

Abstract
Induction of innate immunity, particularly through the induction of interferon and chemokines, by rabies virus (RABV) infection has been reported to be inversely correlated with pathogenicity. To further investigate the association between the expression of chemokines and RABV infection, laboratory-attenuated RABV (B2C) and wild-type (wt) RABV (DRV) were administered to Balb/c mice intramuscularly. Chemokine expression, inflammatory cell infiltration, and blood-brain barrier (BBB) permeability were evaluated at various time points after infection. At day 3 post infection (p.i.) there was very little inflammation in the central nervous system (CNS) and BBB permeability did not change in mice infected with either virus when compared with mock-infected mice. At 6 day p.i., infection with B2C induced the expression of inflammatory chemokines and infiltration of inflammatory cells into the CNS, while these changes were minimal in DRV-infected mice. Furthermore, infection with B2C significantly enhanced BBB permeability comparing to infection with DRV. Among the upregulated chemokines, the expression of IP-10 was best correlated with infiltration of inflammatory cells into the CNS and enhancement of BBB permeability. These data indicate that laboratory-attenuated RABV induces expression of chemokines and infiltration of inflammatory cells into the CNS. Upregulation of chemokines by B2C may have triggered the change in BBB permeability, which helps infiltration of inflammatory cells into the CNS, and thus attenuation of RABV.
Keywords: rabies, innate immunity
1. Introduction
Rabies is one of the most important zoonotic infections and still causes more than 55,000 human deaths each year (Martinez, 2000). Most of the human rabies cases occur in Asia and Africa where dog rabies is prevalent (Anonymous, 1992; Fu, 1997). In the United States, dog rabies has almost been eliminated through massive vaccination during the past 6 decades (Blanton et al., 2006). However, bat (particularly the silver-haired bat) rabies has emerged to be responsible for most of the human rabies cases in the past 20 years (CDC, 2003; Morimoto et al., 1996; Rupprecht et al., 1997). Once clinical signs develop, rabies is always fatal (Anonymous, 1992; Fu, 1997). Despite the lethality of rabies, only mild inflammation and little neuronal destruction were observed in the central nervous system (CNS) of rabies patients (Miyamoto and Matsumoto, 1967; Murphy, 1977). Alternatively, laboratory-attenuated RABV induces extensive inflammation and neuronal degeneration in experimentally infected animals (Miyamoto and Matsumoto, 1967; Murphy, 1977). To understand the differential effects between wildtype (wt) and attenuated viruses, mice were infected with two RABV strains, one laboratory-attenuated RABV and the other wt RABV, and compared the host responses to infection (Wang et al., 2005). It was found that laboratory-attenuated RABV induced extensive inflammation, apoptosis, and neuronal degeneration in the CNS; however, wt RABV caused little or no neuronal damage. Furthermore, laboratory-attenuated RABV induced the expression of genes associated with innate immune responses, particularly type 1 interferon (IFN α and β), chemokines and complements while many of these genes were not activated in mice infected with wt RABV (Wang et al., 2005). The induction of innate immunity has been confirmed by others using laboratory-attenuated viruses to infect mice or neuronal cells (Johnson et al., 2006; Nakamichi et al., 2004; Prehaud et al., 2005). Induced innate immune response genes include inflammatory chemokines (including RANTES, MIP-1α, IP-10) and cytokines (IL-6, IL-1β, and TNF-α), IFN and IFN-related genes (IFN-α/β, STAT1), as well as Toll-like receptors (TLRs) (Johnson et al., 2006; Nakamichi et al., 2004; Prehaud et al., 2005). These observations led to the hypothesis that laboratory-attenuated RABV is a potent inducer of host innate immunity (Wang et al., 2005). Innate immune response, especially proinflammatory cytokines and chemokines can recruit immune cells to the site of infection and remove of pathogens, which might be an important mechanism of RABV attenuation.
Recently it has been reported that wt and laboratory-attenuated RABVs differentially induce changes in the blood-brain barrier (BBB) permeability (Fabis et al., 2008; Phares et al., 2007; Roy and Hooper, 2007; Roy et al., 2007). The BBB was more permeable in mice infected with laboratory-attenuated CVS-F3 than mice infected with silver-haired bat rabies virus (SHBRV). The former is an antibody escape mutant derived from CVS virus (Dietzschold et al., 1983) and the latter is a wt RABV isolated from a human patient (Morimoto et al., 1996). It was reasoned that enhancement of BBB permeability allows immune effectors to cross the BBB and enter the CNS. Indeed, adoptive transfer of immune cells isolated from mice infected with laboratory-attenuated RABV which resulted in clearance of wt RABV from the CNS (Roy et al., 2007). These studies suggest that changes in BBB permeability are associated with clearance of the apathogenic RABV from the CNS (Phares et al., 2007; Roy et al., 2007). Alternatively, failure to increase the permeability of BBB leads to disease in wt RABV-infected mice (Roy et al., 2007).
The present study attempts to determine the contributions of the innate immune response, especially the role of inflammatory chemokines, in the enhancement of BBB permeability and the outcome of RABV infection. If increased BBB permeability is the major contributor towards survival, it is important to determine the mechanism that is responsible for this change in RABV-infected animals. Therefore, expression of chemokines, infiltration of inflammatory cells, and enhancement of BBB permeability were investigated in mice infected with laboratory-adapted or wt RABV. It was found that expression of chemokines was closely associated with infiltration of inflammatory cells and increases in BBB permeability. Among the chemokines investigated, the expression of IP-10 was best correlated with such changes in mice infected with laboratory-attenuated RABV. The data indicate that laboratory-attenuated RABV upregulates the expression of chemokines, which increases infiltration of inflammatory cells into the CNS, triggering changes in the BBB permeability. This in turn helps more infiltration of effector cells into the CNS resulting in attenuation of RABV virulence.
2. Materials and methods
2.1. Viruses, antibodies
Four RABVs were used in this study and they are SHBRV, DRV, B2C, and SN-10. SHBRV is a wt RABV isolated from a human patient (Morimoto et al., 1996). DRV is a wt virus isolated from a dog (Dietzschold et al., 2000). B2C is a laboratory-attenuated virus isolated from challenge virus standard (CVS-24) by serial passaging in BHK cells (Morimoto et al., 1998). SN-10 is a cloned virus derived from the attenuated SAD B19 vaccine strain (Schnell et al., 1994). Virus stocks were prepared as described (Sarmento et al., 2005; Wang et al., 2005). Briefly, one-day-old suckling mice were inoculated with 10 μl of viral inoculum by the intracerebral (IC) route. When moribund, mice were euthanized and brains were removed. A 10% (w/v) suspension was prepared by homogenizing the brain in DMEM. The homogenate was centrifuged to remove debris and the supernatant collected and stored at −80°C. Fluorescein isothiocyanate (FITC)-conjugated antibody against the RABV nucleoprotein (N) protein was purchased from FujiRebio (FujiRebio Diagnostic INC, PA). Anti-RABV N monoclonal antibody 802-2 (Hamir et al., 1995) was obtained from Dr. Charles Rupprecht, Center for Disease Control and Prevention (CDC). Anti-RABV glycoprotein (G) polyclonal antibody was prepared in rabbits as described (Fu et al., 1993) and has been shown to have similar affinity to the G from wt SHBRV and laboratory-adapted N2C (Yan et al., 2001). Anti-CD3 polyclonal antibody was purchased from Dako (Dako North America, CA).
