor 350: trp fluorescence was excited at 290 nm and the emission from Alexa Fluor 350 observed at 438 nm. The reactions, in 5 mm cuvettes, started with mixtures containing 50 nM Alexa-21 (Table 1) and 25 nM SfiI protein in either Ca2+ or Mg2+ fluorescence buffers [10 mM Tris–HCl (pH 7.5), 25 mM NaCl, 5 mM βME and either 2 mM CaCl2 or 2 mM MgCl2 respectively]. After ∼5 min, C-21 was added to a final concentration of 500 nM. The emission was recorded before adding the C-21 and at 1–15 min intervals over the following 3 h: values are cited relative to that on adding the C-21. Parallel measurements were made on the enzyme alone, to correct for photobleaching of the Trp residues.
Fluorescence measurements over short time scales were recorded at 25°C in a SF61-DX2 stopped-flow fluorimeter (Hi-Tech Scientific): excitation was at 290 nm and emission (in arbitrary units) observed through a cut-off filter that transmits wavelengths >455 nm. For each experiment, ≥5 transients were averaged. Association reactions were carried out by mixing Alexa-21 with an equal volume of SfiI protein, both in Mg2+ or in EDTA fluorescence buffer (with 2 mM EDTA in place of MgCl2). For the dissociation of Alexa-21 from the D79A protein, a solution containing 50 nM D79A and 100 nM Alexa-21 in Mg2+ fluorescence buffer was mixed in the stopped-flow with an equal volume of 1 µM C-21. The subsequent decrease in the FRET signal was monitored.
RESULTS
Mutagenesis
In the crystal structure of the SfiI endonuclease bound to its recognition sequence in the presence of Ca2+ (45), the active site contains one Ca2+ ion, but this is located too far away from the target bond for any direct role in phosphodiester hydrolysis (Figure 1a). The Ca2+ is coordinated by Asp79 and Asp100 and by water molecules. In contrast, the crystal structure of BglI (46), a closely-related restriction enzyme, shows two Ca2+ ions per active site, ideally positioned for phosphodiester hydrolysis, one of which is coordinated by the aspartate residues that are analogous to Asp79 and Asp100 in SfiI (Figure 1b). Asp79 and Asp100 in SfiI correspond to the second and third carboxylates in the (E/D) … . PD … . (D/E)XK motif commonly found at the active sites of restriction enzymes. Hence, though Asp79 and Asp100 are too far away in the crystal structure for any direct role in catalysis, the crystal structure may reflect a pre-catalytic state (45) and that a metal bound to Asp79 and Asp100 may become involved in catalysis after conformational changes. To test this possibility, two mutants of the SfiI enzyme were constructed, in which Asp79 and Asp100 were replaced separately with alanine, to give D79A and D100A respectively.
The SfiI nuclease displays its optimal activity on DNA with two copies of its recognition sequence, in reactions with equimolar concentrations of enzyme and substrate (40,43). It interacts more readily with two sites in cis, on the same DNA, than with sites in trans, on separate DNA molecules (37,39), but excess enzyme over DNA leads to separate (inactive) tetramers of the enzyme at each site rather than a single (active) tetramer spanning two sites. The activities of wt SfiI and both the D79A and the D100A mutants were tested under these optimal conditions using as a substrate a 7.6 kb linear DNA with two SfiI sites separated by 1 kb (Figure 2). Linear rather than circular DNA was used so that the only products detected were those with double-strand breaks at one or both SfiI sites: products with single-strand breaks, of the sort made by non-specific nuclease contaminants, are not detected. Under these conditions, wt SfiI converted the entire DNA into the final product cut at both sites within 0.5 min, as expected (40). In contrast, neither the D79A nor the D100A mutants gave any detectable cleavage, even after overnight incubations. Hence, Asp79 and Asp100 are both essential for catalysis by SfiI, and substituting these residues with alanine destroys activity.
Figure 2.

Enzyme activities. The reactions contained, in reaction buffer at 50°C, 5 nM [3H]-labelled DNA (the linear form of pGB1, a plasmid with two SfiI sites), and 5 nM enzyme: (a), wt SfiI; (b), D79A; (c), D100A. Aliquots were taken at various times after adding the enzyme to the DNA and quenched immediately. The quenched samples analysed by electrophoresis through agarose and the amounts of each form of the DNA were determined as in the ‘Materials and Methods’ section. For wt SfiI (a), the following are shown: linear DNA substrate (marked S), filled circles; DNA cut at one SfiI site (marked P1), open triangles; the mean of two products after cutting both SfiI sites (marked P2), open squares. For D79A (b) and D100A (c), the following are shown: linear DNA substrate (marked S), filled circles; the sum of all cleaved products (P1 + P2), open inverted triangles. All three panels employ discontinuous time scales.
Molecular weights for the D79A and D100A proteins were determined, in parallel with the wt enzyme, by sedimentation equilibrium in the analytical ultracentrifuge. MW values of 117, 116 and 112 kDa were obtained for the wt, the D79A and the D100A proteins, respectively (data not shown). The subunit MW for SfiI is 31 kDa so both the D79A and D100A proteins are, like the wt, tetramers. The D79A and D100A variants thus seem to be suitable systems with which to explore the effects of Ca2+ and Mg2+ on DNA binding by SfiI.
Association by gel shifts in Ca2+
DNA binding by wt and mutant SfiI proteins was initially examined by the gel shift method. Two substrates of different lengths were used: HEX-35 and HEX-21 (Table 1), 35 and 21 bp long, respectively. Both contained the recognition sequence for SfiI and carried at the 5′ end of one strand a hexachlorofluorescein (HEX) moiety, so that after electrophoresis the labelled DNA could be detected by fluorescence imaging. Previous gel shift studies with wt SfiI had revealed DNA–protein complexes only with cognate duplexes and only in the presence of Ca2+ (18,39). Under these conditions, the addition of wt SfiI to HEX-21, or to HEX-35, gave in each case a single retarded complex; the 21 bp complex was retarded less than the 35 bp complex (Figure 3). However, when wt SfiI was added to mixtures of HEX-21 and HEX-35, it yielded—as before (18,39)—three complexes: one with the mobility of the 21 bp complex, another equivalent to the 35 bp complex and a third with intermediate mobility (Figure 3). The yields of the three complexes varied with the ratio of the two duplexes in a binomial manner. The most retarded complex must therefore be the SfiI tetramer bound to two 35 bp duplexes and the least retarded the tetramer with two 21 bp duplexes, while the intermediate carries one of each: these will be noted as EA2, EB2 and EAB respectively.
Figure 3.

DNA binding. Samples were prepared by adding SfiI enzyme (in the lanes marked +), or a buffer blank (lanes marked −), to the following preparations of DNA in Ca2+ binding buffer: HEX-21 alone; mixtures of HEX-21 and HEX-35; HEX-35 alone. The SfiI protein, either wt, D100A or D79A, as indicated above the panel (in red, blue and fuscia, respectively), was at a final concentration of 5 nM. The DNA was at a total concentration of 10 nM: the individual concentrations of HEX-21 and HEX-35 in each sample are as indicated above the corresponding lane. After 30 min at room temperature, the samples were subjected to electrophoresis through polyacrylamide and the gels analysed in a PhosphorImager to record the HEX fluorescence. The electrophoretic mobilities of free HEX-21 and HEX-35 are indicated by arrows on the left of the gel. The mobilities of three DNA–protein complexes are also marked by the cartoons on the left: in order of increasing mobility, the SfiI tetramer bound to two molecules of HEX-35, to one HEX-35 and one HEX-21, and to two molecules of HEX-21.
