

Abstract
AMP activated protein kinase (AMPK) plays an important role in regulating myocardial metabolism and protein synthesis. Activation of AMPK attenuates hypertrophy in cultured cardiac myocytes, but the role of AMPK in regulating the development of myocardial hypertrophy in response to chronic pressure overload is not known. To test the hypothesis that AMPKα2 protects the heart against systolic overload induced ventricular hypertrophy and dysfunction, we studied the response of AMPKα2 gene deficient (KO) mice and wild type (WT) mice subjected to 3 weeks of transverse aortic constriction (TAC). Although AMPKα2 KO had no effect on ventricular structure or function under control conditions, AMPKα2 KO significantly increased TAC-induced ventricular hypertrophy (ventricular mass increased 46% in WT mice compared to 65% in KO mice), while decreasing left ventricular ejection fraction (ejection fraction decreased 14% in WT mice compared to a 43% decrease in KO mice). AMPKα2 KO also significantly exacerbated the TAC–induced increases of atrial natriuretic peptide, myocardial fibrosis and cardiac myocyte size. AMPKα2 KO had no effect on total S6 Ribosomal Protein (S6), p70 S6 kinase (70S6K), eukaryotic initiation factor 4E (eIF4e) and 4E binding protein-1 (4EBP1) or their phosphorylation under basal conditions, but significantly augmented the TAC-induced increases of p-70S6KThr389, p-S6Ser235, and p-eIF4eSer209. AMPKα2 KO also enhanced the TAC-induced increase of p-4EBP1Thr46 to a small degree and augmented the TAC-induced increase of p-AktSer473. These data indicate that AMPKα2 exerts a cardiac protective effect against pressure overload induced ventricular hypertrophy and dysfunction.
Keywords: hypertrophy, congestive heart failure, mTOR
Introduction
Increases of cardiac work resulting from systolic overload necessitate an increase of ATP utilization in proportion to the increase in LV systolic wall stress 1, 2. In response to chronic systolic overload cardiac myocyte hypertrophy occurs, characterized by increased protein synthesis, while myocardial oxygen consumption and carbon substrate utilization are increased to accommodate the need for increased energy availability. This initially occurs with no change in high energy phosphate levels, but with the development of pathologic hypertrophy and congestive heart failure, ATP levels fall and cytosolic free ADP levels increase (as indicated by a decrease of the myocardial phosphocreatine/ATP ratio) 2, 3. In this situation, the adenylate kinase reaction can catalyze the reaction of two molecules of ADP to produce one molecule of ATP and one molecule of AMP. An increased AMP/ATP ratio results in activation of the energy stress sensor known as AMP activated protein kinase.
AMPK is composed of one catalytic α subunit (either α1 or α2) and two regulatory subunits (β andγ). AMPKα2 is the dominant catalytic subunit in the heart3, 4 where it is predominantly expressed in cardiac myocytes. AMPK is activated by metabolic stresses that deplete cellular ATP or increase AMP 3, 4. Recent studies have demonstrated that activation of AMPK attenuates signaling through the mTOR pathway, which plays a critical role in activating translational machinery during cell growth or hypertrophy 5. While genetic deletion of AMPKα2 6 or AMPKα17, or overexpression of dominant negative AMPKα, has no effect on ventricular mass or function under unstressed conditions, the role of AMPK in the response of the heart to systolic overload has not been directly tested. As AMP/ATP ratios increase during systolic overload, AMPK would be predicted to play a more significant role under these conditions than under basal conditions. Consequently, this study was designed to determine the effect of decreasing AMPK activity by AMPKα2 KO on LV mass and function in mice exposed to chronic pressure overload produced by TAC. Here we report that AMPKα2 KO had no effect on LV structure or function during basal conditions, but significantly exacerbated TAC-induced ventricular hypertrophy, fibrosis and dysfunction, and this was associated with significantly greater increases of p-70S6KThr389, p-S6Ser235 and p-eIF4eSer209 in the KO mice as compared with WT mice. In addition, we demonstrated that activation of AMPK with either 5-aminoimidazole-4-carboxy-amide-1-d-ribofuranoside (AICAR) or metformin, or overexpression of constitutively active AMPK alpha 2(CA-AMPKα2) attenuated phenylephrine induced cardiac myocyte hypertrophy and reduced -phosphorylation of p70s6k(Thr389) in cultured neonatal rat cardiac myocytes. The data indicate that AMPKα2 negatively regulates pressure overload induced ventricular hypertrophy and dysfunction.
Materials and methods
Mice
Heterozygous AMPKα2 KO mice of C57BL6 background (crossed back to C57BL6 mice at least 8 times) were generated as previously described 6, 7. The AMPKα2 KO mice and WT littermates generated from the heterozygotes were used as breeders to generate KO and WT mice for the study. This study was approved by the Institutional Animal Care and Use Committee of University of Minnesota.
