


Abstract
Sugar phosphate stress in Escherichia coli is sensed and managed by the transcriptional regulator SgrR and the small RNA (sRNA) SgrS. SgrS is a dual function RNA that performs base pairing-dependent regulation of mRNA targets and encodes a small protein, SgrT. Homologs of SgrR were analyzed for gene synteny and inter-homolog identity to identify those that are likely to be functionally analogous. These 22 SgrR homologs were used to manually locate adjacent sRNAs functionally analogous to SgrS. SgrS homologs shared little sequence identity with E. coli SgrS, but most shared several structural features. The most conserved feature of SgrS homologs was the base pairing region while the most variable feature was the sgrT-coding sequence. Analyses of predicted interactions between SgrS:ptsG mRNA pairs in different organisms revealed interesting differences in the patterns of base pairing interactions. RNA pairs with more interrupted regions of complementarity had a higher proportion of G:C base pairs than those with longer contiguous stretches of complementarity. The identification of this set of homologous sRNAs and their targets sets the stage for future studies to further elucidate the molecular requirements for regulation by SgrS.
INTRODUCTION
Small RNAs (sRNAs) are ubiquitous and have been identified in the genomes of organisms from all three domains of life. Numerous studies using a variety of global approaches (1–3) have identified more than 80 sRNAs in the genome of Escherichia coli K12. The most abundant class of sRNAs is comprised of those that post-transcriptionally regulate expression of target genes by a base pairing-dependent mechanism that requires the RNA chaperone Hfq (referred to as riboregulation). The most extensively studied member of this class is RyhB, which was first identified in E. coli K12 and has been shown to regulate numerous genes involved in cellular iron homeostasis (4–6). RyhB homologs have been identified in Erwinia chrysanthemi (7), Shigella dysenteriae (8), Vibrio cholerae (9,10) and Salmonella (11,12) species on the basis of nucleotide sequence identity with the E. coli RyhB. Functionally analogous sRNAs have been identified by other means in more distantly related species such as Pseudomonas aeruginosa (13) and Bacillus subtilis (14). These RyhB-like sRNAs are expressed under the same conditions (iron limitation) and regulate the same types of target genes as the RyhB sRNAs in E. coli and related organisms yet share little sequence similarity. How widely RyhB and other base pairing-dependent sRNAs occur in the bacterial world remains to be determined, and this is no small task given the divergence of primary sequences of sRNAs and the inherent difficulty of locating sRNAs in genomes of distantly related organisms. Nevertheless, elucidating the conserved structural and functional features of bacterial sRNAs is a crucial step toward understanding how they evolve.
In this study, the species distribution and conservation of the sRNA SgrS was investigated. SgrS is an Hfq-dependent sRNA that was first identified in E. coli (15,16). SgrS expression is induced by a metabolic stress referred to as glucose-phosphate stress, so named because stress is associated with intracellular accumulation of glucose-6-phosphate (G6P) or its analog α-methyl glucoside-6-phosphate (αMG6P) (15,17). SgrS is unique from other characterized Hfq-dependent sRNAs because it has two distinct functions. The first is a base pairing dependent riboregulation function that results in translational repression and degradation of mRNAs that encode glucose transporters of the phosphoenolpyruvate phosphotransferase system (PTS) (15,18). The second is an mRNA function whereby SgrS encodes the 43-amino-acid polypeptide SgrT. SgrT inhibits glucose transport by a mechanism independent of the riboregulation activity (19). Together, these two functions serve to prevent further uptake of G6P or αMG6P and thus promote recovery from glucose-phosphate stress (20).
The goals of the present study were 2-fold: (i) to define the distribution of SgrS and SgrT homologs and (ii) to analyze the conservation of determinants known from other studies (15,18,19) to be important for function. The genetic linkage of sgrS to sgrR, the gene encoding its regulator, greatly facilitated identification of SgrS homologs. Since sRNAs are typically not conserved at the primary nucleotide sequence level except between very closely related organisms, most homologs are not identifiable by BlastN searches. However, we were able to utilize sgrR as a genomic ‘handle’ to identify SgrS homologs that we would not have found by BlastN searches. This approach was used successfully to identify homologs of the sRNA GcvB, which, like SgrS, is encoded divergently from the gene encoding its transcriptional regulator, GcvA (21,22). In another study (C. S. Wadler and C. K. Vanderpool, submitted for publication), the functions of a subset of the SgrS homologs identified here were tested in a heterologous host (E. coli) in order to confirm their function and gain insight into the requirements for riboregulation and SgrT in the glucose-phosphate stress response.
Here we describe the identification of SgrR, SgrS and SgrT homologs in a variety of bacterial species. SgrR homologs are found in bacterial species belonging to a number of different phyla, however, those that we believe are most likely functionally analogous to E. coli SgrR are distributed only among the γ-Proteobacteria, primarily in enteric species. Likewise, our findings suggest that SgrS and SgrT homologs are mainly confined to enteric bacteria. Predictions for SgrS:target mRNA interactions suggest that the riboregulation function is relatively conserved, while comparison of SgrT homologs revealed that the mRNA function of SgrS is more variably distributed.
MATERIALS AND METHODS
Identification of SgrR homologs
The SgrR amino-acid sequence was retrieved from EchoBASE (23) and used as a query in a BlastP search against the non-redundant protein sequence databases at NCBI, UniProt database at EBI and the OMNIOME pep database at CMR. Additional BlastP searches using the same databases were performed using a subset of proteins obtained in the results of the primary search as queries. Results from all searches were curated to remove redundancy, which produced a list of 60 unique SgrR homologs. The Blast searches were completed in November 2008. The amino-acid sequences of the 60 SgrR homologs were aligned using ClustalW (Gonnet protein weight matrix) (The FASTA format amino-acid sequence alignment is available in Supplementary Figure S1). Phylogenetic and molecular evolutionary analyses were conducted using MEGA version 4 with the UPGMA, Neighbor Joining and Minimum Evolution algorithms (24). Homologs were divided into clades (Figure 1A and Supplementary Figure S2) based on the consensus among the trees. For analyses of gene synteny, chromosomal sequences were obtained from NCBI Entrez and EBI Genomes for each species containing a SgrR homolog. These GenBank files were analyzed using Artemis (Version 10) (25) from the Sanger Centre to examine gene context surrounding the sgrR homolog and obtain DNA sequences of the region between the nearest conserved upstream and downstream genes, tbpA and leuD, respectively. Where tbpA and/or leuD could not be found in close proximity to a sgrR homolog, Artemis (or WebACT) and coliBASE were used to identify their locations elsewhere on the chromosome. To further investigate the putative functions of genes in the sgrR neighborhood in different organisms, both coliBLAST (26) and EchoBASE (23) were used to find the corresponding E. coli homolog of the gene.