2.2. Mice
Female ICR mice at the age of 4-6 weeks were purchased from Harlan (Harlan, IN) and Balb/c mice at 6-8 weeks of age were purchased from NCI (NCI-Frederick, MD). Mice were housed in temperature- and light-controlled quarters in the Animal Facility, College of Veterinary Medicine, University of Georgia. All animal experiments were carried out as approved by the Institutional Animal Care and Use Committee.
2.3. Real-time SYBR Green PCR and Real-time quantitative RT-PCR
Brains and spinal cords were removed from infected mice at indicated time points and flash frozen on dry ice before being stored at -80°C. RNA was extracted from these tissues using Trizol following the manufacturer's instructions and used for real-time PCR as described previously (Wang et al., 2005). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an endogenous reference gene. Amplification primers of the proinflammatory genes are listed in a previous paper (Wang et al., 2005). To determine virus replication and RNA transcription in each sample, virus specific N mRNA and genomic RNA were measured by quantitative RT-PCR. cDNA standards were made from RABV N gene. Standard curves were constructed with Ct values obtained using dilutions of the synthetic standards. The mRNA and genomic RNA copy numbers in each sample were normalized to the respective copy number derived from the standard curve.
2.4. BBB integrity
BBB permeability was assessed using a modification of a previously described technique in which Na-fluorescein (NaF) is utilized as a tracer molecule (Phares et al., 2007). Mice received 100 μl of 10% NaF in PBS intravenously under anesthesia. After 10 min to allow circulation of the NaF, cardiac blood was collected and the animals were transcardially perfused with PBS. Spinal cord or brain tissues were homogenized in cold 7.5% trichloroacetic acid (TCA) and centrifuged for 10 min at 10,000 g to remove insoluble precipitates. After the addition of 0.25 ml 5N NaOH, the fluorescence of a 100 μl supernatant sample was determined using a BioTek Spectrophotometers (Bio-Tek Instruments, INC) with excitation at 485 nm and emission at 530 nm. Standards (125 to 4000 μg/ml) were used to calculate the NaF content of the samples. NaF uptake into tissue is expressed as (μg fluorescence spinal cord/mg tissue)/(μg fluorescence sera/ml blood) to normalize values for blood levels of the dye at the time of tissue collection (Phares et al., 2007).
2.5. Histopathology and immunohistochemistry
For histopathology and immunohistochemistry, animals were anesthetized with ketamine-xylazine at a dose of 0.1 ml/10g body weight and then perfused by intracardiac injection of PBS followed by 10% neutral buffered formalin as described previously (Li et al., 2005). Brains, spinal cords and dorsal root ganglia (DRG) were removed and paraffin embedded for coronal sections (4 μm). To de-paraffin, slides were heated at 60°C for 25 min and then dipped in CitriSolv (Fisher Scientific, PA) three times for 5 min and dried until chalky white. After de-paraffinization, slides were stained with hematoxylin and eosin (H&E). Slides were heated in antigen unmasking solution (Vector Laboratories, CA) above 90°C for 20 min and naturally cooled down to room temperature. Anti-RABV N monoclonal antibody 802-2 was used to detect the viral antigen. The primary antibody and then secondary antibody (biotinylated) were used for immunological reaction as described (Yan et al., 2001). The avidin-biotin-peroxidase complex (Vector Laboratories,CA) was then used to localize the biotinylated antibody. Finally, diaminobenzidine (DAB) was used as a substrate for color development. The intensity of DAB signals corresponding to RABV N or G antigen were measured by Image-pro Plus software (Media Cybernetics, Inc. Bethesda, MD).
2.6. Enzyme-linked immunosorbent assay (ELISA)
ELISA was used to quantify the amount of MIP-1α, IP-10 and RANTES in mouse brain suspensions by using the murine ELISA Kit (R&D Systems, Minneapolis, MN) according to the manufacture's protocol.
3. Result
3.1. Differential induction of chemokine expression after IC infection with laboratory-attenuated and wt RABVs
Previously it was shown that infection of mice with laboratory-attenuated B2C virus activated innate immunity in the mouse CNS, while wt SHBRV did so to a much lesser degree (Wang et al., 2005). To extend these studies, groups of ICR mice were infected with larger doses of wt RABV (SHBRV or DRV) or laboratory-attenuated RABV (B2C or SN-10) by the intracerebral (IC) route with a virus dose of 10 ICLD50. At the onset of severe paralysis, the mice were sacrificed and their brains removed for immunohistochemistry to quantify the expression of nascent N or for realtime-PCR to quantify expression of innate immunity genes. The level of viral N expression was measured by the intensity of DAB signals by Image-pro Plus Software. As shown in Fig 1, the levels of N expression were similar in mice regardless of virus phenotype, indicating that the level of viral replication in the CNS is similar for all the viruses. On the other hand, significantly more G was detected in mice infected with laboratory-attenuated than with wt RABV, an observation similar to those reported previously (Sarmento et al., 2005; Wang et al., 2005). To determine the expression of genes relevant to the innate immunity, real-time PCR was used to measure the expression of 8 chemokine genes. As shown in Table 1, the expression of these genes was upregulated after RABV infection; however, the upregulation was usually higher in mice infected with laboratory-attenuated than with wt RABV. MCP1, MIP1β, RANTES, MCP3, MIG, IP-10 are more upregulated in B2C and SN-10 infected mice than in DRV and SHBRV infected mice. Most of these genes are upregulated 2- to 1000- folds in B2C-infected mice than in DRV-infected mice or 2- to 20- folds in B2C-infected mice than in SHBRV-infected mice. The upregulation of these genes in mice infected with SN-10 are 2- to 50- fold or 2- to 61- fold higher than that in mice infected with DRV or SHBRV, respectively. The most upregulated gene was IP-10, which increased 2394-fold in animals infected with B2C virus when compared with uninfected mice. These data are consistent with our previous findings (Wang et al., 2005) and suggest that differential induction of chemokines by wt and laboratory-attenuated RABV may be contributing to pathogenicity and/or attenuation of the respective RABV infections.