Under the same conditions with Ca2+ present, the addition of the D100A mutant of SfiI to either HEX-21 or HEX-35, or to mixtures of the two, gave the same series of retarded complexes as the native enzyme, as did also the D79A mutant (Figure 3). Hence, like wt SfiI, both mutants exist as tetramers that can bind two DNA molecules at the same time. Moreover, the concentration of tetrameric protein required to bind essentially the entire DNA preparations in the mixtures of HEX-21 and HEX-35 was one-half of the total DNA concentration (10 nM). Hence, in the presence of Ca2+, both mutant and wt proteins bind the duplexes with high affinities (KD << 10 nM).
Dissociation by gel shifts in Ca2+
To measure the dissociation of DNA bound to SfiI in the presence of Ca2+ ions, the enzyme—either wt SfiI or D100A—was first incubated with a labelled DNA that contained the recognition sequence (HEX-35): the labelled DNA was at a lower concentration than DNA-binding sites in the SfiI tetramer, to ensure almost all of it was bound. A 10-fold excess of an unlabelled DNA, that also had the recognition site (C-21: Table 1), was then added. The samples were analysed by electrophoresis through polyacrylamide to separate the various forms of the labelled DNA: the enzyme carrying two molecules of HEX-35, or one HEX-35 and one C-21, or free HEX-35 after its dissociation from the enzyme (Figure 4a). The additions of the unlabelled DNA to the samples of the labelled DNA-SfiI mix were made at various times before loading the samples onto the gel, and the amounts of each form of HEX-35 evaluated at each time point tested (Figure 4b).
Figure 4.

Dissociation in Ca2+: gel shifts. (a) Each sample contained, in Ca2+ binding buffer, 7.5 nM enzyme (wt SfiI, left-hand lanes; D100A, right-hand lanes) and 10 nM HEX-35. After 30 min at room temperature, C-21 was added to a final concentration of 100 nM and the incubation at room temperature continued until the sample was loaded onto a polyacrylamide gel. The interval (in h) between the time of addition of C-21 to each sample and the time when all the samples were loaded onto the gel is recorded above each lane: for wt SfiI, 0–24 h; for D100A, 0–3 h. After electrophoresis, the gel was analysed in a Phosphorimager to record the HEX fluorescence. The electrophoretic mobilities of SfiI bound to two molecules of HEX-35 (EA2), to one HEX-35 and one C-21 (EAB) and that of the free HEX-35 (A) are marked on the right of the gels. (b) The fluorescence intensity from each band in the gel with D100A was measured to assess the amounts of HEX-35 in the following species: D100A bound to two HEX-35 molecules, open circles; D100A bound to one HEX-35 and one C-21, closed triangles; free HEX-35, open squares. The decline in the concentration of the species with two HEX-35 molecules with time was fitted to a single and to a double exponential decline to an end-point of 1.0 nM (to account for the 10-fold excess of C-21 over HEX-35). The best fit to a single exponential (red line) gave a rate constant of 2.2(±0.2) × 10−3 s−1 and the best fit to a double exponential (blue line) the following parameters: fast phase, 3.8 nM at 1.4(±0.2) × 10−3 s−1; slow phase, 5.2 nM at 1.7(±0.1) × 10−4 s−1.
When C-21 was added to wt SfiI bound to HEX-35, a small fraction (<10%) of the doubly-bound complex (Figure 4a) was converted in <30 min to the hybrid complex, with one HEX-35 and one C-21, and to free HEX-35. However, virtually no further dissociation of the HEX-35 occurred over the following 24 h. This behaviour contrasts steeply with that observed after the addition of wt SfiI to a mixture of 21 and 35 bp DNA molecules, which gave close to a binomial distribution between the bound species (Figure 3), which in turn shows that the enzyme must have approximately equal affinities for the 21 and the 35 bp duplexes. Hence, the inability of the 21 bp DNA to displace the 35 bp duplex from wt SfiI cannot be due to too low an affinity. The binding of C-21 must instead be limited kinetically by the prior dissociation of HEX-35, which for wt SfiI with Ca2+ must have a half-time (τ½) of >>24 h.
Conversely, the addition of C-21 to the complex of D100A with HEX-35 led eventually to the displacement of about 90% of the HEX-35 from the mutant, the level expected given the 10-fold excess of C-21. However, the decline in the concentration of the species carrying two molecules of HEX-35 showed biphasic kinetics (the best fit to a single exponential decay deviated substantially from the data: Figure 4b): about 33% of the decline occurred at a relatively rapid rate (1.4 × 10−3 s−1, τ½ = 8 min) and the remaining 67% about 10 times more slowly (1.7 × 10−4 s−1, τ½ = 68 min). If the dissociation rate is limited by a single step, for example
then the decline in the concentration of EA2 will follow a single exponential (Supplementary Figure 1). The simplest scheme to account for the biphasic decline is that it is governed by a slow equilibration between two forms of the EA2 complex,
so that one fraction of the decline in the concentration of the species with two HEX-35 molecules occurs at the relatively fast rate of the EA2 → EA step while the remainder is limited by the slower E * A2 → EA2 transition (Supplementary Figure 1). The partition between the fast and slow phases can then be assigned to the initial ratio of [EA2] to [E*A2].
This scheme implies that, for D100A bound to HEX-35 in the presence of Ca2+, ∼33% of the DNA-protein complex is in the EA2 state, from which the DNA dissociates with a τ½ of 8 min, while ∼67% starts from the E*A2 state, from which the rate of dissociation of the DNA is limited by the E*A2 → EA2 transition, which occurs with τ½ at 68 min. Moreover, the same scheme can accommodate the behaviour of wt SfiI with Ca2+. With the wt enzyme, only a small fraction (∼10%) of the DNA–protein complex appears to be in its EA2 state, from which the DNA can dissociate within 30 min, while the remainder is trapped in an E*A2 state. For wt SfiI in Ca2+, the E*A2 → EA2 step fixes the half-time for DNA dissociation at >>24 h.
Ca2+ ions thus cause wt SfiI to bind almost irreversibly to its recognition sequence (Figure 4a). However, Ca2+ has a less severe effect on DNA binding by the D100A mutant of SfiI, as the dissociation from the DNA from the mutant proceeds to completion over a finite—albeit extended—time scale, the complete process taking ∼3 h.
FRET in Ca2+
As in previous studies with EcoRV (17), the dynamics of the interactions of the D100A and D79A proteins with DNA were characterized by FRET. The dye Alexa Fluor 350 was attached to the 5′ end of a 21 bp DNA via a C6 linker (Alexa-21: Table 1). Each subunit in the SfiI tetramer contains three Trp residues (45) and the fluorescence emission from tryptophan overlaps the excitation spectrum of Alexa Fluor 350. Following excitation of the Trps, the emission from the Alexa Fluor 350 will be enhanced whenever the dye on the DNA is located close to the Trp residues in the protein. A 21 bp substrate was used, rather than the 35 bp duplex, so that when the DNA is bound to the protein, the label is within energy transfer distance of the Trp residues in the adjacent subunit of the protein; it will be 25–40 Å away. However, an Alexa Fluor moiety attached to one end of the 21 bp DNA will be within 65 Å of the Trps in all four subunits, still inside the distance for at least some transfer, so the FRET signal will not necessarily vary linearly with the extent of binding. For example, the FRET signal from one bound Alexa-21 may be >50% of that from two, as a single dye may be able to capture >50% of the total energy transfer from all 12 Trp residues. Consequently, no attempt was made here to evaluate donor-acceptor distances from the amplitudes of the FRET changes and instead this signal was used solely to monitor the kinetics of DNA binding.