Figure 1.

(A) Summary of SgrR homologs belonging to the seven major clades. One representative homolog from each clade is described with its gene context shown. Coding sequences are color-coded as follows: sgrR homologs: blue; putative sugar transport or metabolic genes: green; unknown protein-coding sequences (CDS): orange. The gene synteny of all members has been investigated for sgrR (Clade 1) and ybaE (Clade 2) and the genes in purple are boundary genes that encode proteins that are not predicted to be functionally related. The HTH score and standard deviation (31) for proteins in a given clade are listed in the right-most column. (B) Two-way comparison between the representative members of each clade. Alignments were performed using ClustalW; inter-homolog identity (left value) and similarity (right value) are shown.
Identification of SgrS and SgrT homologs
The homologs of SgrR that appeared to be functionally analogous to E. coli K12 SgrR based on the above analyses were used as landmarks to look for a small RNA. The DNA sequence upstream of and on the opposite strand from the sgrR coding sequence through the next annotated gene was analyzed. All open reading frames within the retrieved sequences were compared against the SgrT amino-acid sequence of E. coli K12 using Bl2Seq (27). In all genomes where a peptide with identity to SgrT was found, Artemis was used to define the start and stop codons for the open reading frame. Promoter elements and sgrS transcription start sites were identified primarily by alignment of sequences with the E. coli K12 sgrS gene and upstream sequences for which these elements have been experimentally determined (15,28). In addition, promoter predictions were generated using the Softberry BPROM network server and the Neural Network Promoter Prediction by the BDGP using prokaryotic settings. The sequences analyzed by these programs encompassed the region between sgrR and sgrT homologs. The predictions generated by the BPROM network server supported predictions derived from sequence alignments more frequently than predictions generated by the BDGP algorithm. Putative intrinsic terminators for sgrS were identified by examining sequences downstream of the sgrT stop codon manually and searching for an inverted repeat followed by a run of Ts. Mfold (29) and RNAalifold (30) were used to determine the secondary structure of this region to confirm the predicted stem loop structure.
Identification of base pairing determinants in SgrS homologs
Homologs of ptsG were identified in each genome containing an sgrR homolog by BlastP using the E. coli PtsG protein as the query. (The FASTA format alignment is given in Supplementary Figure S3.) Gene synteny around these homologs was analyzed using coliBASE for localized genome alignments and WebACT for genome wide alignments between pairs of genomes downloaded from GenBank.
ClustalW alignments were performed to localize base pairing determinants in SgrS homologs. These analyses identified a highly conserved region near the 3′ end corresponding to the region required for the base pairing function of E. coli K12 SgrS (15,18). To define interactions between this region of SgrS and ptsG mRNA, a Microsoft Excel Macro (created by R.S.P.H., available upon request) was used to produce an alignment that yielded the maximal complementarity between the base pairing region of SgrS and the 5′ untranslated region (UTR) of ptsG mRNA (defined for this purpose as a region from −100 through +30 with respect to the start codons). The macro determined maximal complementarity by optimizing both the total number of interactions and the maximum length of contiguous interactions over a 50-nt window.
Alignments of SgrT and sgrR–sgrS intergenic region
The amino-acid sequences of the SgrT homologs were aligned using ClustalW. DNA sequences of the intergenic region between the initiation codons of SgrR and SgrT were aligned using ClustalW. (This alignment is given in FASTA format in Supplementary Figure S4.)
RESULTS AND DISCUSSION
Identification of homologs of SgrR
To determine the extent of conservation of SgrR, SgrS and SgrT, a systematic search for homologs was undertaken. This search identified a total of 60 potential SgrR homologs. Alignments and phylogenetic analyses allowed us to divide these 60 members into seven clades that contained homologs from more than one genus (Figure 1A and B; Supplementary Figure S2); an additional clade contained two homologs found only in Chromobacterium violaceum. Representative proteins from each of the seven main clades show between 24% and 39% identity to SgrR of E. coli K12 (DR_B042 from Deinococcus radiodurans and STM3860 from Salmonella enterica serovar typhimurium, respectively; Figure 1B). Within the clade defined by E. coli SgrR (Clade 1, Figure 1A), identity with SgrR of E. coli K12 ranges from 57% to 98% (plu3677 from Photorhabdus luminescens and SSON_0075 from Shigella sonnei, respectively). Clade 2 members are homologous to an E. coli protein of unknown function, YbaE, and can be found in a variety of other enteric species. Proteins belonging to Clade 3 are more closely related to those of Clade 1 (SgrR) than those in other clades (Figure 1B and Supplementary Figure S2); these proteins, found in Salmonella species, Chromobacterium violaceum, Yersinia enterocolitica and Aeromonas species, may have originated from the duplication of an ancestral sgrR-like gene. Homologs belonging to Clades 4, 5 and 6 are narrowly distributed among gram negative enteric bacteria and Clade 7 encompasses proteins from gram positive organisms (Figure 1A).
The helix-turn-helix (HTH) motif weight and frequency tables of Dodd and Egan (31) revealed that homologs from Clades 5 and 7 were not predicted to contain a functional HTH motif characteristic of DNA-binding proteins (indicated in Figure 1A). Thus, these proteins may not be transcription factors like E. coli SgrR. Alternatively, they may be bona fide DNA-binding proteins that are too divergent from the proteins in the training set (31) to be accurately analyzed. Of the SgrR homologs that contain a conserved HTH motif, several are located on the opposite DNA strand divergent from genes that encode proteins with putative functions related to sugar transport or metabolism. By analogy with the E. coli sgrR-sgrS organization and pattern of regulation, we hypothesize that the genes encoded divergently from sgrR homologs are targets of transcriptional regulation by these homologs.