TAC procedure
TAC was performed in male wild type mice (n=45) and KO mice (n=42) at ~2 months of age by using the minimally invasive suprasternal approach as previously described 8-10.
LV function measurements
Echocardiography was performed 3 weeks after TAC or sham surgery under anesthesia with 1.5% isoflurane by inhalation as previously described 9, 10. LV and aortic hemodynamics were determined by a 1.2 Fr. pressure catheter (Scisense Inc. Ontario Canada) in wild type and KO mice under control conditions as previously described 9.
Western Blots, and measurement of myocardial fibrosis and myocyte hypertrophy
Western Blots were performed as previously described9. Tissue sections (8μm) from the central portion of the LV were stained with picrosirius red for detection of fibrosis9, 10, and FITC-conjugated wheat germ agglutinin (Alexa Fluor-488, Invitrogen) to evaluate myocyte size9, 10. Percent fibrosis volume density was quantified on the picrosirius red stained sections using point counting as described in Unbiased Stereology 11. For the average myocyte size, the mean short diameter and cross sectional area of ~120 cells/sample of 4 samples in each group were examined.
Myocardial AMPK activity
Isoform-specific activities of AMPK were measured as previously described 12. Please see http://hyper.ahajournals.org.
Neonatal rat cardiomyocyte isolation and culture, and cell area determination
Neonatal rat cardiomyocyte were isolated from 2-day-old Sprague-Dawley rats by enzymatic digestion13 and separated from non-muscle cells on a discontinuous Percoll gradient according to a modified protocol from Dr. U. Mende 13 with minor modification as previously described 14. Please see http://hyper.ahajournals.org
Data and statistical analyses
All values were expressed as mean ± standard error of the mean (SEM). Statistical significance was defined as P < 0.05. Two-way analysis of variance (ANOVA) was used to test each variable for differences among the treatment groups with SigmaSTAT. If ANOVA demonstrated a significant effect, individual comparisons between groups were made using Tukey’s Test.
Results
AMPKα2 KO has no effect on LV function under control conditions
Under control conditions ventricular mass, lung mass, and the ratio of ventricular mass or lung mass to tibia length were not different between AMPKα2 KO and WT mice matched for age, body weight (24.8±0.4 gram in WT mice as compared with 25.0±0.8 gram in KO mice) and gender (Figure 1A, B). Histological examination showed no cardiac myocyte hypertrophy and no increase in myocardial fibrosis in the AMPKα2 KO mice. Echocardiographic imaging showed normal LV size and systolic shortening in the KO mice (Table 1), which is consistent with previous reports demonstrating that disruption of AMPK activity by overexpressing mutated AMPKα2 had no impact on LV structure or function under control conditions in young mice 15, 16. However, LV dP/dtmax and LV dP/dtmin were significantly decreased in the KO mice as compared with WT mice, indicating a subtle decrease in contractility, although LV systolic pressure and mean aortic pressure were not different between KO and WT mice under control conditions (Table 1).
Figure 1.
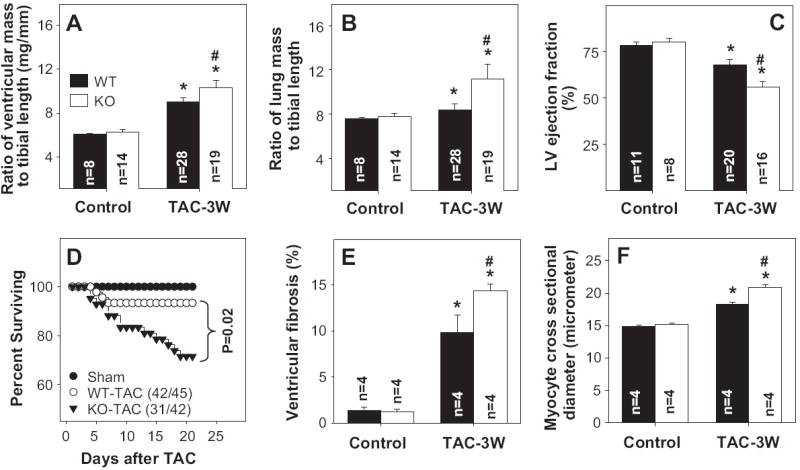
AMPKα2 KO exacerbates TAC-induced ventricular hypertrophy (A), pulmonary congestion (B), decrease of LV ejection fraction (C), increase of mortality rate (D), myocardial fibrosis (E), and cardiac myocyte hypertrophy (F). *p<0.05 compared to the corresponding control; #p<0.05 compared to WT-TAC mice.
Table 1.