Figure 1.
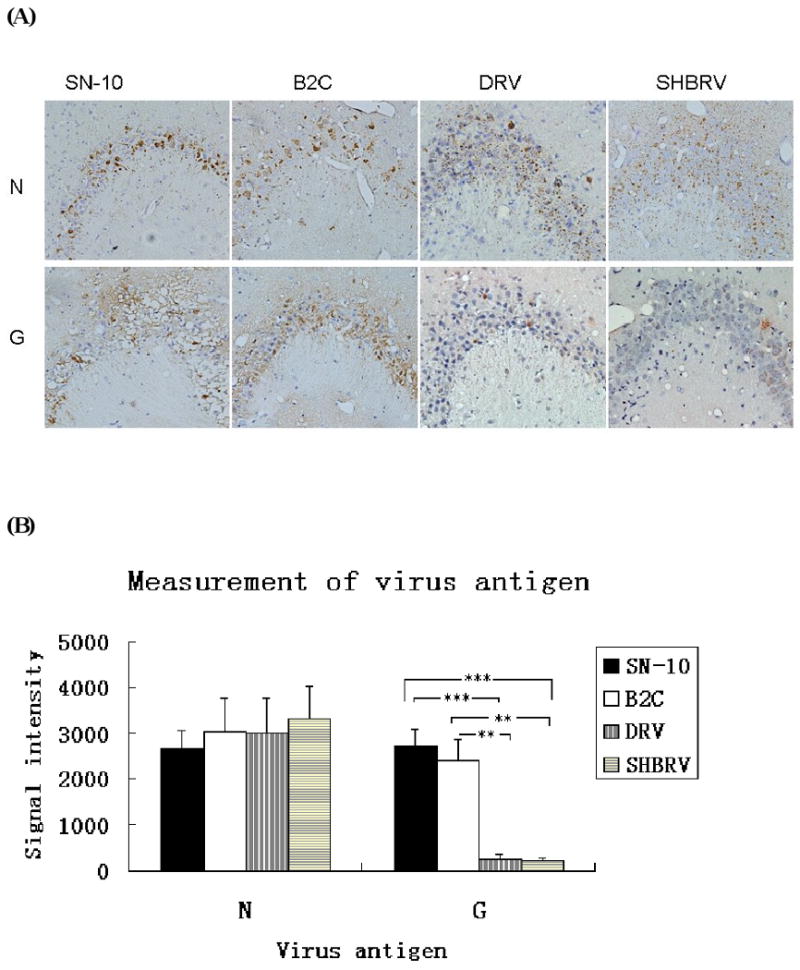
Detection of viral antigen in brains of mice infected with different RVs. Viral antigens RABV-N and RABV-G were detected by immunohistochemistry (A). The intensity of DAB signals corresponding to each viral antigen was measured in the hippocampus area of 3 animals by Image-pro plus software. The average of signal intensity and standard deviation are shown (B). Statistically significant differences between the viruses in infected mice were determined by the Mann-Whitney U test, *** (p<0.0001) and ** (p<0.01). Magnificant 20×.
Table 1. Expression of inflammatory genes in mouse brain infected with RABV.
| Realtime-PCR (Fold change) | ||||
|---|---|---|---|---|
| DRV | SHBRV | B2C | SN-10 | |
| MCP1 (CCL2) | 8.5 | 4.6 | 11.0 | 283.0 |
| MIP1α (CCL3) | 50.5 | 61.0 | 44.0 | 702.0 |
| MIP1β (CCL4) | 5.8 | 13.0 | 15.4 | 67.6 |
| RANTES (CCL5) | 17.9 | 267.0 | 413.0 | 305.0 |
| MCP3 (CCL7) | 18.4 | 101.0 | 226.0 | 242.0 |
| MCP5 (CCL12) | 2.6 | 158.0 | 155.0 | 82.2 |
| Mig (CXCL9) | 3.0 | 2.4 | 43.1 | 24.7 |
| IP-10 (CXCL10) | 2.5 | 819.0 | 2394.0 | 56.6 |
3.2. Evaluation of viral antigen and viral RNA in mice after IM infection with laboratory-attenuated and wt RABV
Since most of the RABV infections are caused by animal bites, wt DRV and laboratory-attenuated B2C each at 10 IMLD50 were inoculated intramuscularly (IM) into Balb/c mice. At days 3, 6 and 9 p.i., animals were sacrificed and brains, spinal cords, as well as DRG were collected for evaluation of viral antigen by immunohistochemistry and/or viral RNA by realtime-PCR. Viral antigen was detected in the DRG in most of the infected animals at day 3 p.i., and in all the infected animals by day 6 p.i. although the level of viral antigen was low (Fig 2A). By day 9 p.i., a higher level of viral antigen was detected in the DRG, particularly in B2C-infected mice (Fig 2A). Virus antigen could not be detected in either the spinal cord or the brain infected with DRV or B2C at day 3 p.i. At days 6 and 9 p.i., virus antigen was only detected sparsely in the spinal cord or the brain in B2C- or DRV-infected mice (Fig. 2A). Thus viral replication (measuring mRNA and genomic RNA) in the CNS was evaluated by real-time PCR using RABV gene-specific primers. As shown in Fig 2B, more mRNA and genomic RNA was detected in mice infected with B2C compared with DRV at day 6 p.i. On the other hand, more mRNA and genomic RNA were detected in the CNS of mice infected with DRV compared with B2C at day 9 p.i.
Figure 2.