To narrow the list of homologs to those likely to be functionally analogous to E. coli SgrR, we focused on members of Clades 1 and 3 (Supplementary Figure S2) and further analyzed gene synteny and identity to SgrR of E. coli K12. We also examined genes divergently transcribed from each of these homologs and looked for characteristics of small RNAs. This analysis cut the list of proteins likely to be functional analogs of SgrR to 22. (Hereafter, when referring to ‘SgrR homologs’, we mean this subset likely to be functionally analogous to E. coli K12 SgrR.) The gene neighborhoods of putative SgrR homologs are depicted in Figure 2. Two genes that flank the sgrR region in all but three genomes and have no obvious link to the function of SgrRST are tbpA, which encodes the extracytoplasmic solute receptor of the thiamine ABC transporter, and leuD, encoding a leucine biosynthesis enzyme. Both of these genes were present near the location of all putative sgrR homologs, except those of Aeromonas species and Photorhabdus luminescens. In Aeromonas genomes, the tbpA and leuD genes are present in close proximity to one another but distant from the sgrR homolog. In P. luminescens, the leuD gene is encoded upstream of the sgrR homolog, but a genome rearrangement appears to have translocated tbpA/thiPQ and mutH/ygdB (Figure 2). The sgrR homologs of these organisms have the lowest levels of identity to E. coli K12 SgrR out of the set of 22 homologs. Although there are contradictory indicators for the sgrR homologs from Aeromonas species and P. luminescens, we retained them on the list of putative functional analogs because we found signatures of sgrS and/or sgrT adjacent to them.
Figure 2.

Gene synteny of the region centered on sgrRST. The gene synteny of each specific SgrR homolog is shown with the homolog in blue. The genes-encoding TbpA and LeuD are considered boundaries of the region and are shown in purple. CDS are open reading frames encoding proteins of unknown function (and absent in E. coli K12). Genes-encoding proteins whose functions are unknown or unlikely to be involved in sugar metabolic or transport processes are shown in orange, setA [encoding a sugar efflux pump (42,43)] is shown in pink. If SgrT is present, it is shown as a green arrow within the larger yellow SgrS arrow. The sizes of SgrS and SgrT are represented in proportion to each other and between species. Other CDS are not represented to scale. In the Klebsiella and Erwinia genomes the gene-encoding LeuD is further downstream of SgrS, shown by the dashed line and stated distance; the additional genes between sgrS and leuD are not individually represented. In the Aeromonas genomes, sgrR is not near leuD and tbpA; these are represented by a dotted line that indicates the genes are located elsewhere on the genome at the stated distance from sgrRST. The synteny around P. luminescens sgrR indicates that while leuD is encoded upstream of SgrR, an inversion has swapped the downstream gene, tbpA, for mutH and ygdQ that are normally encoded elsewhere on the chromosome as indicated for E. coli K12. The gene arrangements for Citrobacter rodentium and Serratia marcescens are not shown as genome sequencing and annotation is not complete.
Analysis of regulatory sequences adjacent to sgrR homologs supported our assignment of this subset of 22 proteins as likely functionally analogous to E. coli SgrR. The region upstream of the sgrR coding sequence is highly conserved (Supplementary Figure S5A and B). This is particularly true for the sequences that we have previously shown are important for transcriptional activation of sgrS (15,28). Furthermore, based on the conservation of the sequences that comprise the −10 element of the sgrS promoter in E. coli (15), we could assign a putative transcription start site for sgrS homologs from all organisms except Y. enterocolitica (Supplementary Figure S5A).
Identification of homologs of SgrS and SgrT
To find SgrS homologs, the region between sgrR and leuD was examined for features indicative of a small RNA. These features included: (i) a promoter that could not be assigned to any other known gene, (ii) a small open reading frame encoding a protein with identity to SgrT, (iii) a region with complementarity to E. coli ptsG mRNA or with the species’ cognate ptsG mRNA and (iv) an inverted repeat followed by a run of T residues (putative intrinsic terminator). We identified putative SgrS homologs that met at least three of the four criteria in all of the genomes containing SgrR homologs (Figure 2, Table 1). In several cases, not all features were present, for example several putative SgrS homologs did not encode SgrT. Alignments of all SgrS homologs showed significant levels of identity between SgrS sequences only from organisms most closely related to E. coli (Supplementary Figure S6A). Although sequence identity across the whole length of sgrS homologs was minimal, a short stretch (13 nt) near the 3′ end was nearly invariant. This region corresponds to the sequences of E. coli K12 SgrS that constitute the determinants for base pairing with ptsG mRNA. The consensus sequence for this region is 5′- CUGAGUAuUGGUG-3′ where the central ‘u’ is the only position that varies among all SgrS homologs except those from Aeromonas and Photorhabdus species. The 22 SgrS homologs are found exclusively in species of γ-Proteobacteria, and with the exception of Aeromonas species, all are members of the Enterobacteriaceae. This distribution is similar to that of several other Hfq-dependent sRNAs (32–34), but is narrower than that of the GcvB sRNA (21) which is found in a broader range of γ-Proteobacteria.
Table 1.
Characteristics of SgrS homologs and base pairing to ptsG
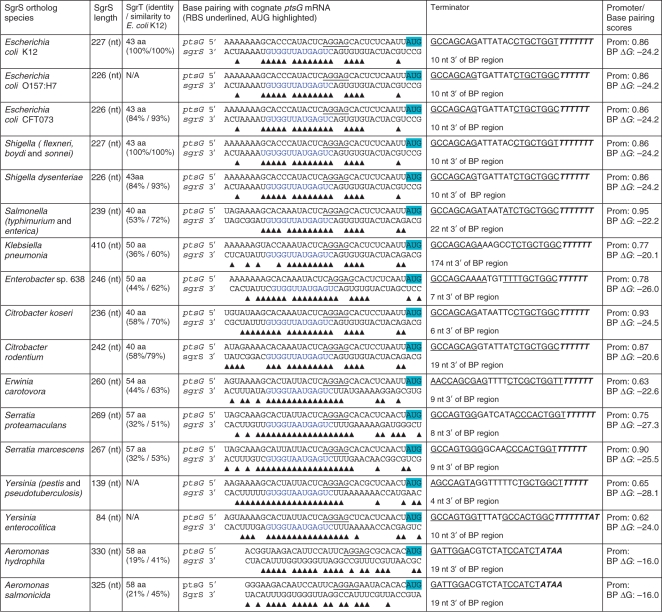 |
Twenty-two homologs of SgrS were identified; 17 were unique as indicated by the rows in the table that contain more than one strain name. The SgrS homolog of P. luminescens was excluded from the Table as it does not encode SgrT and the genome does not encode a ptsG homolog. SgrT identity is relative to E. coli K12 SgrT. Base pairing between SgrS and the cognate ptsG mRNA was predicted by comparison with the base pairing interactions experimentally demonstrated for E. coli K12 (18) and automated alignment of the two RNA sequences (this study). For ptsG mRNA: the ribosome binding site is underlined and initiation codon is highlighted; for SgrS: sequences in blue represent the conserved base pairing region. The predicted terminator sequence is shown with the inverted repeat of the SL underlined; the distance from the end of the predicted base pairing region is indicated. The promoter scores were determined by Neural Network Promoter Prediction algorithm at BDGP. A score closer to 1.00 indicates a stronger promoter; this algorithm did not predict a promoter at the correct position relative to the probable +1 of SgrS in Aeromonas (based on alignment with E. coli K12 SgrS). The base pairing (BP) score was determined using UNAfold at the DINAMelt Server (http://dinamelt.bioinfo.rpi.edu/hybrid2.php) to predict ΔG for each pairing interaction.