Anatomic and functional data for wild type and AMPKα2-/- mice.
| Parameter | Wild type control | AMPKα2-/- control | Wild type TAC | AMPKα2-/- TAC |
|---|---|---|---|---|
| Anatomic data (number of mice) | n = 8 | n = 14 | n = 28 | n = 19 |
| Body weight before TAC procedure | Not applicable | Not applicable | 22.1 ± 0.56 | 23.0 ± 0.56 |
| Body weight before sample collection (g) | 24.8 ± 0.4 | 24.4 ± 0.8 | 25.2 ± 0.44 | 24.2 ± 0.61 |
| Body weight gain | Not applicable | Not applicable | 3.07 ± 0.27 | 1.25 ± 0.45† |
| Ventricular mass (mg) | 106 ± 1.5 | 105 ± 3.7 | 156 ± 5.7* | 172 ± 6.7*,† |
| Ratio of ventricular mass to body weight (mg/g) | 4.27 ± 0.06 | 4.32 ± 0.10 | 6.17 ± 0.16 | 7.14 ± 0.25*† |
| Lung mass (mg) | 133 ± 2.2 | 133 ± 4.6 | 145 ± 7.4* | 191 ± 16*,† |
| Ratio of lung mass to body weight (mg/g) | 5.34 ± 0.08 | 5.45 ± 0.17 | 5.76± 0.25* | 7.92 ± 0.70*,† |
| Tibia length (mm) | 17.3 ± 0.10 | 16.8 ± 0.12 | 17.3 ± 0.09 | 17.1 ± 0.08 |
| Echocardiographic data (number of mice) | n = 11 | n = 8 | n = 20 | n = 16 |
| Heart rate (beats per minute) | 546 ± 28 | 538 ± 19 | 519 ± 13 * | 514 ± 13 * |
| LV end systolic diameter (mm) | 2.21 ± 0.13 | 2.23 ± 0.12 | 2.69 ± 0.13* | 3.05 ± 0.14*† |
| LV end diastolic diameter (mm) | 3.69 ± 0.12 | 3.85 ± 0.10 | 3.96 ± 0.09* | 4.09 ± 0.12*, |
| LV fractional shortening (%) | 64.2 ± 2.1 | 66.1 ± 2.5 | 53.8 ± 2.8* | 43.2 ± 3.9*,† |
| LV posterior wall thickness at end diastole (mm) | 0.70 ± 0.01 | 0.68 ± 0.01 | 0.94 ± 0.03* | 0.99 ± 0.02* |
| LV posterior wall thickness at end systole (mm) | 1.08 ± 0.04 | 1.13 ± 0.03 | 1.31 ± 0.02* | 1.30 ± 0.02* |
| Hemodynamic data (number of mice) | n = 10 | n = 7 | ||
| Mean aortic pressure (mm Hg) | 91.7±2.2 | 92.7±6.4 | not available | not available |
| Systolic LV pressure (mm Hg) | 112±1.9 | 106±6.8 | not available | not available |
| LV dP/dtmax (mmHg/s) | 8692±434 | 7529±591† | not available | not available |
| LV dP/dtmini (mmHg/s) | -7944 ±310 | -6740±622† | not available | not available |
p<0.05 as compared with corresponding control conditions.
p<0.05 as compared with corresponding wild type mice.
AMPKα2 KO exacerbates TAC-induced myocardial hypertrophy, fibrosis and dysfunction
In response to TAC for 3 weeks AMPKα2 KO mice developed significantly greater increases of ventricular weight and the ratio of ventricular weight to body weight or tibia length as compared to WT mice (Figure 1), indicating that AMPKα2 deficiency exacerbated TAC-induced myocardial hypertrophy. In addition, lung weight and the ratio of lung weight to tibia length were significantly greater in KO mice as compared to WT mice, indicating more pulmonary congestion in the KO mice (Figure 1). Furthermore, KO mice had a higher mortality following TAC as compared to wild type mice (Figure 1D). The mean body weight and tibia length were not different between WT and KO mice (Table 1).
Histological analysis demonstrated that TAC resulted in more ventricular fibrosis (Figure 1E) and a greater increase in cardiac myocyte diameter (Figure 1F) in KO mice as compared with WT mice, indicating that the greater ventricular mass in the KO mice after TAC was due to both myocyte hypertrophy and an increase of ventricular fibrosis.
TAC resulted in significantly more impairment of LV systolic function in the KO mice, as demonstrated by a greater reduction of systolic fractional shortening and a significant increase in LV end-systolic diameter as compared to WT mice (Figure 1, Table 1). The heart rates were not significantly different among these groups (Table 1).
AMPKα2 KO attenuated myocardial AMPK activity and increased TAC-induced p70S6K-S6 activation
Total myocardial AMPKα and phosphorylated acetyl CoA carboxylase (p-ACCSer79) were significantly decreased in AMPKα2 KO mice both under control conditions and after TAC as compared with the WT mice (Figure 2A,B,C). Consistent with the decreased myocardial total AMPKα in KO mice, myocardial AMPKα2 activity was diminished in KO mice under both control conditions and after TAC (Figure 2D), while AMPKα1 activity was not significantly different under control conditions (7.6±0.2pmol/mg protein/min in WT mice as compared with 8.2±0.3pmol/mg protein/min in KO mice). After 3 weeks TAC, AMPKα1 activity was significantly increased in AMPKα2 KO mice (8.7±0.42 pmol/mg protein/min) as compared with WT mice (7.8±0.22 pmol/mg protein/min), indicating a compensatory increase of AMPKα1 in KO mice. The greater increase of AMPKα1 activity and AMPKα1 protein content in the AMPKα2 KO mice following TAC was not able to fully compensate for the AMPKα2 deficiency, as demonstrated by significantly lower levels of p-ACC2 (Figure 2A,C) and greater ventricular hypertrophy in the KO mice. In addition, myocardial ANP protein (Figure 2A,F) and mRNA (Figure 2G) were significantly greater in the KO mice as compared to wild type mice 3 weeks after TAC.