The FRET method was tested by first applying it to a reaction that had been characterized previously by gel retardation, the displacement of labelled DNA from D100A with an unlabelled competitor in the presence of Ca2+ (Figure 5a). The labelled DNA was Alexa-21 while C-21 was again employed as the competitor. The addition of the competitor resulted in a decrease in the emission from the Alexa-21 over a ∼3 h time period. The decrease followed similar kinetics to those for the decline in the concentration of the complexes with two labelled duplexes that had been measured by gel retardation (Figure 4). Both gave biphasic curves that could be fitted to double (but deviated from single) exponentials, with virtually the same parameters for both fast and slow phases: in both cases, ∼40% of the decline occurred at a relatively rapid rate (1.4 × 10−3 to 1.9 × 10−3 s−1) and ∼60% at a ∼10-fold slower rate (1.6 × 10−4 to 1.7 × 10−4 s−1). However, since the FRET signal may not be related linearly to the degree of saturation of the SfiI tetramer with Alexa-21, the biphasic decline in the fluorescence emission cannot be correlated to a reaction mechanism. Consequently, in all of the following, the changes in the FRET signal with time are related to a single rate constant, to indicate the overall time scale. For the dissociation of Alexa-21 from D100A in Ca2+, the best fit to a single constant gave a value of 5.4 × 10−4 s−1 (Figure 5a).
Figure 5.
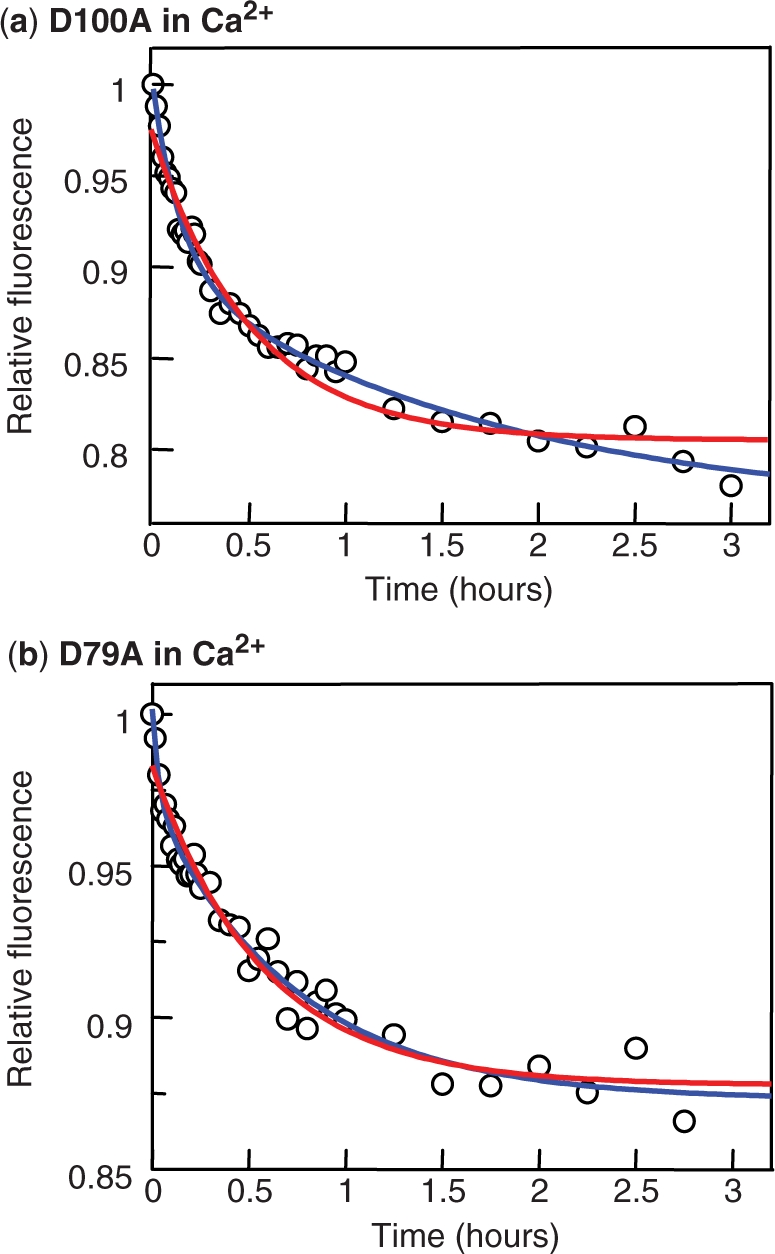
Dissociation in Ca2+: FRET. Reactions were conducted by first incubating, in a cuvette in the spectrofluorimeter, 50 nM Alexa-21 and 25 nM SfiI protein [in (a) D100A; in (b) D79A] in Ca2+ fluorescence buffer at 25°C before adding C-21 to a final concentration of 500 nM. Fluorescence excitation was at 290 nm and emission observed at 438 nm: values cited are relative to that directly after the addition of C-21. In both panels, the decrease in emission with time was fitted to a single exponential decline (red line) and to a double-exponential (blue line), both with offsets. For D100A (a), the best fits were with the following parameters: single exponential, ΔF (change in relative fluorescence) = −0.17(±0.01), k (rate constant) = 5.4(±0.5) × 10−4 s−1; double exponential fast phase, ΔF1 = –0.10(±0.01), k1 = 1.9(±0.3) ×10−3 s−1; double exponential slow phase, ΔF2 = −0.13(±0.01), k2 = 1.6(± 0.6) × 10−4 s−1. For D79A (b), the best fits were as follows: single exponential, ΔF = −0.11(±0.004), k = 4.9(± 0.5) × 10−4 s−1; double exponential fast phase, ΔF1 = –0.03 (±0.01), k1 = 7.8(±3.4) × 10−3 s−1; double exponential slow phase, ΔF2 = −0.10(±0.004), k2 = 3.8(±0.5) × 10−4 s−1.
When the complex of the D79A variant of SfiI with Alexa-21 formed in the presence of Ca2+ was challenged with excess C-21 (Figure 5b), the FRET signal decreased over the same time scale, again taking ∼3 h to reach completion. In this case, unlike D100A, the double exponential fit was no significant improvement over the single-exponential scheme. The dissociation of DNA from D79A fitted best to a single rate constant, whose value (4.9 × 10−4 s−1) was similar to the single constant for the dissociation from D100A (5.4 × 10−4 s−1). Hence, while DNA bound to wt SfiI in the presence of Ca2+ remains more or less glued to the protein forever, DNA bound to the D79A and D100A mutants can under these conditions dissociate from the protein, though the dissociation is slow: both mutant take ∼3 h to reach completion.