The structure of a riboregulator plays a major role in its function, particularly for regions that interact with target mRNAs. Although there are few bacterial riboregulators whose structures have been mapped, for those that have, base pairing sequences have often been localized to single-stranded regions. These single-stranded regions are sometimes localized to loop regions of stem-loop (SL) structures and are important for making initial interactions with loop sequences of their targets through formation of so-called ‘kissing complexes’ (35). Loop sequences of sRNAs can also interact with non-loop single-stranded regions of their mRNA targets (33). Alternatively, single-stranded regions of sRNAs important for interactions with targets may be located adjacent to SL structures (21). Structural rearrangements promoted by the RNA chaperone Hfq may subsequently allow more extensive sRNA–mRNA interactions beyond those made by initial contacts (36).
Structural predictions for SgrS homologs were made by the RNA folding algorithms MFOLD (29), which calculates secondary structure based only on thermodynamic properties and RNAalifold (30), which predicts a consensus structure based on a set of aligned RNA sequences (Supplementary Figure S6A) and uses information from covariation to compute the secondary structure and energy score. The predictions of the two folding programs were highly similar, and revealed several conserved structural features that are shown schematically in Figure 3B (Mfold outputs are shown). The most conserved structural feature is the terminator SL at the 3′ end. Most of the homologs are predicted to have an additional SL preceding the terminator. The base pairing sequences of most homologs (Table 1) are localized to a single-stranded region adjacent to this hairpin; in a number of cases some of the predicted base pairing residues extend into one side of the stem of this hairpin (Figure 3B). Only in the case of the Enterobacter SgrS homolog is the base pairing region predicted to be fully contained within a hairpin structure. Several SgrS homologs, including those of E. coli K12 and Shigella and Citrobacter species, have an additional small hairpin at the 3′ end of the sgrT-coding sequence; the significance of this structure, if any, is not known. The 5′ region of SgrS encompassing the sgrT coding sequence is represented as an unstructured dashed line because it is expected that translation of sgrT would result in unfolding of the mRNA region of SgrS.
Figure 3.

(A) Mfold prediction for secondary structure of E. coli K12 SgrS. SgrT and the base pairing region are highlighted in blue and green, respectively. The arrow indicates the 5′ end of the sRNA. (B) Diagrammatic representation of the secondary structures of SgrS homologs as predicted by Mfold. Each unique structure diagram represents the SgrS described in one row of Table 1; only SgrS from E. coli 0157:H7 is missing because it is identical to E. coli CFT073 except for the point mutation that removes SgrT. For simplicity, the sgrT CDS is shown as a dashed light blue line without secondary structure since translation of sgrT would unfold this region of the sRNA. The base pairing region is shown in green and the terminator is shown in red. The number of base pairs in the stem and loop components of the hairpin structures are shown proportionally to each other.
Most SgrS homologs are in the size range of 200–300 bases, which is considerably longer than the typical Hfq-dependent riboregulator (usually 60–100 bases). This additional length for SgrS homologs is due to the presence of the sgrT coding sequence (CDS) (ranging from 120 to 171 bases) at the 5′ end of most homologs. Homologs from Yersinia species, Photorhabdus luminescens and Klebsiella pneumoniae deviate from this size range for reasons that will be discussed below. SgrT homologs range in size from 40 to 57 amino acids. There are a number of conserved leucines and a repeating motif of a polar residue followed by an aromatic residue, but minimal conservation of other residues in SgrT (Figure 4 and Supplementary Figure S4). Despite the relatively low level of primary amino-acid sequence identity with SgrT of E. coli K12 (Table 1), SgrT homologs from S. typhimurium, K. pneumoniae and E. carotovora complement an E. coli sgrST mutant (C. S. Wadler and C. K. Vanderpool, submitted for publication), indicating conservation of SgrT function.
Figure 4.

ClustalW alignment of SgrT homologs. The 14 unique SgrT sequences identified are aligned. (Of the 22 SgrS homologs, five do not encode SgrT and three others encode SgrT homologs identical to one of those shown.) The conserved leucines (l), and repeating polar (p) and aromatic (a) residue motifs are indicated above the alignment. The ClustalW consensus in red indicates the conserved residues when SgrT sequences from Aeromonas sp. are excluded; the consensus in black includes alignment of Aeromonas SgrTs. Abbreviations are as follows: EcK12, Escherichia coli K12; EcCFT, Escherichia coli CFT073; Shidy, Shigella dysenteriae; Shifl, Shigella flexneri; Salty, Salmonella typhimurium; Citko, Citrobacter koseri; Citro, Citrobacter rodentium; Klebp, Klebsiella pneumoniae; Enter, Enterobacter sp. 638; Serma, Serratia marcescens; Serpr, Serratia proteamaculans; Erwca, Erwinia carotovora; Aeroh, Aeromonas hydrophila; Aeros, Aeromonas salmonicida.
Descriptions of individual SgrS and SgrT homologs
While most SgrS homologs share major functional elements such as the presence of the sgrT CDS sequence, base pairing region and terminator, there are a number of notable differences, particularly for homologs from organisms more distantly related to E. coli. Below and in Table 1 we have summarized the characteristics of individual homologs that may have relevance for their physiological functions. We begin the descriptions with E. coli SgrS and proceed to homologs from organisms that are increasingly more distantly related to E. coli (37).