Figure 2.
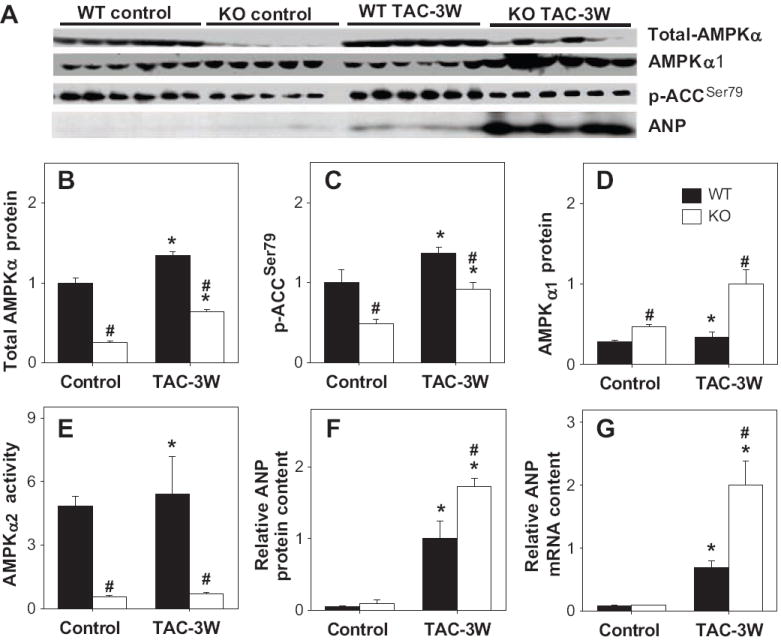
Alterations of myocardial total-AMPKα (A,B), AMPKα1 (A,C), p-ACCSer79 (A,D), AMPKα2 activity (E), and ANP protein (F) and mRNA content (G) under control conditions and after TAC for 3 weeks. *P<0.05 compared to the corresponding control; #p<0.05 compared to corresponding WT mice. Mean value was obtained from 5 to 6 samples each group.
It is reported that activation of AMPK attenuates protein synthesis by inhibiting the mTOR signaling pathway. p70S6K and 4EBP1 are two well defined downstream targets of mTOR; activation of mTOR increases p-70S6KThr389 and p-4EBP1Thr46, which enhances the translation initiation process and protein synthesis. We found that AMPKα2 KO had no significant effect on myocardial total p70S6K, S6 or eIF4e under both control conditions and after TAC. TAC caused a significant increase of total 4EBP1 in WT mice but not in KO mice. Three weeks after TAC, p-70S6KThr389, p-S6Ser235, p-elf4eSer209 and p-4EBP1Thr46 were increased in both WT and KO mice (Figure 3A,B). However, the increases of p-70S6KThr389, p-S6Ser235 and p-elf4eSer209 were significantly greater in the AMPKα2 KO animals (Figure 3A,B), indicating that AMPKα2 negatively regulates the phosphorylation of 70S6K and its down stream targets. AMPKα2 KO resulted in a small but significant increase of p-4EBP1Thr46 in response to TAC (Figure 3A,B).
Figure 3.