FRET in Mg2+
When tested for DNA cleavage in the presence of Mg2+, neither the D79A nor the D100A mutants showed any detectable level of activity (Figure 2). Provided their lack of activity is not due to an inability to bind DNA in buffers containing Mg2+, these variants might allow for an analysis of the effect of Mg2+ on DNA binding by SfiI. To determine whether these proteins could bind DNA when Mg2+ was present, varied concentrations of either mutant were mixed with Alexa-21 in the stopped-flow fluorimeter. Upon excitation of the Trp residues, any binding of the duplex to the protein should lead to enhanced emission from the Alexa-21. In all cases, an enhanced emission was observed. Both the rate and the amplitude of the increase varied with the protein concentration, higher concentrations leading to faster rates and larger amplitudes (Figure 6). However, the amplitude reached its maximal value, when all of the Alexa-21 was bound, at a lower concentration of D100A (Figure 6a) than of D79A (Figure 6b). Hence, in Mg2+-buffer, the D100A protein has a higher affinity than D79A for cognate DNA. In addition, the rate of association at each concentration tested was faster with D100A than D79A. The variations in the rate with protein concentration indicate that, under these conditions, the bimolecular rate constant for DNA binding by D100A is of the order of 6 × 108 M−1s−1 while that for D79A is about 2 × 108 M−1s−1.
Figure 6.
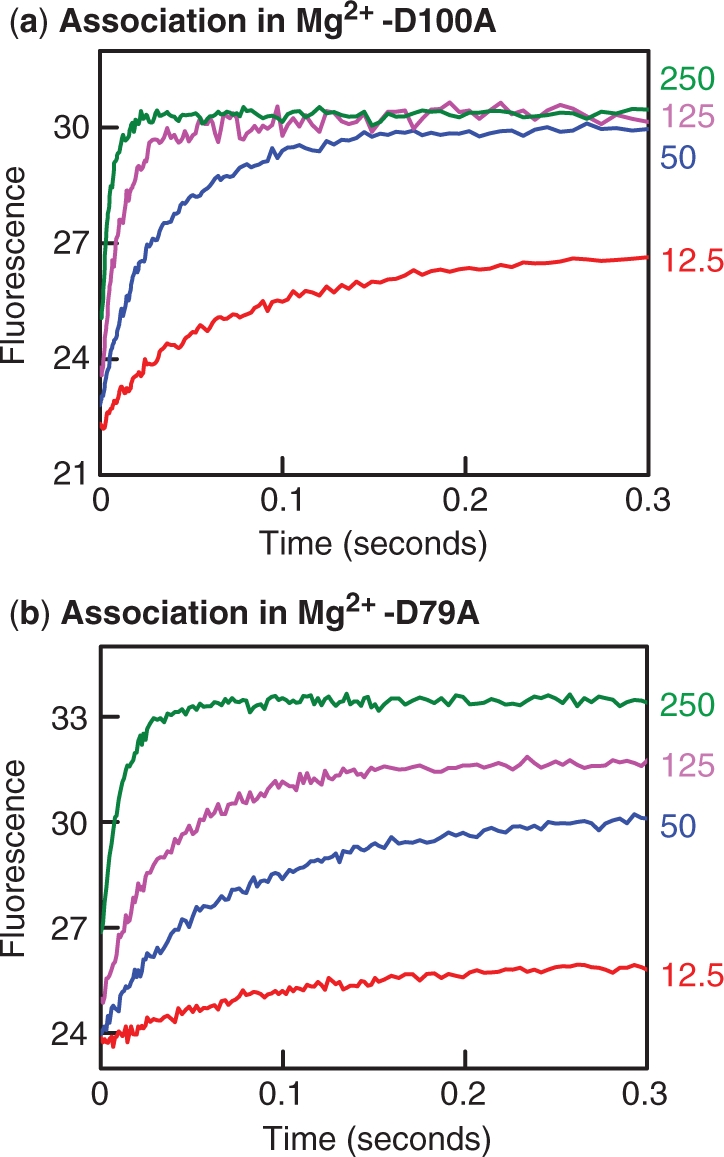
Association in Mg2+. Equal volumes of Alexa-21 and a variant of the SfiI endonuclease, D79A (a) or D100A (b), were mixed in the stopped-flow fluorimeter to give reactions in Mg2+ fluorescence buffer at 25°C that contained 20 nM Alexa-21 and one of the following concentrations of SfiI protein (as indicated on the right): 12.5 nM, in red; 50 nM, in blue; 125 nM in fuchsia; 250 nM, in green. The increase in emission from Alexa-21, following Trp excitation was monitored with time and is shown in arbitrary units.
The effect of Mg2+ on the dissociation of DNA from the D100A and the D79A proteins was examined by the same method as used for the dissociation in Ca2+ (Figure 5), by using the FRET signal to monitor the displacement of Alexa-21 from the protein after adding an excess of an unlabelled competitor DNA, C-21 (Figure 7). For the D100A protein in Mg2+-buffer, the decrease in fluorescence again occurred over a 3 h time scale (Figure 7a), similar to that in Ca2+-buffer (Figure 5a). In marked contrast, the displacement of Alexa-21 from D79A in Mg2+-buffer occurred too rapidly to measure by manual mixing in a standard spectrofluorimeter. The decrease in fluorescence was monitored instead by using a stopped-flow device to add the competitor to the DNA–protein complex (Figure 7b): the displacement was complete in <1 min.
Figure 7.
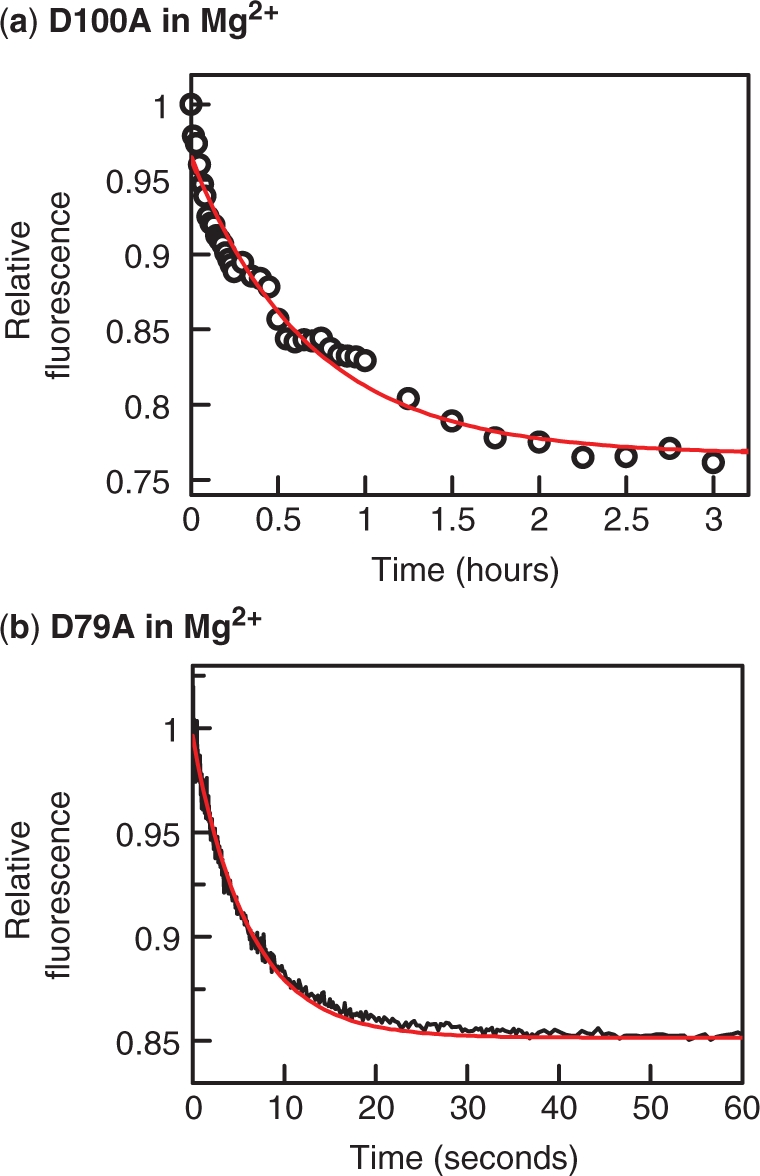
Dissociation in Mg2+. Reactions, in Mg2+ fluorescence buffer at 25°C, were carried out by first incubating together Alexa-21 and SfiI protein and then adding C-21: after the addition, the final concentrations were 50 nM Alexa-21, 25 nM protein and 500 nM C-21. In (a), the SfiI protein was D100A and the C-21 was added by pipette to a cuvette containing the other components: the subsequent change in fluorescence was recorded in a spectrofluorimeter over a 3-h period. In (b), the protein was D79A and the C-21 was added by using the stopped-flow fluorimeter: the subsequent change in fluorescence was recorded over a 1 min period. The red lines in both panels indicate the best fits to single exponentials, which gave rate constants of 4.1(±0.4) × 10−4 s−1 for the dissociation from D100A (a) and 0.16(±0.1) s−1 for that from D79A (b). Fluorescence readings are cited relative to that directly after the addition of the C-21.