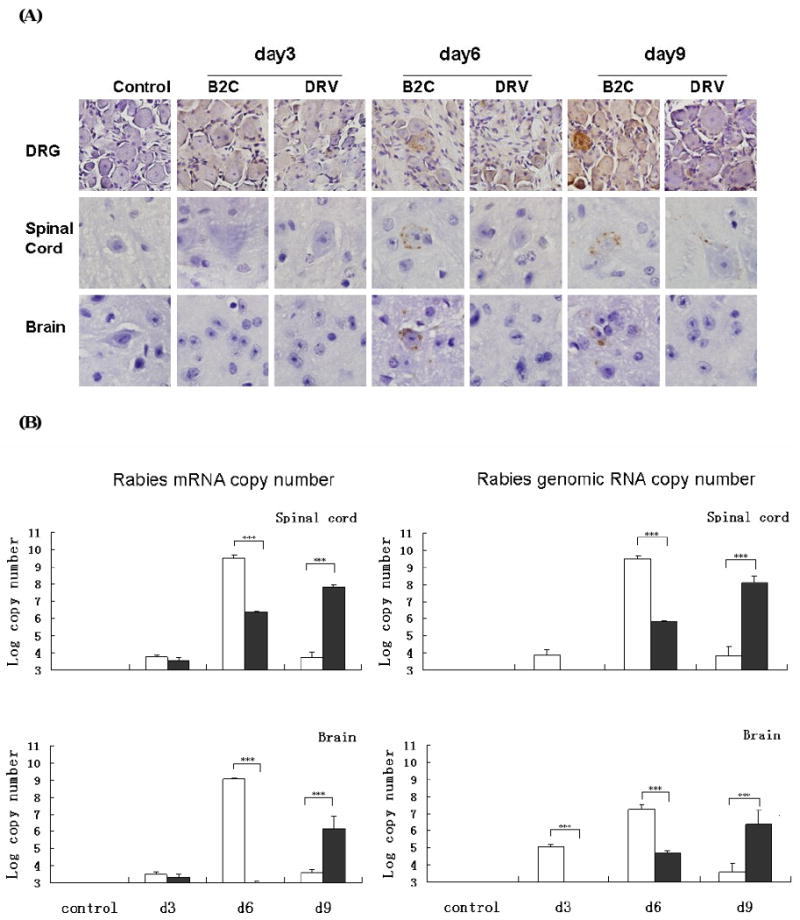
Detection of viral antigen in mice infected with B2C or DRV at different time points with 10 IMLD50. RABV N was detected by immunohistochemistry in mouse DRG, spinal cord and brain section (A). By using viral gene-specific primers, RABV mRNA and genomic RNA were detected in B2C (white histogram)- and DRV (black histogram)-infected mice (C). Statistically significant differences between the infected and uninfected mice were determined by the Mann-Whitney U test, *** (p<0.0001), ** (p<0.001), * (p<0.005), and · (p<0.01). Magnificant 40× in DRG, 100× in spinal cord and brain (N antigen staining).
3.3. Changes in BBB permeability in mice after IM infection with laboratory-attenuated and wt RABV
To investigate if infection with each RABV induces changes in BBB permeability, the leakage of the NaF from circulation into the CNS tissues was measured in the spinal cord, cerebellum, and cerebrum of mice infected with 10 IMLD50 of each virus. As shown in Fig. 3A, no significant change in BBB permeability was observed in mice infected with either virus at day 3 p.i. BBB permeability was significantly enhanced in all brain regions of the CNS by 6 days after infection with B2C, but not with DRV when compared to sham-infected mice. By day 9 p.i., BBB permeability in mice infected with B2C returned to the level as seen in DRV-infected mice, which is not significantly different from sham-infected mice. To ensure that the changes in BBB permeability are not affected by the virus concentration, mice were infected with an increased dose of DRV or decreased dose of B2C. As shown in Fig. 3B, increasing the DRV dose to 100 IMLD50 did not result in enhanced BBB permeability while decreasing the B2C dose to 1 IMLD50 led to enhanced BBB permeability. At day 6 p.i., mice infected with 1 IMLD50 B2C show significant higher BBB permeability changes than mice infected with either 10 or 100 IMLD50 DRV. These data indicate that enhancement of BBB permeability is not dose-dependent, but virus-dependent.
Figure 3.

BBB permeability in RABV infected mice. BBB permeability changes are presented as the fold increase in NaF uptake in the cerebellum, cerebrum, and spinal cord in 10 IMLD50 infected mice over the level of NaF in uninfected mice (A). BBB permeability change is also detected with different virus inoculate dose (B). Statistically significant differences between the infected and uninfected mice were determined by the Mann-Whitney U test, ** (p<0.01).
3.4. Induction of inflammatory chemokines in mice after IM infection with laboratory-attenuated and wt RABV
To determine what innate immune responses occur following RABV infection, spinal cords and brains from mice infected with 10IMLD50 of DRV or B2C were collected on day 3, 6 and 9 p.i. and homogenates were subsequently assayed for the expression of cytokines and chemokines by real-time PCR. At day 3 p.i., the expression of TLR-3, IRF-7, IFNα4, IFNγ, IL-6, and Mx-1 was upregulated in the spinal cord, while up-regulation in the expression of TLR3, IRF7, IFNα4, IFNα5, IFNγ, IL-6, MMP9, Mx-1,and OAS-1g was observed in the brain of mice infected with B2C (data not shown). None of these genes was found to be upregulated in either the spinal cord or the brain of mice infected with DRV. By 6 days p.i., expression of cytokines and chemokines was highly increased in the CNS of mice infected with B2C (Fig. 4A). The most upregulated gene in expression is IP-10 in the spinal cord (data not shown) and MIP-1α in the brain when compared to sham-infected mice. In mice infected with DRV, expression of these chemokines genes was either not changed when compared to sham-infected mice or upregulated to a much lesser extent than that observed in B2C-infected animals. These data are consistent with our previous findings (Wang et al., 2005) and suggest that laboratory-attenuated RABV activates the expression of the genes involved in the innate immunity while wt RABV does not. Furthermore, the expression pattern of IP-10, MIP-1α, and RANTES are parallel to the changes in BBB permeability (Fig. 4A). These data suggest that expression of chemokines may play an important role in the enhancement of BBB permeability. The expression of TNF-α in the brain is also parallel to the changes in BBB permeability; however, it has been reported that TNF-α is not a determinant of changes in BBB integrity (Phares et al., 2007).
Figure 4.