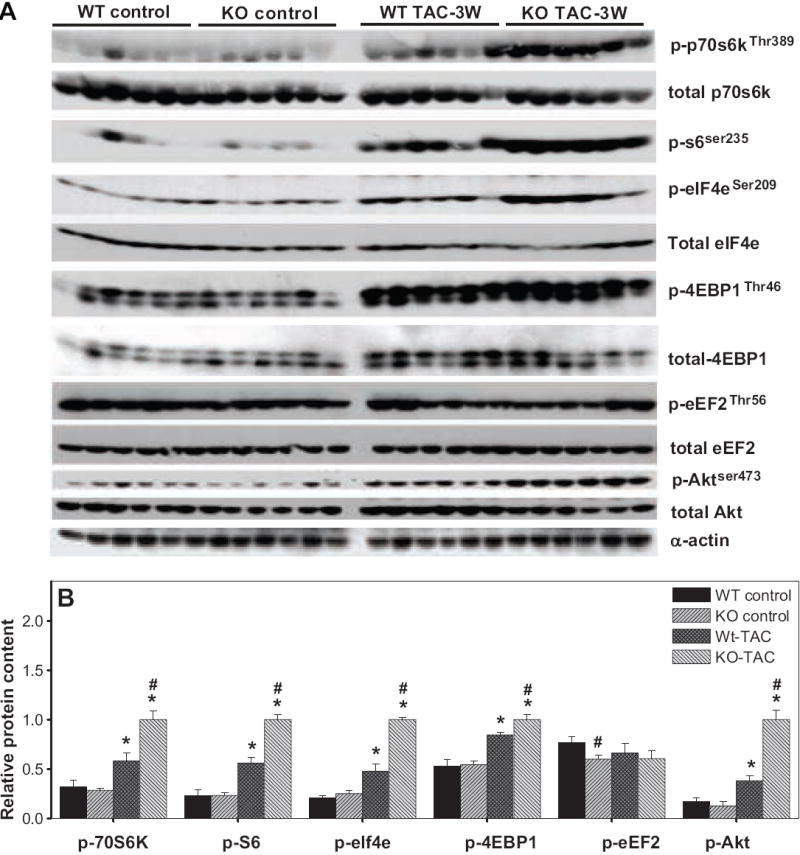
Effect of AMPKα2 KO on TAC-induced increases of p-p70s6kThr389, p-s6Ser235/236, p-elf4eSer209, p-Akt Ser473, p-eEF2 Thr56, and p-4EBP1Thr46. *P<0.05 compared to the corresponding control; #p<0.05 compared to corresponding WT mice. Mean value was obtained from 5 to 6 samples each group.
Eukaryotic elongation factor-2 (eEF2) is responsible for mediating the step of peptide-chain elongation. Phosphorylation of eEF2 at the Thr56 site results in inactivation of eEF2 (or decreased eEF2 activity) and a decrease of protein synthesis 17, 18. AMPKα2 KO had no effect on total eEF2 protein content either under control conditions or after TAC. Myocardial p-eEF2Thr56 content was not different between wild type and AMPKα2 KO under control conditions, but was significantly decreased in the AMPKα2 KO mice after TAC, indicating an increase of eEF2 activity in these mice (Figure 3A, B).
Pressure overload caused an increase of myocardial p-Akt Ser473, which is generally considered to be an upstream target of AMPK. Interestingly, we found that AMPKα2 KO also significantly augmented the TAC-induced increase of p-Akt Ser473 (Figure 3A,B).
AMPK activation or overexpression of constitutively active AMPKα2 attenuated phenylephrine (PE) induced cardiac myocyte hypertrophy
We examined the effect of overexpression of constitutively active AMPKα2 or activation of AMPK with AICAR and metformin on PE-induced hypertrophy of isolated neonatal cardiomyocytes (For the supplementary Figures, please see http://hyper.ahajournals.org). PE significantly increased the size of the cardiac myocytes over 48 hours of treatment, while overexpression of constitutively active AMPKα2 (CA-AMPK, Figure S1), metformin (0.2 to 5 mM) (Figure S2) and AICAR (0.2 mM) (Figure S3) all significantly attenuated PE-induced cardiac myocyte hypertrophy and increase of p-p70s6kThr389 (Figure S4, S5), indicating that activation of AMPK attenuates PE-induced activation of p70s6k and cardiac myocyte hypertrophy in the cultured cells. Furthermore, to examine whether AMPK acts upstream or downstream of Akt activation, and to limit the influence of other signaling pathways activated by AICAR or metformin, we used constitutively active Akt (CA-Akt: myristoylated, 5 pfus/ce//) in combination with CA-AMPK (5pfus/cell). Our results demonstrate that p70s6k phosphorylation is activated by CA-Akt, but this activation is significantly blunted by CA-AMPK (Figure S6). In parallel, we examined protein synthesis (H3-Leucine incorporation) in response to CA-Akt. While CA-AMPK had little effect on basal protein synthesis, it significantly reduced CA-Akt induced protein synthesis (Figure S7). These results together demonstrate that AMPK acts downstream of Akt to reduce mTOR/p70s6k signaling in the in vitro system.
Discussion
The major new findings of this study are that (i) TAC resulted in more hypertrophy and fibrosis in AMPKα2 KO hearts than in wild type hearts, with a greater increase of LV diameter at end systole and a greater decrease of LV ejection fraction (indicative of LV dysfunction); (ii) TAC caused greater increases of myocardial 70S6KThr389, p-S6Ser235, p-elf4eSer209 and p-4EBP1Thr46, indicating that AMPKα2 KO augmented the response of these well defined downstream targets of mTOR to chronic systolic overload; (iii) AMPKα2 KO resulted in an unanticipated increase of p-Akt Ser473. To the best of our knowledge, these findings provide the first evidence that AMPKα2 exerts a protective effect against pressure overload-induced LV remodeling through attenuating the activation of downstream targets of mTOR.