The rate constant for dissociation from the D79A protein is thus very much faster in Mg2+ (0.16 s−1: Figure 7b) than in Ca2+ (4.9 × 10−4 s−1: Figure 5b), while the dissociation from D100A occurs at similar rates in Mg2+ (4.1 × 10−4 s−1: Figure 7a) and Ca2+ (5.4 × 10−4 s−1). While D79A and D100A behave similarly in solutions containing Ca2+, they differ markedly in buffers with Mg2+. Under the latter conditions, D79A binds the cognate sequence with a lower affinity than D100A, due in part to a slower association rate but in the main to a much faster dissociation rate.
FRET in EDTA
In several restriction enzymes, mutations at the active site carboxylates enhance the affinity of the protein for DNA and allow for sequence-specific binding in the absence of divalent metal ions (13,16,23,29). To investigate whether this might also apply to SfiI, DNA binding and displacement experiments were carried in a buffer containing EDTA, to remove divalent metal ions. The DNA used here was Alexa-21 so that the existence—or otherwise—of a DNA–protein complex could again be detected by FRET.
When the D79A mutant of SfiI was mixed with Alexa-21 in EDTA-buffer, an increase in FRET was noted (Figure 8a). Both the rate and the amplitude of the increase were similar to those in comparable reactions in Mg2+-buffer (Figure 6b): they again gave an association rate constant of ∼2 × 108 M−1s−1. The D79A protein must therefore be able to bind DNA in the absence of divalent metal ions. However, when the complex formed between D79A and Alexa-21 in EDTA was challenged with excess C-21, the FRET signal declined rapidly (Figure 8b), to give a fast rate constant for the dissociation step (1.35 s−1). The half-time for the release of Alexa-21 from D79A thus changes from 0.5 s in EDTA (Figure 8b) to 4 s in Mg2+ (Figure 7b) to 1400 s in Ca2+ (Figure 5b). Hence, even though D79A can bind DNA in the absence of metal ions, its affinity for DNA is lower than with either Mg2+ or Ca2+ present, as the faster dissociation rate creates a higher KD.
Figure 8.
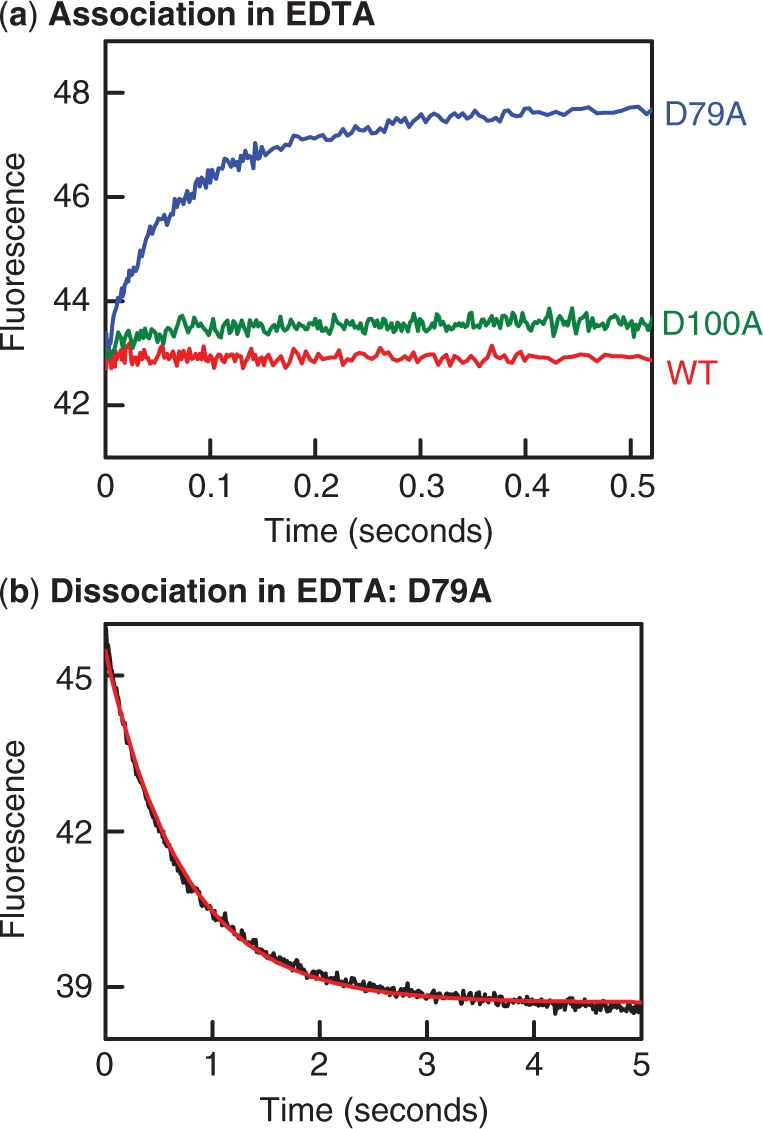
Binding and dissociation in EDTA. (a) Reactions to examine the binding of SfiI to DNA were carried out by mixing in the stopped flow fluorimeter equal volumes of Alexa-21 and SfiI protein, to give reactions at 25°C that contained 50 nM Alexa-21 and 25 nM protein in EDTA fluorescence buffer. The changes in fluorescence observed during the reactions with each protein are shown as follows: wt SfiI, red trace; D100A, green trace; D79A, blue trace. (b) The dissociation of DNA from the D79A protein was examined by mixing in the stopped flow fluorimeter one solution of Alexa-21 and D79A in EDTA fluorescence buffer with an equal volume of C-21 (also in EDTA fluorescence buffer), to give a reaction at 25°C that contained 25 nM D79A, 50 nM Alexa-21 (initially bound to the protein) and 500 nM C-21. The change in fluorescence was monitored: the red line indicates the best fit to a single exponential, to give a rate constant of 1.35(±0.01)s−1. In (a) and (b), fluorescence intensities are cited in arbitrary units.