Among complete and partially sequenced Escherichia genomes, we identified approximately 20 SgrS homologs. These homologs range in identity with E. coli K12 SgrS from 99% for E. coli strains 55989, SE11, ATCC8739, E24377A and HS to 81% for Escherichia fergusonii. Further analyses concentrated on SgrS homologs from three E. coli species whose genomes are fully sequenced and annotated, namely MG1655 (K12), CFT073 and 0157:H7 Sakai. These three E. coli SgrS homologs are highly similar with 88% identity between K12 SgrS and the SgrS homolog of either pathogenic E. coli strain. The identity between the homologs of the two pathogenic strains is 99%, and this is explained by two single nucleotide changes in the sgrS sequence of E. coli 0157:H7 Sakai, one of which results in mutation of the sgrT start codon. Strikingly, this point mutation is present in both 0157:H7 strains whose genomes are fully sequenced as well as numerous 0157:H7 strains with partially sequenced genomes, suggesting that the SgrS made by these strains does not encode a functional SgrT. Shigella SgrS homologs also show a high degree of identity to E. coli SgrS and Shigella SgrT sequences are 100% identical to E. coli K12 SgrT. The E. coli K12 SgrS has one feature present in only 3 out of 15 E. coli strains (K12, 53638 and 101.1) and not present in any other Escherichia or Shigella species: this is the presence of a small SL structure immediately before the sgrT CDS (Figure 3A and B). This SL sequesters the ribosome-binding sequence and was shown in another study to inhibit sgrT translation (C. S. Wadler and C. K. Vanderpool, submitted for publication).
Salmonella enterica serovar typhimurium LT2 and S. enterica serovar choleraesuis (referred to hereafter as S. typhimurium and S. enterica, respectively) contain identical SgrS and SgrT homologs. The Salmonella SgrT homolog is slightly shorter (40 aa) than E. coli SgrT (43 aa). The predicted secondary structure, shown schematically in Figure 3B, shows that the Salmonella homologs lack the SL structure that occurs directly upstream of sgrT in the E. coli K12 SgrS homolog. This suggests that the ribosome-binding site of Salmonella SgrS would not be occluded, a prediction supported by experimental data showing higher levels of SgrT production from S. typhimurium SgrS compared with E. coli SgrS (C. S. Wadler and C. K. Vanderpool, submitted for publication). The SL terminator for Salmonella SgrS is positioned 22-nt downstream of the base pairing region, rather than the 10 nt that is more common among the homologs; the significance of this is unknown. The additional nucleotides are predicted to participate in formation of the SL preceeding the terminator (Figure 3B).
The K. pneumoniae SgrS homolog contains a longer SgrT (50 aa) than E. coli; the well-conserved base pairing region is located in the usual position 3′ of the sgrT CDS. However, the K. pneumoniae homolog does not contain a terminator SL in the typical position. Termination at the nearest predicted factor-independent terminator downstream would result in an sRNA of 410 nt. When the K. pneumoniae homolog is expressed in E. coli, an RNA of ∼400 nt is produced (C. S. Wadler and C. K. Vanderpool, submitted for publication), consistent with the idea that transcription of this sgrS homolog terminates at the distal structure, which is 174-nt downstream of the base pairing region. There are no additional ORFs that would encode peptides longer than 15 aa located within this sequence, and the physiological role of this additional sequence is unknown.
The Enterobacter SgrS homolog also encodes a longer SgrT peptide (50 aa) compared with E. coli SgrT, but otherwise contains the same functional elements as E. coli SgrS. The base pairing region is positioned at the usual location 3′ of sgrT, however, one predicted structural difference is the localization of base pairing elements to an SL structure. This differs from most homologs where the base pairing nucleotides are mostly unpaired (Figure 3B). In some cases, Hfq binding to sRNAs has been shown to remodel their secondary structure (36,38). Perhaps the Enterobacter SgrS undergoes more extensive Hfq-induced conformational changes than other homologs in order to interact with its cognate ptsG mRNA.
The two homologs from the Citrobacter genus both encode a 40 aa SgrT and have similar base pairing sequences complementary to their cognate ptsG mRNAs (Table 1). The position of the terminator relative to the base pairing region differs between these two homologs: the Citrobacter rodentium SgrS has a 19-nt spacer between these elements in contrast to the 6-nt spacer in Citrobacter koseri SgrS.
The SgrS homolog found in Erwinia carotovora encodes the second longest SgrT homolog at 54 aa. Structural analyses indicate a short SL structure upstream of the sgrT CDS similar to the one identified in E. coli K12 SgrS (Figure 3B). However, unlike the GC-rich stem in E. coli, the putative stem in E. carotovora contains two A:U base pairs that should weaken the interaction. In vivo complementation studies of an E. coli sgrST mutant with the E. carotovora sgrST strongly suggest that E. carotovora's sgrT is well-translated and functional (C. S. Wadler and C. K. Vanderpool, submitted for publication). These data argue against any significant translational inhibition mediated by this structure. The base pairing region and terminator of the E. carotovora sgrS are located at the typical 3′ position.
SgrS homologs found in Yersinia species range in size from 84 to 139 nt owing to the lack of an sgrT. For Y. enterocolitica, this appears to be caused by a 5′ truncation of sgrS resulting from insertion of two genes, ybiY and ybiW, in the sgrR-sgrS intergenic region (Figure 2). The ybiYW genes are uncharacterized, but they are predicted to encode a pyruvate formate lyase and its activating enzyme (23). Notably, the Y. enterocolitica sgrS homolog is the only one that is not divergently transcribed from sgrR. Whether ybiYW expression is controlled by SgrR is not known, though the conservation of regulatory sequences adjacent to sgrR and upstream of ybiY (Supplementary Figure S5A) suggests that this might be the case. Examination of the region immediately upstream of the Y. enterocolitica sgrS homolog (downstream of ybiW) did not reveal any additional putative SgrR regulatory sequences, suggesting that if Y. enterocolitica sgrS is expressed, it may be operonic with ybiYW. For Y. pestis and Y. pseudotuberculosis the cause of the 5′ sgrS truncation is not obvious; there are no apparent insertion sequences or other signatures of mobile elements in this region. However, it has been reported that Yersinia genomes are extremely plastic and extensive rearrangements are common (39). Despite extensive differences at their 5′ ends, the base pairing and terminator elements of Yersinia sgrS homologs are conserved and located at typical distances from one another. Another interesting characteristic of Yersinia SgrS homologs is their extensive pattern of predicted complementarity with their cognate ptsG mRNAs. The predicted interactions for Y. pestis and Y. pseudotuberculosis SgrS: ptsG mRNA pairs represent the longest contiguous stretches of complementarity for any homologous pair. In another study, it was shown that the Y. pestis sgrS homolog can complement an E. coli sgrST mutant, indicating that this truncated SgrS is still a fully functional riboregulator (C. S. Wadler and C. K. Vanderpool, submitted for publication).