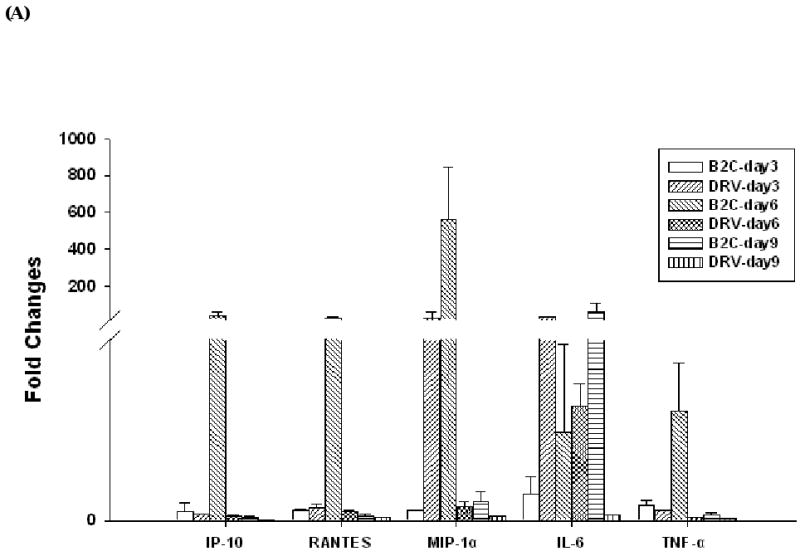
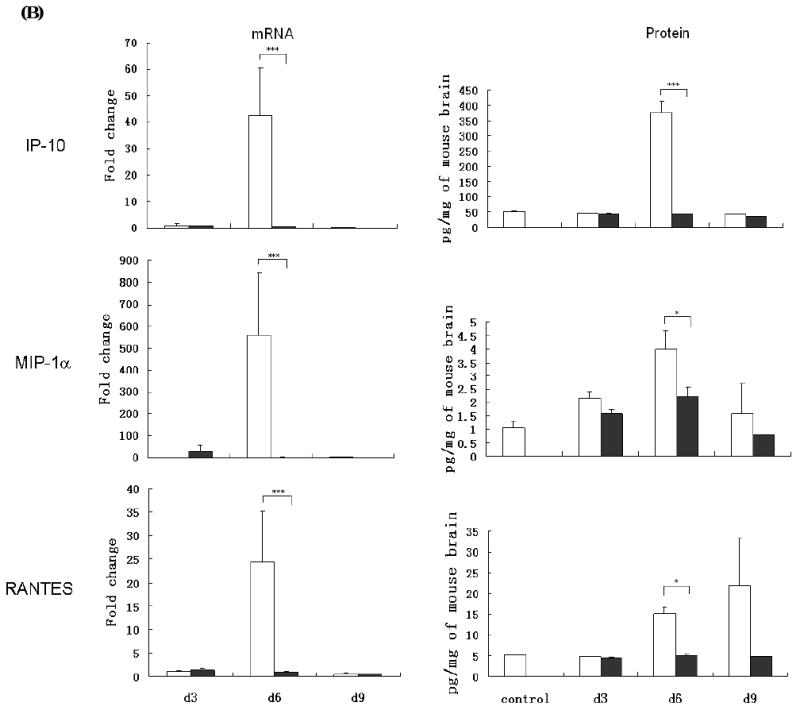
Determination of proinflammatory cytokine and chemokine levels in RABV infected mice. Balb/c mice were infected with 10 IMLD50 of B2C or DRV. Chemokine and cytokine levels in brain (A) were assayed by realtime PCR as described in Material and Methods. The mRNA data are expressed as the mean ± SEM of smRNA for specific chemokine genes over the mRNA for a specific endogenous housekeeping gene (GAPDH) in infected mouse tissue minus the background levels from uninfected mouse tissue. IP-10, MIP-1α, and RANTES mRNAs (left) and protein levels (right) were assayed by realtime PCR or ELISA respectively, as described in Material and Methods (C). Mice infected with B2C or DRV were shown in white or black histogram, respectively.
Since the most-upregulated genes are chemokines, particularly IP-10, RANTES, and MIP-1α, ELISA was carried out to determine protein expression in brains of mice infected with 10IMLD50 of B2C or DRV. As shown in Fig. 4B, the expression of IP-10 MIP-1α, and RANTES proteins was significantly upregulated at day 6 p.i. in the brains of mice infected with B2C, correlating with the enhancement of BBB permeability. On the other hand, the expression level of IP-10, MIP-1α, and RANTES in mice infected with DRV was similar to that in sham-uninfected mice at all three time points. The expression patterns of IP-10 and MIP-1α proteins in the brain were parallel to those observed in the expression of mRNA and in the changes in BBB permeability. The expression of RANTES increased at days 6 and reached a peak by day 9 p.i., however there is no significant difference between B2C- and DRV-infected mice at day 9 p.i. Furthermore, the greatest increased in the level of protein expression is IP-10 with level reaching 400 pg per mg of mouse brain. Thus, the data suggest that differential induction of chemokines, particularly IP-10, by B2C and DRV may contribute to the observed differences in innate immune responses and BBB permeability changes.
3.5. Inflammatory response in mice after IM infection with laboratory-attenuated and wt RABV
To investigate if upregulation of chemokine expression led to infiltration of inflammatory cells into the nervous system of infected animals, DRGs, spinal cords and brains were collected for histopathology and immunohistochemistry. At day 3 p.i., infiltration of inflammatory cells into the CNS was minimal in mice infected with either virus (Fig. 5A). Severe inflammation was observed in the spinal cord and brain of mice infected with B2C at 6 days p.i., which subsided by day 9 p.i. Only mild inflammation was observed in DRV-infected mice. To quantfy the inflammatory response in the CNS, CD3-positive cells were measured by using anti-CD3 antibodies in the thalamus/hypothalamus. As shown in Figs. 5B and 5C, no significant difference in the number of CD3-positive cells was found between B2C-, DRV- or mock-infected mice at day 3 p.i. By day 6 p.i., significantly more CD3-positive cells were found in B2C- than in DRV- or mock-infected mice. By day 9 p.i., the number of CD3-positive cells in B2C-infected mice declined, but was still more than that found in DRV-infected mice (Figs. 5B and 5C). The patterns of CD3-positive cells in DRG or spinal cord are similar to those in the brain. The results thus suggested that laboratory-attenuated RABV induces more inflammation than wt RABV.
Figure 5.