When added to Alexa-21 in EDTA, the D100A mutant of SfiI caused only a small increase in FRET, while wt SfiI gave no detectable change (Figure 8a). Hence, at the concentrations tested, only a small amount of complex was generated with D100A, and seemingly none with wt SfiI. The D100A protein thus binds DNA very much more readily in the presence of divalent metal ions than in their absence while DNA binding by wt SfiI is undetectable without divalent metal ions. The latter concurs with previous gel-shift studies (18), in which wt SfiI had bound specifically to its recognition sequence in the presence of Ca2+ but showed no binding without divalent metals.
DISCUSSION
Enzymes that act on nucleic acids often need Mg2+ or a similar metal ion for their catalytic reactions (1–3,7–9), though they generally (but not always: 10) have no activity with Ca2+ in place of Mg2+. In many instances, Mg2+ also plays a role in binding the enzyme to the nucleic acid (29). Ca2+ ions have been widely used as mimics of Mg2+ in equilibrium binding studies, to promote DNA binding without catalysis (13,18–24) and in structural studies, to give an inactive enzyme–Ca2+–DNA complex whose structure can be determined and which may reflect the structure of the transient enzyme–substrate complex with Mg2+ (1–3,45,46). The structures of nucleases bound to a nucleic acid and Ca2+ often show two Ca2+ ions at the active site, in appropriate positions to catalyse the hydrolysis of the target phosphodiester bond by a two-metal-ion mechanism (1,25), though some contain the Ca2+ ion(s) in inappropriate positions (45,49,50). In binding studies, Ca2+ generally acts as a faithful mimic of Mg2+ (13,21,22). For example, with EcoRV, different duplexes with various base analogues and different mutant enzymes yielded a wide range of binding constants but in all cases the ΔΔG° values were the same with Ca2+ as with Mg2+ (24).
Under the reaction conditions used for the binding studies reported here but with Mg2+ present (40,48), the SfiI endonuclease binds rapidly to two recognition sites and proceeds to cleave the DNA at both sites within 30 s, to generate the enzyme–product complex. At 25°C, the enzyme–product complex dissociates slowly and this limits the turnover rate of the enzyme to about 1 mol (two-site) DNA per mol enzyme tetramer per hour. [SfiI has a much higher turnover rate at 50°C (40), the standard temperature for its reactions (42).] As in previous studies (18), the native enzyme failed to bind to its recognition site in the absence of divalent metals (Figure 8) but bound avidly to its cognate sequence in the presence of Ca2+ (Figure 3). Hence, as with many other restriction enzymes, DNA binding by wt SfiI requires divalent metal ions. However, when the Ca2+ complex was challenged with excess competitor DNA, the majority of the DNA already bound to the enzyme was still bound 24 h later (Figure 4a). Even though the enzyme–product complex generated by cleaving the DNA in the presence of Mg2+ has a remarkably long lifetime, about 1 h at 25°C, the lifetime of the enzyme–substrate complex in Ca2+, >>24 h, is very much longer than that for any intermediate in the reaction pathway with Mg2+.
The SfiI restriction enzyme thus binds DNA in the presence of Ca2+ to give an aberrant complex that bears no resemblance, at least in kinetic terms, to any complex from its reaction pathway with Mg2+. It is tempting to speculate that the crystal structure of the SfiI–DNA complex formed with Ca2+ reflects this aberrant complex as the metal ion and/or the DNA in that structure is out of position for catalysis (45). Still, the indefinite stability of this enzyme–Ca2+–DNA complex accounts for why Ca2+ blocked the native enzyme from releasing the loops it traps on plasmids with two SfiI sites, even after 7 h (44). This behaviour contrasts with that of another tetrameric restriction enzyme, Cfr10I: in the presence of Ca2+, DNA loops trapped on a plasmid with two Cfr10I sites were released in <2 min (44). In the following paper (48), a single molecule assay reveals that wt SfiI can form a loop on DNA with two cognate sites but that, in the presence of Ca2+, loop breakage is never observed.
For wt SfiI, in contrast to many other Mg2+-dependent enzymes acting on DNA, Ca2+ is clearly an invalid analogue of Mg2+. Ca2+ may promote DNA binding by SfiI but the SfiI–Ca2+–DNA complex is not a facsimile of the SfiI–Mg2+–DNA complex that just lacks catalytic activity. On the other hand, the D100A mutant of SfiI also binds to DNA more readily in the presence of divalent metal ions (Figure 6a) than in their absence (Figure 8a) but in this case the lifetime of the DNA–protein complex formed in Ca2+ (Figures 5a) is similar to that with Mg2+ (Figure 7a). Hence, with D100A, Ca2+ duplicates the behaviour of Mg2+ and so is a faithful mimic of Mg2+ for this particular protein. However, with either metal, D100A takes >3 h to dissociate from specific DNA, though this rate is still much faster than that for wt SfiI with Ca2+.
In the wt enzyme, Asp79 and Asp100 are the second and third carboxylates in the (E/D) … PD … (D/E)XK motif (Figure 1a). Of the SfiI proteins tested here, only D79A bound DNA readily in the absence of divalent metal ions (Figure 8a). Any electrostatic repulsion that might exist in the absence of metal ions between the active-site carboxylates and the DNA phosphates, of the type seen with BamHI and MunI (13,22), must therefore stem mainly from Asp79 rather than Asp100. Though the rate constants for the association of D79A with the SfiI recognition sequence were approximately the same in either the absence of divalent ions or in the presence of Mg2+ (∼2 × 108 M−1s−1 in both cases: Figures 8a and 6b, respectively), it dissociated from the specific DNA much more rapidly in EDTA (τ½ = 0.5 s: Figure 8b) than in either Mg2+ (τ½ = 4 s: Figure 7b) or Ca2+ (τ½ = 1400 s: Figure 5b).
While both Mg2+ and Ca2+ enhance the binding of D79A to DNA, the D79A protein behaves like wt SfiI in forming a more stable complex with Ca2+ than with Mg2+. Even so, the fact that Ca2+ is not equivalent to Mg2+ for DNA binding by D79A is largely immaterial as the inactivity of this mutant (Figure 2) means that its equilibrium binding to DNA can be analysed in the presence of the natural cofactor for SfiI, Mg2+. The D79A protein dissociates from its recognition site in the presence of Mg2+ over a relatively rapid time scale, ∼10 s, so with D79A and Mg2+ the DNA-binding process will reach equilibrium within an accessible time period. In contrast, for wt SfiI with Ca2+ or for D100A with either Ca2+ or Mg2+, the equilibration between free and bound DNA occurs over highly extended time scales, too slow for practicable measurements.