Our finding of greater ventricular hypertrophy in the AMPKα2 KO mice after TAC is consistent with the notion that AMPKα2 negatively regulates protein synthesis 4. This is in agreement with reports 19, 20 that adiponectin attenuated hypertrophic growth in mice subjected to TAC via activation of AMPK and an AMPK-dependent signaling mechanism. The finding that AMPKα2 KO had no effect on LV structure and function in the present study is consistent with a previous report in which a decrease of myocardial AMPK activity produced by overexpression of mutated AMPKα2 also had no effect on LV function under unstressed conditions 16.
The increase of myocardial total-AMPKα, AMPKα1 and p-ACC observed in the WT mice 3 weeks after TAC in the present study is consistent with a previous report that pressure overload produced by clipping the ascending aorta of rats caused increases of myocardial AMPK activity, p-AMPK and total AMPKα1 protein content 3. The increased myocardial AMPKα1 protein content was not associated with a proportional increase of AMPKα1 activity in these mice, suggesting that some AMPKα1 protein modification might occur to attenuate its activity. The increase in myocardial AMPKα1, total-AMPKα and p-ACC in the AMPKα2 KO mice after TAC in the present study indicates that compensatory up-regulation of AMPKα1 was not sufficient to fully compensate for the loss of AMPKα2. Furthermore, the greater myocardial hypertrophy/fibrosis and dysfunction in the KO animals implies that AMPKα2 contributed to the response by which the heart adjusted to the increased hemodynamic load produced by TAC.
Our finding that AICAR attenuated cardiomyocyte hypertrophy produced by phenylephrine stimulation in cultured rat cardiomyocytes reaffirms the previous observation 18. The finding that constitutively active AMPKα dose-dependently attenuated phenylephrine-induced myocyte hypertrophy is new but also consistent with the findings obtained from activation of AMPK by AICAR or metformin (both of which result in activation of AMPK) as previously reported 18.
The rapamycin-sensitive mTOR complex has two well defined primary downstream targets, ribosomal S6 kinase (p70S6K) and eukaryotic initiation factor 4E binding protein (4EBP; 4E-BP1 is the dominant isoform in heart) 5. The increase of 70S6KThr389, p-S6Ser235 and p-4EBP1Thr46 in the mice exposed to TAC indicates mTOR complex activation. Activation of the mTOR signaling pathway exacerbates myocardial hypertrophy 21, while inhibition of mTOR signaling with rapamycin attenuated the development of ventricular hypertrophy in mice exposed to ascending aortic banding 21, 22. The finding that AMPKα2 deficiency enhanced TAC-induced phosphorylation of a group of downstream targets of mTOR such as p-70S6KThr389, p-S6Ser235 and p-elf4eSer209 reaffirms the concept that AMPKα2 is a negative regulator of the mTOR/p-70S6K signaling pathway in cardiac myocytes under conditions of hemodynamic overload. The finding that AMPKα2 KO did not affect the phosphorylation of 70S6KThr389, S6Ser235 and elf4eSer209 under control conditions, but significantly enhanced the TAC-induced increase of p-70S6KThr389, p-S6Ser235 and p-elf4eSer209 likely reflects the role of AMPKα2 as a negative regulator of these pathways particularly under stress conditions. Because it has been demonstrated that upregulation of myocardial p-70S6KThr389 and p-S6Ser235 activity by expression of either wild type or rapamycin-resistant 70S6K causes moderate ventricular hypertrophy, the greater increase of p-70S6KThr389, p-S6Ser235 and p-elf4eSer209 in the AMPKα2 KO after TAC likely contributed to the increased ventricular hypertrophy in these mice. As compared to the increase of myocardial p-70S6KThr389 and p-S6Ser235 in the AMPKα2 KO after TAC, AMPKα2 KO enhanced the TAC-induced increase of p-4EBP1Thr46 to a relatively smaller degree, indicating that AMPKα2 KO did not equally affect the phosphorylation of all of its downstream targets, suggests that p-70S6KThr389 and p-4EBP1Thr46 are differentially regulated. Activation of AMPK 18 or rapamycin 23 attenuates phenylephrine or endothelin-1 induced protein synthesis, cardiac myocyte hypertrophy, and the increase of p-eEF2Thr56. The decrease of myocardial p-eEF2Thr56 in the AMPKα2 KO mice after TAC in the present study is consistent with the finding of more ventricular hypertrophy in these mice, and suggests that activation of eEF2 (by a decrease of p-eEF2Thr56) may also have contributed to the greater hypertrophy in these mice.