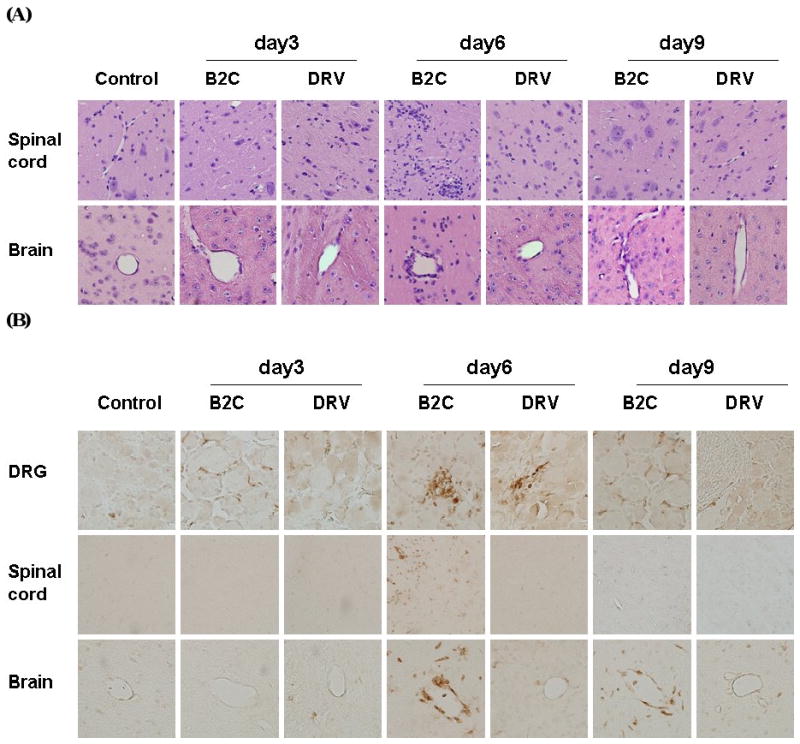
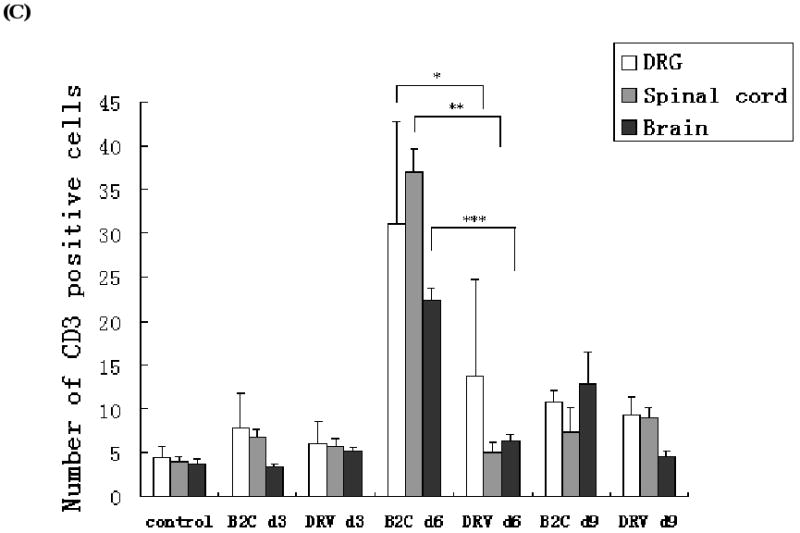
Detection of inflammation and CD3-positive cells in the mouse nervous tissue. Mice were infected with B2C or DRV with 10IMLD50 and were transcardially perfused with 10% formalin at days 3, 6, or 9 p.i. Paraffin sections were subjected to HE staining (A) or immunohistochemistry for detecting CD3-positive cells (B). Three serial sections and two vessels were selected from each mouse for quantification, and the average numbers of CD3-positive cells obtained were used for statistical analysis (C). Statistically significant differences between the infected with uninfected mice were determined by the Mann-Whitney U test, as *** (p<0.0001), ** (p<0.01), * (p<0.05), and · (p<0.1). Magnificant 20× in the DRG and spinal cord sections, 40× in the brain.
4. Discussion
Previously we reported that laboratory-attenuated RABV is a potent inducer of innate immune response while wt RABV is not, which led us to hypothesize that induction of innate immunity is an important mechanism of RABV attenuation (Wang et al., 2005). Recently, it has been reported that change in BBB permeability is important in virus clearance and thus play a decisive role in the virus attenuation (Phares et al., 2007; Roy and Hooper, 2007; Roy et al., 2007). In this study, we attempted to determine if induction of innate immunity, especially induction of chemokines, is related to, or associated with changes in BBB permeability. Our results suggest that these two events are closely related, at least as a function of time after RABV infection.
Our previous studies with microarray analysis and real-time PCR showed that attenuated B2C induced higher levels of chemokine expression than wt SHBRV (Wang et al., 2005). In the present study, we extended our previous study by including more laboratory-attenuated and wt viruses. It was found that laboratory-attenuated RABVs (B2C and SN-10) consistently induced higher levels of expression of chemokiness than wt viruses (SHBRV and DRV) irrespective the route of infection (IC vs. IM) (Table 1). Furthermore, infection with attenuated RABV as well as RABV-like viruses upregulates chemokine expression (Johnson et al., 2006; Mansfield et al., 2008; Nakamichi et al., 2004; Prehaud et al., 2005). Together, these studies further confirm that infection with laboratory-attenuated RABV induces the expression of genes involved in innate immunity, particularly chemokines, which may be one of the mechanisms by which RABV is attenuated.
A series of recent studies in Hooper's laboratory showed that changes in BBB permeability are associated with clearance of the apathogenic RABV isolates (CVS-F3, ERA, and PM) from the CNS, while pathogenic RABV strains (DRV, Skunk RV, etc) fail to change BBB permeability and deliver immune effectors into CNS (Phares et al., 2007; Roy and Hooper, 2008; Roy et al., 2007). Adoptive transfer of immune cells isolated from mice infected with attenuated RABV resulted in clearance of wt RABV from the CNS (Roy et al., 2007). Enhancement of the BBB permeability with myelin basic protein (MBP) can also increase the survivorship of mice after infection with wt RABV (Roy and Hooper, 2007). The PLSJL mouse strain which is easier to initiate CNS inflammation than the normal 129 mouse strain was more resistant to the death caused by pathogenic RABV infection (Roy and Hooper, 2008). All these studies indicate that failure to open the BBB leads to disease in RABV infections. However, the mechanism leading to increased BBB permeability after infection with laboratory-attenuated RABV is not entirely clear. TNF-α, an inflammatory cytokine, has not been found to be important in enhancing BBB permeability although it is upregulated after infection with laboratory-attenuated RABV (Phares et al., 2007). Peroxynitrite (ONOO-) has been reported to play a role in this process (Phares et al., 2007). Inhibition of peroxynitrite-dependent radicals by urate, however, did not change the outcome of RABV infection (Fabis et al., 2008). Thus, it is not clear if peroxynitrite is the main trigger of BBB opening in RABV infection.
In this study, an attempt was made to determine the contribution of different chemokines in the enhancement of BBB permeability and the outcome of RABV infection. Two viruses (wt DRV and laboratory-attenuated B2C) were used to infect Balb/c mice by IM route and the induction of chemokine expression and changes in BBB permeability were monitored. It was found that laboratory-attenuated B2C induced the expression of many chemokines, particularly at day 6 p.i. In addition, infiltration of inflammatory cells to the CNS was the highest at this time of infection. Enhancement of BBB permeability in the CNS was found only in mice infected with B2C at day 6 p.i. Therefore the induction of chemokine expression correlates with the enhancement of BBB permeability at least as a function of time post infection. Detailed analysis indicates that upregulation of chemokine genes and the increased chemokine protein expression correlates with change in BBB permeability in B2C-infected mice. Among these, the upregulation of IP-10 was the highest and the timing of IP-10 upregulation corresponded in parallel to the enhancement of BBB permeability. It has been reported that chemokines play a major role in increasing BBB permeability (Glabinski and Ransohoff, 1999; Man et al., 2007; Zozulya et al., 2007). Increased expression of MIG, IP-10 and RANTES is observed within the cerebral spinal fluid (CSF) and CNS tissue of multiple sclerosis (MS) patients during periods of clinical attack that correlates with infiltration of leukocytes into the CNS (Sorensen et al., 1999). Administration of rabbit antisera specific for either MIG or IP-10 to mouse hepatitis virus (MHV)-infected mice correlated with a dramatic decrease of infiltrating cells (Liu et al., 2001). Man et al (2007) found that the interaction of MIP-1α and CCR5 (MIP1α receptor) triggered the opening of endothelial tight junctions in the BBB. Zozulya et al (2007) demonstrated that MIP-1α increases the transmigration of dendritic cells across the brain's microvessel endothelial monolayer. Thus upregulated expression of chemokines, particularly IP-10, may be responsible for the enhancement of BBB after infection with attenuated RABV.