The SfiI restriction endonuclease is now well established as a paradigm for DNA looping (31) but, for the wt enzyme with Mg2+, any loop that the enzyme might form on a DNA with two SfiI sites will be followed by DNA cleavage and the subsequent loss of the loop. In the presence of Ca2+, wt SfiI forms stable loops but never releases the loop (44,48), doubtless due to its infinitely slow dissociation from DNA (Figure 4). Consequently, wt SfiI with Ca2+ cannot be used to examine the dynamics of DNA loop formation and breakdown. The catalytically-inactive mutants tested here, D79A and D100A, also displayed slow dissociation rates from DNA in the presence of Ca2+ (Figures 4 and 5), so neither of these systems make suitable tests for DNA looping. However, the association and dissociation of the inactive mutants to/from the recognition sequence could be studied in the presence of Mg2+ (Figures 6 and 7). One of the inactive mutants, D100A, gave the same slow dissociation kinetics in Mg2+ as it had in Ca2+, so the D100A protein with Mg2+ remains inappropriate for looping dynamics. In contrast, Mg2+ resulted in a relatively rapid dissociation of D79A from its recognition sequence, so D79A with Mg2+ appears to be uniquely well-suited to the analysis of DNA loop formation and breakage by the SfiI restriction enzyme. The following paper (48) utilizes this system to explore the fundamental dynamics of DNA looping processes. Moreover, it is shown there (48) that, in the presence of Mg2+, the rate of loop formation by D79A is similar to that by wt SfiI, which shows that D79A is indeed a valid surrogate for the native enzyme in its DNA looping reactions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Wellcome Trust [grant 078794/Z/06]; Funding for open access charge: Wellcome Trust.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Susan Retter for technical support, Marks Dillingham and Szczelkun for aid and advice, and Gijs Wuite, Guus Harms and Niels Laurens for extensive collaborations.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint first authors.
REFERENCES
- 1.Brautigam CA, Steitz TA. Structural and functional insights provided by crystal structures of DNA polymerases and their substrate complexes. Curr. Opin. Struct. Biol. 1998;8:54–63. doi: 10.1016/s0959-440x(98)80010-9. [DOI] [PubMed] [Google Scholar]
- 2.Galburt EA, Stoddard BL. Catalytic mechanisms of restriction and homing endonucleases. Biochemistry. 2002;41:13851–13860. doi: 10.1021/bi020467h. [DOI] [PubMed] [Google Scholar]
- 3.Yang W, Lee J, Nowotny M. Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol. Cell. 2006;22:5–13. doi: 10.1016/j.molcel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 4.Lagunavicius A, Sasnauskas G, Halford SE, Siksnys V. The metal-independent Type IIS restriction enzyme BfiI is a dimer that binds two DNA sites but has only one catalytic centre. J. Mol. Biol. 2003;326:1051–1064. doi: 10.1016/s0022-2836(03)00020-2. [DOI] [PubMed] [Google Scholar]
- 5.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE—enzymes and genes for DNA restriction and modification. Nucleic Acids Res. 2007;35:D269–D270. doi: 10.1093/nar/gkl891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, Blumenthal RM, Degtyarev SK, Dryden DTF, Dybvig K, et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003;31:1805–1812. doi: 10.1093/nar/gkg274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts RJ, Halford SE. Type II restriction enzymes. In: Linn SM, Lloyd RS, Roberts RJ, editors. Nucleases. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1993. pp. 35–88. [Google Scholar]
- 8.Aggarwal AK. Structure and function of restriction endonucleases. Curr. Opin. Struct. Biol. 1995;5:11–19. doi: 10.1016/0959-440x(95)80004-k. [DOI] [PubMed] [Google Scholar]
- 9.Perona JJ. Type II restriction endonucleases. Methods. 2002;28:353–364. doi: 10.1016/s1046-2023(02)00242-6. [DOI] [PubMed] [Google Scholar]
- 10.Saravanan M, Vasu K, Kanakaraj R, Rao DN, Nagaraja V. R.KpnI, an HNH superfamily REase, exhibits differential discrimination at non-canonical sequences in the presence of Ca2+ and Mg2+ Nucleic Acids Res. 2007;35:2777–2786. doi: 10.1093/nar/gkm114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halford SE, Johnson NP. The EcoRI restriction endonuclease with bacteriophage lambda DNA. Equilibrium binding studies. Biochem. J. 1980;191:593–604. doi: 10.1042/bj1910593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terry BJ, Jack WE, Rubin RA, Modrich P. Thermodynamic parameters governing interaction of EcoRI endonuclease with specific and nonspecific DNA sequences. J. Biol. Chem. 1983;258:9820–9825. [PubMed] [Google Scholar]
- 13.Engler LE, Sapienza P, Dorner LF, Kucera R, Schildkraut I, Jen-Jacobson L. The energetics of the interaction of BamHI endonuclease with its recognition site GGATCC. J. Mol. Biol. 2001;307:619–636. doi: 10.1006/jmbi.2000.4428. [DOI] [PubMed] [Google Scholar]
- 14.Taylor JD, Badcoe IG, Clarke AR, Halford SE. EcoRV restriction endonuclease binds all DNA sequences with equal affinity. Biochemistry. 1991;30:8743–8753. doi: 10.1021/bi00100a005. [DOI] [PubMed] [Google Scholar]
- 15.Zebala J, Choi J, Barany F. Characterization of steady state, single-turnover, and binding kinetics of the TaqI restriction endonuclease. J. Biol. Chem. 1992;267:8097–8105. [PubMed] [Google Scholar]
- 16.Lagunavicius A, Siksnys V. Site-directed mutagenesis of putative active site residues of MunI restriction endonuclease: replacement of catalytically essential carboxylate residues triggers DNA binding specificity. Biochemistry. 1997;36:11086–11092. doi: 10.1021/bi963125i. [DOI] [PubMed] [Google Scholar]
- 17.Erskine SG, Halford SE. Reactions of the EcoRV restriction endonuclease with fluorescent oligodeoxynucleotides: identical equilibrium constants for binding to specific and non-specific DNA. J. Mol. Biol. 1998;275:759–772. doi: 10.1006/jmbi.1997.1517. [DOI] [PubMed] [Google Scholar]
- 18.Embleton ML, Williams SA, Watson MA, Halford SE. Specificity from the synapsis of DNA elements by the SfiI endonuclease. J. Mol. Biol. 1999;289:785–797. doi: 10.1006/jmbi.1999.2822. [DOI] [PubMed] [Google Scholar]
- 19.Vanamee ES, Santagata S, Aggarwal AK. FokI requires two specific DNA sites for cleavage. J. Mol. Biol. 2001;309:69–78. doi: 10.1006/jmbi.2001.4635. [DOI] [PubMed] [Google Scholar]
- 20.Daniels LE, Wood KM, Scott DJ, Halford SE. Subunit assembly for DNA cleavage by restriction endonuclease SgrAI. J. Mol. Biol. 2003;327:579–591. doi: 10.1016/s0022-2836(03)00143-8. [DOI] [PubMed] [Google Scholar]
- 21.Vipond IB, Halford SE. Specific DNA recognition by EcoRV restriction endonuclease induced by calcium ions. Biochemistry. 1995;34:1113–1119. doi: 10.1021/bi00004a002. [DOI] [PubMed] [Google Scholar]