Previous studies have demonstrated that chronic pressure overload in response to TAC or myocardial infarction increases myocardial p-Akt Ser473 24, 25. Since AMPK is generally regarded as a downstream target of p-Akt Ser473, the greater increase of myocardial p-Akt Ser473 in the AMPKα2 KO mice after TAC was not initially anticipated. Our finding that CA-AMPK attenuated CA-Akt induced increase of p-p70s6kThr389 and cardiac myocyte hypertrophy suggests that AMPKα2 acts downstream of Akt to reduce mTOR/p70s6k signaling and protein synthesis at least in the in vitro system. Nevertheless, a recent study demonstrated that activation of AMPK with AICAR or metformin increased p-Akt Ser473 in cultured rat cardiac myocytes 18, indicating that AMPK activity can also regulate p-Akt Ser473 at least in cultured cardiac myocytes. However, due to the complex physiological role of Akt in ventricular hypertrophy and function, it is not clear whether the increase of myocardial p-Akt Ser473 in the AMPKα2 KO mice after TAC was a compensatory event, or whether it contributed to the hypertrophy and dysfunction in these mice.
AMPK also plays an important role in regulating glucose and fatty acid oxidation under stress conditions. Activation of AMPK causes an increase of p-ACCSer79 to attenuate ACC activity and malonyl-CoA production. A decrease of p-ACCSer79 increases ACC activity to cause an increase of malonyl-CoA 26, 27, an endogenous inhibitor of CPT1 (the rate-limiting enzyme for entry of long-chain acyl-CoA into the mitochondria). Recent studies have demonstrated that decreased malonyl-CoA production by ACC2 gene KO not only enhanced fatty acid oxidation but also increased glucose oxidation 28, suggesting that the decreased p-ACCSer79 in AMPKα2 KO mice has the potential to impair myocardial energy production through inhibition of both fatty acid and glucose oxidation by accumulation of malonyl-CoA. Future studies measuring metabolic genes, and fatty acid and glucose uptake and oxidation in vivo, will be needed to determine the contribution of abnormalities of myocardial metabolism in the AMPKα2 KO mice.
Perspective
AMPK plays an important role in regulating energy balance, protein synthesis and cell growth. AMPK activation enhances fatty acid and glucose metabolism to augment ATP production and attenuates protein synthesis to preserve ATP. Using AMPKα2 KO mice in combination with transverse aortic constriction, we demonstrated that the reduction of myocardial AMPK activity exacerbated the development of ventricular hypertrophy, fibrosis and dysfunction in response to chronic pressure overload. AMPKα2 KO also significantly augmented the pressure overload induced increases of p-70S6KThr389, p-S6Ser235, p-elf4eSer209, and 4EBP1Thr46. Our findings provide the first direct evidence that AMPKα2 plays an important role in regulating pressure overload induced LV remodeling, and suggest that regulation of AMPKα2 may be a potential therapeutic approach to attenuate pressure overload induced ventricular hypertrophy.
Supplementary Material
Acknowledgments
Sources of Funding: This study was supported by NHLBI Grants HL71790 (YC) and HL021872 (RJB) from the NIH, ADA RA-12 (DAB). Drs. Zhang and Fassett are supported by Scientist Development Awards from the AHA-National Center.
Footnotes
Disclosures: None
References
- 1.Starling RC, Hammer DF, Altschuld RA. Human myocardial ATP content and in vivo contractile function. Molecular and cellular biochemistry. 1998;180:171–177. [PubMed] [Google Scholar]
- 2.Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circulation research. 2004;95:135–145. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- 3.Tian R, Musi N, D’Agostino J, Hirshman MF, Goodyear LJ. Increased adenosine monophosphate-activated protein kinase activity in rat hearts with pressure-overload hypertrophy. Circulation. 2001;104:1664–1669. doi: 10.1161/hc4001.097183. [DOI] [PubMed] [Google Scholar]
- 4.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell metabolism. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 5.Kwiatkowski DJ, Manning BD. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Human molecular genetics. 2005;14:R251–258. doi: 10.1093/hmg/ddi260. [DOI] [PubMed] [Google Scholar]
- 6.Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JF, Kahn A, Carling D, Schuit FC, Birnbaum MJ, Richter EA, Burcelin R, Vaulont S. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. The Journal of clinical investigation. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JF. Knockout of the alpha2 but not alpha1 5’-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. The Journal of biological chemistry. 2004;279:1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- 8.Hu P, Zhang D, Swenson L, Chakrabarti G, Abel ED, Litwin SE. Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signaling during pressure overload. American journal of physiology. 2003;285:H1261–1269. doi: 10.1152/ajpheart.00108.2003. [DOI] [PubMed] [Google Scholar]