The Serratia proteamaculans and S. marcescens SgrS homologs encode the longest SgrT proteins at 57 aa. While the sRNA is slightly longer to compensate for the longer CDS, the base pairing region and terminator occur at typical positions relative to one another. The predicted base pairing interactions with the cognate ptsG mRNA are striking, as they are comprised of a single long stretch of complementarity similar to that predicted for Yersinia species (Table 1).
The SgrS homologs of Aeromonas species and P. luminescens are the most divergent from E. coli SgrS. The sgrT CDS is not present in the P. luminescens SgrS while the SgrT encoded by A. hydrophila and A. salmonicida shows very low identity with E. coli SgrT (Figure 4, Supplementary Figure S4). The genome of P. luminescens does not contain a ptsG homolog; however, it does contain genes encoding another PTS capable of transporting glucose, manXYZ. Results from our laboratory have shown that E. coli SgrS also regulates manXYZ mRNA (J. B. Rice and C. K. Vanderpool, unpublished results), so perhaps this regulation is conserved in P. luminescens. However, due to the poor similarity between P. luminescens and Aeromonas SgrS and E. coli SgrS, we cannot claim with certainty that these homologs are functionally analogous to E. coli SgrS.
Base pairing of SgrS homologs to mRNA targets
One of the great challenges in bacterial small RNA research has been to define rules that govern RNA:RNA interactions that result in positive or negative regulation of mRNA targets. Since we have identified a set of homologous sRNAs, we analyzed interactions between these sRNAs and their cognate targets in order to gain some insight into the requirements for regulation by SgrS. In order to conduct these analyses, we first identified ptsG homologs in the genomes where we had already identified SgrR, SgrS and SgrT homologs. For each of these genomes, with the sole exception of P. luminescens, a ptsG homolog with strong identity (Supplementary Figure S3) and gene synteny to E. coli ptsG was identified.
We previously predicted (15) that for E. coli, the SgrS:ptsG mRNA base pairing interactions occurred through short, interrupted segments of complementarity that we will refer to as a 5-8-4 pattern (referencing the number of contiguous base pairs in each segment). The 5-8-4 pattern of complementarity sequesters most of the ribosome-binding site (RBS) of ptsG mRNA resulting in translation inhibition of ptsG (40). The base pairing predictions were supported by genetic evidence obtained by another group (18). These results suggested that the central eight contiguous base pair segment was particularly important for regulation. However, one caveat to these experiments is that they only measured the effect on steady state levels of ptsG mRNA at 20 min after SgrS expression was induced. Thus, subtle contributions of the flanking regions of complementarity to the initial kinetics of regulation would not have been elucidated. At this time there have been no structural probing experiments to validate or refute the base pairing predictions.
The relative position of interacting sequences is conserved among all homologous pairs, i.e. 3′ of the sgrT CDS for SgrS and at least partially encompassing the RBS of ptsG. This strongly suggests that the regulatory outcome of translation inhibition and destabilization of ptsG mRNA is also conserved. The 5-8-4 pattern is conserved for SgrS:ptsG mRNA partners in E. coli and Shigella species, and is very similar (4-9-4) for Salmonella species (Table 1). Interestingly, the more divergent SgrS:ptsG mRNA pairs are predicted to have more extensive interactions. The Erwinia, Yersinia and Serratia homologs have a longer contiguous segment of complementarity that encompasses between 15 and 21 base pairs. For the most divergent SgrS homologs in Aeromonas species, the predicted interactions are somewhat shorter and more interrupted, but are still predicted to occlude the RBS.
There is currently little data to shed light on how the base composition of the base pairing region affects regulation by small RNAs. The mutations with the strongest negative effects on regulation by SgrS alter G:C base pairs in the central 8-bp region of SgrS:ptsG mRNA interactions in E. coli (18). As G:C base pairs form three hydrogen bonds versus the two hydrogen bonds contributed by A:U or G:U pairs, it would not be surprising if G:C base pairs make the most substantial contribution to the riboregulation. The E. coli, Shigella and Salmonella interaction regions are all composed of ∼50% G:C base pairs. Interestingly, for homologous pairs with more extensive complementarity, e.g. Erwinia, Yersinia and Serratia, G:C base pairs comprise only ∼35% of the interactions. Perhaps a more A:U/G:U-rich base pairing region requires a greater number of consecutive base pairs to achieve a rapid interaction in vivo.
To provide another qualitative way to compare these predicted sRNA:mRNA complexes, we utilized the DINAMelt server (41) to predict melting profiles for the short segments of SgrS and ptsG mRNA shown in Table 1. The ΔG values reported for each pair (Table 1) are meant only as a means to compare the relative stabilities of these short regions of complementarity for each homologous sRNA:mRNA pair. The ΔG values ranged from −16 to −28, with the majority falling between −22 and −26. The native E. coli K12 SgrS:ptsG mRNA pair had a ΔG value of −24.2. A single point mutation in SgrS that alters one G:C base pair in the central segment of complementarity reduced the ΔG value to −19; an additional mutation in sgrS that alters a second G:C base pair in this region further reduces the ΔG value to −10. We have shown that the E. coli SgrS single point mutant has a partial defect in regulating ptsG mRNA while the double point mutant is strongly defective for ptsG regulation [(18) and C. S. Wadler and C. K. Vanderpool, submitted for publication]. Given this information, it is interesting to note that K. pneumoniae and C. rodentium SgrS:ptsG mRNA pairs have a ΔG value of −20 while the A. hydrophila ΔG value is only −16, suggesting that SgrS may not regulate ptsG in A. hydrophila. It will be interesting in the future to examine regulation by these SgrS homologs and determine how efficiently they regulate their cognate ptsG mRNAs.