Expression of IP-10 within the brain is often associated with increased infiltrating cells in MHV- or herpes simplex virus (HSV)- infected animals (Klein, 2004) (Molesworth-Kenyon et al., 2005). Treatment of MHV-infected mice with anti-IP-10 resulted in a significant decrease in numbers of CD4+ and CD8+ T lymphocytes infiltrating into the CNS (Liu et al., 2001). IP-10 receptor CXCR3 deficient mice infected with Dengue virus (Hsieh et al., 2006) or West Nile Virus (Zhang et al., 2008) exhibited fewer effecter T cells recruited to the CNS. IP-10 recruits inflammatory cells (mainly T and NK cells) into the CNS and this function is intimately linked to BBB integrity. In our study, increased expression of IP-10 correlates with the enhancement of BBB permeability as a function of time in mice infected with laboratory-attenuated RABV, suggesting that IP-10 may be responsible for the increased infiltration of inflammatory cells into the CNS, which in turn enhances the BBB permeability.
In conclusion, the results from the present study confirm our previous studies that laboratory-attenuated RABV is more active in inducing innate immune responses than pathogenic wt RABV. Overall the induction of innate immunity, particularly the upregulation of chemokine expression in and infiltration of inflammatory cells into the CNS, correlates with the enhancement of BBB permeability in mice infected with laboratory-attenuated B2C. It is possible that laboratory-attenuated RABV induces the expression of these chemokines which leads to infiltration of inflammatory cells into the CNS. Infiltration of inflammatory cells enhances the BBB permeability, which ultimately allows immune effectors into the CNS to clear the infecting virus. Among these chemokines, IP-10 is the most upregulated chemokine tested and its upregulation correlates closely with the enhancement of BBB permeability. Thus, it is possible that expression of IP-10 (and to a lesser extent, MIP-1α) plays a major role in the enhancement of BBB permeability. Further studies are warranted to confirm such findings, for example, using knockout mice or in mice treated with anti-chemokine antibodies or siRNA. These studies should revel if induction of innate immune responses is an important mechanism or a consequence of RABV attenuation.
Acknowledgments
This work is supported partially by Public Health Service grant AI-051560 from the National Institute of Allergy and Infectious Diseases. The authors thank Xiaqing Li, Yuru Liu, and Yongjun Wen for technical help.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anonymous. WHO Technical Report Series 824. WHO Tech Rep Ser(824) 1992:1–84. [Google Scholar]
- Blanton JD, Krebs JW, Hanlon CA, Rupprecht CE. Rabies surveillance in the United States during 2005. J Am Vet Med Assoc. 2006;229(12):1897–911. doi: 10.2460/javma.229.12.1897. [DOI] [PubMed] [Google Scholar]
- CDC. First human death associated with raccoon rabies--Virginia, 2003. MMWR Morb Mortal Wkly Rep. 2003;52(45):1102–3. [PubMed] [Google Scholar]
- Dietzschold B, Morimoto K, Hooper DC, Smith JS, Rupprecht CE, Koprowski H. Genotypic and phenotypic diversity of rabies virus variants involved in human rabies: implications for postexposure prophylaxis. J Hum Virol. 2000;3(1):50–7. [PubMed] [Google Scholar]
- Dietzschold B, Wunner WH, Wiktor TJ, Lopes AD, Lafon M, Smith CL, Koprowski H. Characterization of an antigenic determinant of the glycoprotein that correlates with pathogenicity of rabies virus. Proc Natl Acad Sci U S A. 1983;80(1):70–4. doi: 10.1073/pnas.80.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabis MJ, Phares TW, Kean RB, Koprowski H, Hooper DC. Blood-brain barrier changes and cell invasion differ between therapeutic immune clearance of neurotrophic virus and CNS autoimmunity. Proc Natl Acad Sci U S A. 2008;105(40):15511–6. doi: 10.1073/pnas.0807656105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu ZF. Rabies and rabies research: past, present and future. Vaccine. 1997;15(Suppl):S20–4. doi: 10.1016/s0264-410x(96)00312-x. [DOI] [PubMed] [Google Scholar]
- Fu ZF, Rupprecht CE, Dietzschold B, Saikumar P, Niu HS, Babka I, Wunner WH, Koprowski H. Oral vaccination of racoons (Procyon lotor) with baculovirus-expressed rabies virus glycoprotein. Vaccine. 1993;11(9):925–8. doi: 10.1016/0264-410x(93)90379-c. [DOI] [PubMed] [Google Scholar]
- Glabinski AR, Ransohoff RM. Sentries at the gate: chemokines and the blood-brain barrier. J Neurovirol. 1999;5(6):623–34. doi: 10.3109/13550289909021291. [DOI] [PubMed] [Google Scholar]
- Hamir AN, Moser G, Fu ZF, Dietzschold B, Rupprecht CE. Immunohistochemical test for rabies: identification of a diagnostically superior monoclonal antibody. Vet Rec. 1995;136(12):295–6. doi: 10.1136/vr.136.12.295. [DOI] [PubMed] [Google Scholar]
- Hsieh MF, Lai SL, Chen JP, Sung JM, Lin YL, Wu-Hsieh BA, Gerard C, Luster A, Liao F. Both CXCR3 and CXCL10/IFN-inducible protein 10 are required for resistance to primary infection by dengue virus. J Immunol. 2006;177(3):1855–63. doi: 10.4049/jimmunol.177.3.1855. [DOI] [PubMed] [Google Scholar]
- Johnson N, McKimmie CS, Mansfield KL, Wakeley PR, Brookes SM, Fazakerley JK, Fooks AR. Lyssavirus infection activates interferon gene expression in the brain. J Gen Virol. 2006;87(Pt 9):2663–7. doi: 10.1099/vir.0.82024-0. [DOI] [PubMed] [Google Scholar]
- Klein RS. Regulation of neuroinflammation: the role of CXCL10 in lymphocyte infiltration during autoimmune encephalomyelitis. J Cell Biochem. 2004;92(2):213–22. doi: 10.1002/jcb.20052. [DOI] [PubMed] [Google Scholar]
- Li XQ, Sarmento L, Fu ZF. Degeneration of neuronal processes after infection with pathogenic, but not attenuated, rabies viruses. J Virol. 2005;79(15):10063–8. doi: 10.1128/JVI.79.15.10063-10068.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MT, Armstrong D, Hamilton TA, Lane TE. Expression of Mig (monokine induced by interferon-gamma) is important in T lymphocyte recruitment and host defense following viral infection of the central nervous system. J Immunol. 2001;166(3):1790–5. doi: 10.4049/jimmunol.166.3.1790. [DOI] [PubMed] [Google Scholar]