- 9.Zhang P, Xu X, Hu X, van Deel ED, Zhu G, Chen Y. Inducible nitric oxide synthase deficiency protects the heart from systolic overload-induced ventricular hypertrophy and congestive heart failure. Circulation research. 2007;100:1089–1098. doi: 10.1161/01.RES.0000264081.78659.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu Z, Xu X, Hu X, Zhu G, Zhang P, van Deel ED, French JP, Fassett JT, Oury TD, Bache RJ, Chen Y. Extracellular superoxide dismutase deficiency exacerbates pressure overload-induced left ventricular hypertrophy and dysfunction. Hypertension. 2008;51:19–25. doi: 10.1161/HYPERTENSIONAHA.107.098186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howard CV, Reed MG. Estimation of component volume and volume fraction. In: Catherine J, editor. Unbiased Stereology. Three-dimensional measurement in microscopy. Abingdon, Oxon: BIOS Scientific Publishers; 2005. pp. 17–64. [Google Scholar]
- 12.Hayashi T, Hirshman MF, Fujii N, Habinowski SA, Witters LA, Goodyear LJ. Metabolic stress and altered glucose transport: activation of AMP-activated protein kinase as a unifying coupling mechanism. Diabetes. 2000;49:527–531. doi: 10.2337/diabetes.49.4.527. [DOI] [PubMed] [Google Scholar]
- 13.Zhang W, Anger T, Su J, Hao J, Xu X, Zhu M, Gach A, Cui L, Liao R, Mende U. Selective loss of fine tuning of Gq/11 signaling by RGS2 protein exacerbates cardiomyocyte hypertrophy. The Journal of biological chemistry. 2006;281:5811–5820. doi: 10.1074/jbc.M507871200. [DOI] [PubMed] [Google Scholar]
- 14.Xu X, Fassett JT, Hu XL, Zhu GS, Schnermann J, Bache RJ, Chen Y. Endogenous adenosine protects the heart from severe systolic overload induced ventricular hypertrophy and congestive heart failure. Hypertension. 2008;51:1557–1564. doi: 10.1161/HYPERTENSIONAHA.108.110833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xing Y, Musi N, Fujii N, Zou L, Luptak I, Hirshman MF, Goodyear LJ, Tian R. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. The Journal of biological chemistry. 2003;278:28372–28377. doi: 10.1074/jbc.M303521200. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Miller EJ, Ninomiya-Tsuji J, Russell RR, 3rd, Young LH. AMP-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circulation research. 2005;97:872–879. doi: 10.1161/01.RES.0000187458.77026.10. [DOI] [PubMed] [Google Scholar]
- 17.Nairn AC, Palfrey HC. Identification of the major Mr 100,000 substrate for calmodulin-dependent protein kinase III in mammalian cells as elongation factor-2. The Journal of biological chemistry. 1987;262:17299–17303. [PubMed] [Google Scholar]
- 18.Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. The Journal of biological chemistry. 2004;279:32771–32779. doi: 10.1074/jbc.M403528200. [DOI] [PubMed] [Google Scholar]
- 19.Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, Kumada M, Sato K, Schiekofer S, Ohashi K, Funahashi T, Colucci WS, Walsh K. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nature medicine. 2004;10:1384–1389. doi: 10.1038/nm1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao Y, Takashima S, Maeda N, Ouchi N, Komamura K, Shimomura I, Hori M, Matsuzawa Y, Funahashi T, Kitakaze M. Exacerbation of heart failure in adiponectin-deficient mice due to impaired regulation of AMPK and glucose metabolism. Cardiovascular research. 2005;67:705–713. doi: 10.1016/j.cardiores.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 21.Shioi T, McMullen JR, Tarnavski O, Converso K, Sherwood MC, Manning WJ, Izumo S. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation. 2003;107:1664–1670. doi: 10.1161/01.CIR.0000057979.36322.88. [DOI] [PubMed] [Google Scholar]
- 22.McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, Izumo S. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050–3055. doi: 10.1161/01.CIR.0000130641.08705.45. [DOI] [PubMed] [Google Scholar]
- 23.Wang L, Proud CG. Regulation of the phosphorylation of elongation factor 2 by MEK-dependent signalling in adult rat cardiomyocytes. FEBS letters. 2002;531:285–289. doi: 10.1016/s0014-5793(02)03536-6. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Z, Teng CT. Interplay between estrogen-related receptor alpha (ERRalpha) and gamma (ERRgamma) on the regulation of ERRalpha gene expression. Molecular and cellular endocrinology. 2007;264:128–141. doi: 10.1016/j.mce.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Deel ED, Lu Z, Xu X, Zhu G, Hu X, Oury TD, Bache RJ, Duncker DJ, Chen Y. Extracellular superoxide dimutase protects the heart against oxidative stress and hypertrophy after myocardial infarction. Free Radic Biol Med. 2008;44:1305–1313. doi: 10.1016/j.freeradbiomed.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, Wakil SJ. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science. 2001;291:2613–2616. doi: 10.1126/science.1056843. [DOI] [PubMed] [Google Scholar]
- 27.Dyck JR, Lopaschuk GD. AMPK alterations in cardiac physiology and pathology: enemy or ally? The Journal of physiology. 2006;574:95–112. doi: 10.1113/jphysiol.2006.109389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi CS, Savage DB, Abu-Elheiga L, Liu ZX, Kim S, Kulkarni A, Distefano A, Hwang YJ, Reznick RM, Codella R, Zhang D, Cline GW, Wakil SJ, Shulman GI. Continuous fat oxidation in acetyl-CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:16480–16485. doi: 10.1073/pnas.0706794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.