CONCLUSIONS
We identified homologs of the transcription factor SgrR and the sRNA SgrS in a variety of enteric bacterial species. Most homologs appear to possess both of the functions that we have previously described for E. coli SgrS (15,19): the riboregulation function and ability to produce the SgrT protein. The bioinformatic analyses described in this study suggest that the riboregulatory function of SgrS is the more conserved of the two functions of SgrS, since in a few homologs the sgrT is missing or rendered non-functional by mutation of the start codon. Homologs of SgrR, SgrS and SgrT are more narrowly distributed than PtsG homologs, which are found in a much wider range of organisms. Not surprisingly, conserved sequences in the 5′ UTR of ptsG are found only in the same genomes where a SgrS homolog is found (Supplementary Figure S3 and data not shown). These observations suggest that either glucose-phosphate stress is not a problem for all organisms that use PtsG homologs to take up glucose, or that other mechanisms have evolved to deal with the stress.
To further test the roles of riboregulation and SgrT in the glucose-phosphate stress response, several of the homologs identified in this study were expressed in a heterologous (E. coli) sgrST mutant host. The experimental results of that study (C. S. Wadler and C. K. Vanderpool, submitted for publication) support the hypotheses generated by the identification and comparison of these homologs. Together, these studies reveal that despite low levels of nucleotide sequence similarity, SgrS and SgrT homologs are functionally interchangeable.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
University of Illinois at Urbana-Champaign; American Heart Association [Scientist Development Grant 0835355N]. Funding for open access charge: American Heart Association (Scientist Development Grant 0835355N).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Rachel Whitaker, Susan Gottesman and Caryn Wadler for critical review of the manuscript and many helpful suggestions. We are especially grateful to Rachel Whitaker for assistance with phylogenetic analyses.
REFERENCES
- 1.Altuvia S. Identification of bacterial small non-coding RNAs: experimental approaches. Curr. Opin. Microbiol. 2007;10:257–261. doi: 10.1016/j.mib.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Rivas E, Klein RJ, Jones TA, Eddy SR. Computational identification of noncoding RNAs in E. coli by comparative genomics. Curr. Biol. 2001;11:1369–1373. doi: 10.1016/s0960-9822(01)00401-8. [DOI] [PubMed] [Google Scholar]
- 3.Wassarman KM, Repoila F, Rosenow C, Storz G, Gottesman S. Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev. 2001;15:1637–1651. doi: 10.1101/gad.901001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Massé E, Escorcia FE, Gottesman S. Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev. 2003;17:2374–2383. doi: 10.1101/gad.1127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Massé E, Gottesman S. A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc. Natl Acad. Sci. USA. 2002;99:4620–4625. doi: 10.1073/pnas.032066599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Massé E, Vanderpool CK, Gottesman S. Effect of RyhB small RNA on global iron use in Escherichia coli. J. Bacteriol. 2005;187:6962–6971. doi: 10.1128/JB.187.20.6962-6971.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boughammoura A, Matzanke BF, Bottger L, Reverchon S, Lesuisse E, Expert D, Franza T. Differential role of ferritins in iron metabolism and virulence of the plant-pathogenic bacterium Erwinia chrysanthemi 3937. J. Bacteriol. 2008;190:1518–1530. doi: 10.1128/JB.01640-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy ER, Payne SM. RyhB, an iron-responsive small RNA molecule, regulates Shigella dysenteriae virulence. Infect. Immun. 2007;75:3470–3477. doi: 10.1128/IAI.00112-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis BM, Quinones M, Pratt J, Ding Y, Waldor MK. Characterization of the small untranslated RNA RyhB and its regulon in Vibrio cholerae. J. Bacteriol. 2005;187:4005–4014. doi: 10.1128/JB.187.12.4005-4014.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mey AR, Craig SA, Payne SM. Characterization of Vibrio cholerae RyhB: the RyhB regulon and role of ryhB in biofilm formation. Infect. Immun. 2005;73:5706–5719. doi: 10.1128/IAI.73.9.5706-5719.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ellermeier JR, Slauch JM. Fur regulates expression of the Salmonella pathogenicity island 1 type III secretion system through HilD. J. Bacteriol. 2008;190:476–486. doi: 10.1128/JB.00926-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Padalon-Brauch G, Hershberg R, Elgrably-Weiss M, Baruch K, Rosenshine I, Margalit H, Altuvia S. Small RNAs encoded within genetic islands of Salmonella typhimurium show host-induced expression and role in virulence. Nucleic Acids Res. 2008;36:1913–1927. doi: 10.1093/nar/gkn050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilderman PJ, Sowa NA, FitzGerald DJ, FitzGerald PC, Gottesman S, Ochsner UA, Vasil ML. Identification of tandem duplicate regulatory small RNAs in Pseudomonas aeruginosa involved in iron homeostasis. Proc. Natl Acad. Sci. USA. 2004;101:9792–9797. doi: 10.1073/pnas.0403423101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaballa A, Antelmann H, Aguilar C, Khakh SK, Song KB, Smaldone GT, Helmann JD. The Bacillus subtilis iron-sparing response is mediated by a Fur-regulated small RNA and three small, basic proteins. Proc. Natl Acad. Sci. USA. 2008;105:11927–11932. doi: 10.1073/pnas.0711752105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vanderpool CK, Gottesman S. Involvement of a novel transcriptional activator and small RNA in post-transcriptional regulation of the glucose phosphoenolpyruvate phosphotransferase system. Mol. Microbiol. 2004;54:1076–1089. doi: 10.1111/j.1365-2958.2004.04348.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhang A, Wassarman KM, Rosenow C, Tjaden BC, Storz G, Gottesman S. Global analysis of small RNA and mRNA targets of Hfq. Mol. Microbiol. 2003;50:1111–1124. doi: 10.1046/j.1365-2958.2003.03734.x. [DOI] [PubMed] [Google Scholar]