- Man SM, Ma YR, Shang DS, Zhao WD, Li B, Guo DW, Fang WG, Zhu L, Chen YH. Peripheral T cells overexpress MIP-1alpha to enhance its transendothelial migration in Alzheimer's disease. Neurobiol Aging. 2007;28(4):485–96. doi: 10.1016/j.neurobiolaging.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Mansfield KL, Johnson N, Nunez A, Hicks D, Jackson AC, Fooks AR. Up-regulation of chemokine gene transcripts and T-cell infiltration into the central nervous system and dorsal root ganglia are characteristics of experimental European bat lyssavirus type 2 infection of mice. J Neurovirol. 2008;14(3):218–28. doi: 10.1080/13550280802008297. [DOI] [PubMed] [Google Scholar]
- Martinez L. Global infectious disease surveillance. Int J Infect Dis. 2000;4(4):222–8. doi: 10.1016/s1201-9712(00)90114-0. [DOI] [PubMed] [Google Scholar]
- Miyamoto K, Matsumoto S. Comparative studies between pathogenesis of street and fixed rabies infection. J Exp Med. 1967;125(3):447–56. doi: 10.1084/jem.125.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molesworth-Kenyon S, Mates A, Yin R, Strieter R, Oakes J, Lausch R. CXCR3, IP-10, and Mig are required for CD4+ T cell recruitment during the DTH response to HSV-1 yet are independent of the mechanism for viral clearance. Virology. 2005;333(1):1–9. doi: 10.1016/j.virol.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Morimoto K, Hooper DC, Carbaugh H, Fu ZF, Koprowski H, Dietzschold B. Rabies virus quasispecies: implications for pathogenesis. Proc Natl Acad Sci U S A. 1998;95(6):3152–6. doi: 10.1073/pnas.95.6.3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto K, Patel M, Corisdeo S, Hooper DC, Fu ZF, Rupprecht CE, Koprowski H, Dietzschold B. Characterization of a unique variant of bat rabies virus responsible for newly emerging human cases in North America. Proc Natl Acad Sci U S A. 1996;93(11):5653–8. doi: 10.1073/pnas.93.11.5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy FA. Rabies pathogenesis. Arch Virol. 1977;54(4):279–97. doi: 10.1007/BF01314774. [DOI] [PubMed] [Google Scholar]
- Nakamichi K, Inoue S, Takasaki T, Morimoto K, Kurane I. Rabies virus stimulates nitric oxide production and CXC chemokine ligand 10 expression in macrophages through activation of extracellular signal-regulated kinases 1 and 2. J Virol. 2004;78(17):9376–88. doi: 10.1128/JVI.78.17.9376-9388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phares TW, Fabis MJ, Brimer CM, Kean RB, Hooper DC. A peroxynitrite-dependent pathway is responsible for blood-brain barrier permeability changes during a central nervous system inflammatory response: TNF-alpha is neither necessary nor sufficient. J Immunol. 2007;178(11):7334–43. doi: 10.4049/jimmunol.178.11.7334. [DOI] [PubMed] [Google Scholar]
- Prehaud C, Megret F, Lafage M, Lafon M. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J Virol. 2005;79(20):12893–904. doi: 10.1128/JVI.79.20.12893-12904.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Hooper DC. Lethal silver-haired bat rabies virus infection can be prevented by opening the blood-brain barrier. J Virol. 2007;81(15):7993–8. doi: 10.1128/JVI.00710-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Hooper DC. Immune evasion by rabies viruses through the maintenance of blood-brain barrier integrity. J Neurovirol. 2008;14(5):401–11. doi: 10.1080/13550280802235924. [DOI] [PubMed] [Google Scholar]
- Roy A, Phares TW, Koprowski H, Hooper DC. Failure to open the blood-brain barrier and deliver immune effectors to central nervous system tissues leads to the lethal outcome of silver-haired bat rabies virus infection. J Virol. 2007;81(3):1110–8. doi: 10.1128/JVI.01964-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupprecht CE, Smith JS, Krebs JW, Childs JE. Molecular epidemiology of rabies in the United States: reemergence of a classical neurotropic agent. J Neurovirol. 1997;3 1:S52–3. [PubMed] [Google Scholar]
- Sarmento L, Li XQ, Howerth E, Jackson AC, Fu ZF. Glycoprotein-mediated induction of apoptosis limits the spread of attenuated rabies viruses in the central nervous system of mice. J Neurovirol. 2005;11(6):571–81. doi: 10.1080/13550280500385310. [DOI] [PubMed] [Google Scholar]
- Schnell MJ, Mebatsion T, Conzelmann KK. Infectious rabies viruses from cloned cDNA. Embo J. 1994;13(18):4195–203. doi: 10.1002/j.1460-2075.1994.tb06739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, Qin S, Rottman J, Sellebjerg F, Strieter RM, Frederiksen JL, Ransohoff RM. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest. 1999;103(6):807–15. doi: 10.1172/JCI5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZW, Sarmento L, Wang Y, Li XQ, Dhingra V, Tseggai T, Jiang B, Fu ZF. Attenuated rabies virus activates, while pathogenic rabies virus evades, the host innate immune responses in the central nervous system. J Virol. 2005;79(19):12554–65. doi: 10.1128/JVI.79.19.12554-12565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Prosniak M, Curtis MT, Weiss ML, Faber M, Dietzschold B, Fu ZF. Silver-haired bat rabies virus variant does not induce apoptosis in the brain of experimentally infected mice. J Neurovirol. 2001;7(6):518–27. doi: 10.1080/135502801753248105. [DOI] [PubMed] [Google Scholar]
- Zhang B, Chan YK, Lu B, Diamond MS, Klein RS. CXCR3 mediates region-specific antiviral T cell trafficking within the central nervous system during West Nile virus encephalitis. J Immunol. 2008;180(4):2641–9. doi: 10.4049/jimmunol.180.4.2641. [DOI] [PubMed] [Google Scholar]
- Zozulya AL, Reinke E, Baiu DC, Karman J, Sandor M, Fabry Z. Dendritic cell transmigration through brain microvessel endothelium is regulated by MIP-1alpha chemokine and matrix metalloproteinases. J Immunol. 2007;178(1):520–9. doi: 10.4049/jimmunol.178.1.520. [DOI] [PMC free article] [PubMed] [Google Scholar]