- 22.Lagunavicius A, Grazulis S, Balciunaite E, Vainius D, Siksnys V. DNA binding specificity of MunI restriction endonuclease is controlled by pH and calcium ions: Involvement of active site carboxylate residues. Biochemistry. 1997;36:11093–11099. doi: 10.1021/bi963126a. [DOI] [PubMed] [Google Scholar]
- 23.Cao W. Binding kinetics and footprinting of TaqI endonuclease: Effects of metal cofactors on sequence-specific interactions. Biochemistry. 1999;38:8080–8087. doi: 10.1021/bi9903796. [DOI] [PubMed] [Google Scholar]
- 24.Martin AM, Horton NC, Luseti S, Reich NO, Perona JJ. Divalent metal dependence of site-specific DNA binding by EcoRV endonuclease. Biochemistry. 1999;38:8430–8439. doi: 10.1021/bi9905359. [DOI] [PubMed] [Google Scholar]
- 25.Horton JR, Blumenthal RM, Cheng X. Restriction endonucleases: Structure of the conserved catalytic core and the role of metal ions in DNA cleavage. In: Pingoud A, editor. Nucleic Acids and Molecular Biology, Restriction Endonucleases. Vol. 14. Berlin: Springer-Verlag; 2004. pp. 361–392. [Google Scholar]
- 26.King K, Benkovic SJ, Modrich P. Glu-111 is required for activation of the DNA cleavage center of EcoRI endonuclease. J. Biol. Chem. 1989;264:11807–11815. [PubMed] [Google Scholar]
- 27.Selent U, Ruter T, Kohler E, Liedtke M, Thielking V, Alves J, Oelgeschlager T, Wolfes H, Peters F, Pingoud A. A site-directed mutagenesis study to identify amino acid residues involved in the catalytic function of the restriction endonuclease EcoRV. Biochemistry. 1992;31:4808–4815. doi: 10.1021/bi00135a010. [DOI] [PubMed] [Google Scholar]
- 28.Engler LE, Welch KK, Jen-Jacobson L. Specific binding by EcoRV endonuclease to its DNA recognition site GATATC. J. Mol. Biol. 1997;269:82–101. doi: 10.1006/jmbi.1997.1027. [DOI] [PubMed] [Google Scholar]
- 29.Thielking V, Selent U, Kohler E, Landgraf Z, Wolfes H, Alves J, Pingoud A. Mg2+ confers DNA binding specificity to the EcoRV restriction endonuclease. Biochemistry. 1992;31:3727–3732. doi: 10.1021/bi00130a001. [DOI] [PubMed] [Google Scholar]
- 30.Mucke M, Kruger DH, Reuter M. Diversity of type II restriction endonucleases that require two DNA recognition sites. Nucleic Acids Res. 2003;31:6079–6084. doi: 10.1093/nar/gkg836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halford SE, Welsh AJ, Szczelkun MD. Enzyme-mediated DNA-looping. Annu. Rev. Biophys. Biomol. Struct. 2004;33:1–24. doi: 10.1146/annurev.biophys.33.110502.132711. [DOI] [PubMed] [Google Scholar]
- 32.Gowers DM, Bellamy SRW, Halford SE. One recognition sequence, seven restriction enzymes, five reaction mechanisms. Nucleic Acids Res. 2004;32:3469–3479. doi: 10.1093/nar/gkh685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirsanova OV, Baskunov, VB, Gromova ES. Type IIE and IIF restriction endonucleases interacting with two recognition sites on DNA. Mol. Biol. (Mosk) 2004;38:886–900. [PubMed] [Google Scholar]
- 34.Gemmen GJ, Millin R, Smith DE. Tension-dependent DNA cleavage by restriction endonucleases: Two-site enzymes are “switched off ” at low force. Proc. Natl Acad. Sci. USA. 2006;103:11555–11560. doi: 10.1073/pnas.0604463103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van den Broek B, Vanzi F, Normanno D, Pavone FS, Wuite GJL. Real-time observation of DNA looping dynamics of Type IIE restriction enzymes NaeI and NarI. Nucleic Acids Res. 2006;34:167–174. doi: 10.1093/nar/gkj432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Embleton ML, Siksnys V, Halford SE. DNA cleavage reactions by type II restriction enzymes that require two copies of their recognition sites. J. Mol. Biol. 2001;311:503–514. doi: 10.1006/jmbi.2001.4892. [DOI] [PubMed] [Google Scholar]
- 37.Wentzell LM, Nobbs TJ, Halford SE. The SfiI restriction endonuclease makes a four-strand DNA break at two copies of its recognition sequence. J. Mol. Biol. 1995;248:581–595. doi: 10.1006/jmbi.1995.0244. [DOI] [PubMed] [Google Scholar]
- 38.Siksnys V, Grazulis S, Huber R. Structure and function of the tetrameric restriction enzymes. In: Pingoud A, editor. Nucleic Acids and Molecular Biology, Restriction Endonucleases. Vol. 14. Berlin: Springer-Verlag; 2004. pp. 237–259. [Google Scholar]
- 39.Bellamy SRW, Milsom SE, Kovacheva YS, Sessions RB, Halford SE. A switch in the mechanism of communication between the two DNA-binding sites in the SfiI restriction endonuclease. J. Mol. Biol. 2007;373:1169–1183. doi: 10.1016/j.jmb.2007.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nobbs TJ, Szczelkun MD, Wentzell LM, Halford SE. DNA excision by the SfiI restriction endonuclease. J. Mol. Biol. 1998;281:419–432. doi: 10.1006/jmbi.1998.1966. [DOI] [PubMed] [Google Scholar]
- 41.Wentzell LM, Halford SE. DNA looping by the SfiI restriction endonuclease. J. Mol. Biol. 1998;281:433–444. doi: 10.1006/jmbi.1998.1967. [DOI] [PubMed] [Google Scholar]
- 42.Qiang B-Q, Schildkraut I. A type II restriction endonuclease with an eight nucleotide specificity from Streptomyces fimbriatus. Nucleic Acids Res. 1984;12:4507–4515. doi: 10.1093/nar/12.11.4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szczelkun MD, Halford SE. Recombination by resolvase to analyse DNA communications by the SfiI restriction endonuclease. EMBO J. 1996;15:1460–1469. [PMC free article] [PubMed] [Google Scholar]
- 44.Milsom SE, Halford SE, Embleton ML, Szczelkun MD. Analysis of DNA looping interactions by type II restriction enzymes that require two copies of their recognition sites. J. Mol. Biol. 2001;311:515–527. doi: 10.1006/jmbi.2001.4893. [DOI] [PubMed] [Google Scholar]
- 45.Vanamee ES, Viadiu H, Kucera R, Dorner L, Picone S, Schildkraut I, Aggarwal AK. A view of consecutive binding events from structures of tetrameric endonuclease SfiI bound to DNA. EMBO J. 2005;24:4198–4208. doi: 10.1038/sj.emboj.7600880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Newman M, Lunnen K, Wilson G, Greci J, Schildkraut I, Phillips SEV. Crystal structure of restriction endonuclease BglI bound to its interrupted DNA recognition sequence. EMBO J. 1998;17:5466–5476. doi: 10.1093/emboj/17.18.5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chmiel AA, Bujnicki JM, Skowronek KJ. A homology model of restriction endonuclease SfiI in complex with DNA. BMC Struct. Biol. 2005;5:2. doi: 10.1186/1472-6807-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laurens N, Bellamy SRW, Harms AF, Kovacheva YS, Halford SE, Wuite GJL. Dissecting protein-induced DNA looping dynamics in real time. Nucleic Acids Res. 2009 doi: 10.1093/nar/gkp570. (this issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perona JJ, Martin AM. Conformational transitions and structural deformability of EcoRV endonuclease revealed by crystallographic analysis. J. Mol. Biol. 1997;273:207–225. doi: 10.1006/jmbi.1997.1315. [DOI] [PubMed] [Google Scholar]
- 50.Etzkorn C, Horton NC. Ca2+ binding in the active site of HincII: implications for the catalytic mechanism. Biochemistry. 2004;43:13256–13270. doi: 10.1021/bi0490082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.