- 17.Morita T, El-Kazzaz W, Tanaka Y, Inada T, Aiba H. Accumulation of glucose 6-phosphate or fructose 6-phosphate is responsible for destabilization of glucose transporter mRNA in Escherichia coli. J. Biol. Chem. 2003;278:15608–15614. doi: 10.1074/jbc.M300177200. [DOI] [PubMed] [Google Scholar]
- 18.Kawamoto H, Koide Y, Morita T, Aiba H. Base-pairing requirement for RNA silencing by a bacterial small RNA and acceleration of duplex formation by Hfq. Mol. Microbiol. 2006;61:1013–1022. doi: 10.1111/j.1365-2958.2006.05288.x. [DOI] [PubMed] [Google Scholar]
- 19.Wadler CS, Vanderpool CK. A dual function for a bacterial small RNA: SgrS performs base pairing-dependent regulation and encodes a functional polypeptide. Proc. Natl Acad. Sci. USA. 2007;104:20454–20459. doi: 10.1073/pnas.0708102104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vanderpool CK. Physiological consequences of small RNA-mediated regulation of glucose-phosphate stress. Curr. Opin. Microbiol. 2007;10:146–151. doi: 10.1016/j.mib.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 21.Sharma C, Darfeuille F, Plantinga T, Vogel J. A small RNA regulates multiple ABC transporter mRNAs by targeting C/A-rich elements inside and upstream of ribosome-binding sites. Genes Dev. 2007;21:2804–2817. doi: 10.1101/gad.447207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urbanowski ML, Stauffer LT, Stauffer GV. The gcvB gene encodes a small untranslated RNA involved in expression of the dipeptide and oligopeptide transport systems in Escherichia coli. Mol. Microbiol. 2000;37:856–868. doi: 10.1046/j.1365-2958.2000.02051.x. [DOI] [PubMed] [Google Scholar]
- 23.Misra RV, Horler RS, Reindl W, Goryanin II, Thomas GH. EchoBASE: an integrated post-genomic database for Escherichia coli. Nucleic Acids Res. 2005;33:D329–D333. doi: 10.1093/nar/gki028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 25.Carver T, Berriman M, Tivey A, Patel C, Bohme U, Barrell BG, Parkhill J, Rajandream MA. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics. 2008;24:2672–2676. doi: 10.1093/bioinformatics/btn529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaudhuri RR, Pallen MJ. xBASE, a collection of online databases for bacterial comparative genomics. Nucleic Acids Res. 2006;34:D335–D337. doi: 10.1093/nar/gkj140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tatusova T, Madden T. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 1999;174:247–250. doi: 10.1111/j.1574-6968.1999.tb13575.x. [DOI] [PubMed] [Google Scholar]
- 28.Vanderpool CK, Gottesman S. The novel transcription factor SgrR coordinates the response to glucose-phosphate stress. J. Bacteriol. 2007;189:2238–2248. doi: 10.1128/JB.01689-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gruber A, Lorenz R, Bernhart S, Neuböck R, Hofacker I. The Vienna RNA Websuite. Nucleic Acids Res. 2008;36:W70–W74. doi: 10.1093/nar/gkn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dodd IB, Egan JB. Improved detection of helix-turn-helix DNA-binding motifs in protein sequences. Nucleic Acids Res. 1990;18:5019–5026. doi: 10.1093/nar/18.17.5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johansen J, Eriksen M, Kallipolitis B, Valentin-Hansen P. Down-regulation of outer membrane proteins by noncoding RNAs: unraveling the cAMP-CRP- and sigmaE-dependent CyaR-ompX regulatory case. J. Mol. Biol. 2008;383:1–9. doi: 10.1016/j.jmb.2008.06.058. [DOI] [PubMed] [Google Scholar]
- 33.Papenfort K, Pfeiffer V, Lucchini S, Sonawane A, Hinton JC, Vogel J. Systematic deletion of Salmonella small RNA genes identifies CyaR, a conserved CRP-dependent riboregulator of OmpX synthesis. Mol. Microbiol. 2008;68:890–906. doi: 10.1111/j.1365-2958.2008.06189.x. [DOI] [PubMed] [Google Scholar]
- 34.Papenfort K, Pfeiffer V, Mika F, Lucchini S, Hinton JC, Vogel J. SigmaE-dependent small RNAs of Salmonella respond to membrane stress by accelerating global omp mRNA decay. Mol. Microbiol. 2006;62:1674–1688. doi: 10.1111/j.1365-2958.2006.05524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Argaman L, Altuvia S. fhlA repression by OxyS RNA: Kissing complex formation at two sites results in a stable antisense-target RNA complex. J. Mol. Biol. 2000;300:1101–1112. doi: 10.1006/jmbi.2000.3942. [DOI] [PubMed] [Google Scholar]
- 36.Geissmann TA, Touati D. Hfq, a new chaperoning role: binding to messanger RNA determines access for small RNA regulator. EMBO J. 2004;23:396–405. doi: 10.1038/sj.emboj.7600058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Letunic I, Bork P. Interactive Tree of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–128. doi: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]
- 38.Møller T, Franch T, Hojrup P, Keene DR, Bachinger HP, Brennan R, Valentin-Hansen P. Hfq: a bacterial Sm-like protein that mediates RNA-RNA interaction. Mol. Cell. 2002;9:23–30. doi: 10.1016/s1097-2765(01)00436-1. [DOI] [PubMed] [Google Scholar]
- 39.Parkhill J, Wren BW, Thomson NR, Titball RW, Holden MTG, Prentice MB, Sebaihia M, James KD, Churcher C, Mungall KL, et al. Genome sequence of Yersinia pestis, the causative agent of plague. Nature. 2001;413:523–527. doi: 10.1038/35097083. [DOI] [PubMed] [Google Scholar]
- 40.Morita T, Mochizuki Y, Aiba H. Translational repression is sufficient for gene silencing by bacterial small noncoding RNAs in the absence of mRNA destruction. Proc. Natl Acad. Sci. USA. 2006;103:4858–4863. doi: 10.1073/pnas.0509638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Markham NR, Zuker M. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res. 2005;33:W577–W581. doi: 10.1093/nar/gki591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu JY, Miller PF, Gosink M, Olson ER. The identification of a new family of sugar efflux pumps in Escherichia coli. Mol. Microbiol. 1999;31:1845–1851. doi: 10.1046/j.1365-2958.1999.01321.x. [DOI] [PubMed] [Google Scholar]
- 43.Liu JY, Miller PF, Willard J, Olson ER. Functional and biochemical characterization of Escherichia coli sugar efflux transporters. J. Biol. Chem. 1999;274:22977–22984. doi: 10.1074/jbc.274.33.22977. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}