

Abstract
We present a facile and inexpensive approach to superhydrophobic polymer coatings. The method involves the in-situ polymerization of common monomers in the presence of a porogenic solvent to afford superhydrophobic surfaces with the desired combination of micro- and nano-scale roughness. The method is applicable to a variety of substrates and is not limited to small areas or flat surfaces. The polymerized material can be ground into a superhydrophobic powder, which, once applied to a surface, renders it superhydrophobic. The morphology of the porous polymer structure can be efficiently controlled by composition of the polymerization mixture, while surface chemistry can be adjusted by photografting. Morphology control is used to reduce the globule size of the porous architecture from micro down to nanoscale thereby affording a transparent material. The influence of both surface chemistry as well as the length scale of surface roughness on the superhydrophobicity is discussed.
Keywords: Porous Polymer, Superhydrophobic, Superhydrophilic, Transparency, Photografting, Surface Modification, Superhydrophobicity, Superhydrophobic surface, Polymer monolith, Porous polymer coating
1. Introduction
Superhydrophobic surfaces, i.e. surfaces possessing high advancing water contact angle and low water contact angle hysteresis, have recently attracted significant attention because of their unique water-repellent and self-cleaning properties and their potential for practical applications ranging from biotechnology to self-cleaning commodity materials.[1–9] Superhydrophobicity is usually explained by the Cassie-Baxter model[1–3] according to which air is trapped in the microgrooves of the rough surface and water droplets rest on a “composite” surface comprising air and the tops of the microprotrusions. Nature utilizes the extreme water repellent properties of superhydrophobic surfaces in many plants and animals. Well-known examples include lotus leaves and their self-cleaning properties and water striders that are able to walk on the surface of water. Inspired by Nature, a number of approaches to artificial superhydrophobic surfaces have been developed.[4–9] However, many of the techniques for the preparation of superhydrophobic surfaces described in the literature involve multi-step procedures and sometimes harsh conditions, or specialized reagents and equipment. Many of the methods are expensive and only applicable to small flat surfaces or specific materials. As a result, practical applications of such functional materials have not been fully realized and there is a clear need for an inexpensive and broadly applicable approach towards superhydrophobic coatings.
In a very practical type of application, self-cleaning superhydrophobic coatings would be used on painted surfaces preventing the deposition of dirt or on optically transparent materials such as the surface of solar cells, etc. However, superhydrophobicity requires very high surface roughness, which leads to extensive scattering of propagated light. Therefore, superhydrophobicity and transparency are generally two conflicting properties that are not easily implemented simultaneously in a single material.
In this article, we present an inexpensive broadly applicable single-step method that facilitates the preparation of large area superhydrophobic surfaces. The method is applicable to a variety of substrates such as glass, metal or aluminum foil. We also demonstrate a simple approach to confer significant transparency to the materials by simply tuning the composition of the mixture used in their preparation.
2. Results and Discussion
2.1. Superhydrophobic porous polymer layers
The smooth surface of a polymer film prepared using the photoinitiated copolymerization of butyl methacrylate and ethylene dimethacrylate shows static (θst), advancing (θadv), and receding (θrec) water contact angles (WCA) of 77°, 89° and 66°, respectively. Therefore, this polymer is only slightly hydrophobic and shows a large contact angle hysteresis. However, when the same monomers are polymerized after mixing them with cyclohexanol and 1-decanol, the surface of the material that is obtained becomes superhydrophobic with θst, θadv and θrec as high as 172°, 174° and 171° respectively (Fig. 1a, Video S1). The reason for the observed superhydrophobicity is that the presence of inert solvents (porogens) - cyclohexanol and 1-decanol - in the polymerization mixture leads to phase separation during polymerization once the growing cross-linked polymer chains achieve a critical size[10] and a highly porous structure consisting of interconnected globules is formed as shown on Fig. 1a. Scanning electron micrographs (SEM) of the polymer also reveal both micro and nanoscale roughness (Fig. 1a), which is important for stabilizing superhydrophobicity[11] and very similar to that observed on Lotus leaves.[12]
Figure 1.


Shape of water droplets formed on porous and nonporous polymer layers and SEM micrographs of the superhydrophobic porous polymers. (a) Poly(butyl methacrylate-co-ethylene dimethacrylate). (b) Poly(styrene-co-divinylbenzene).
The general method for the preparation of such superhydrophobic polymer layers on a flat substrate is straightforward. A polymerization mixture containing monovinyl and divinyl monomers, porogens and an initiator is dropped on a surface to be rendered superhydrophobic and covered with a glass plate. The polymerization is carried out either by UV light irradiation for 15 min or by heating at 70°C for 24 h to afford a thin layer of porous polymer. The thickness of the layer can be controlled between 7.5 μm and about 250 μm by the thickness of Teflon strips that are placed at the edges of the glass plate. The polymer layer is then washed with methanol for 2 min and dried in air. The presence of divinyl monomers in the polymerization mixture leads to highly cross-linked, insoluble materials that are significantly more rigid and mechanically more stable than noncross-linked polymers with the same porous structure.
While many methods developed for the fabrication of superhydrophobic surfaces based on the generation of roughness are limited to the particular material being used and cannot be simply applied to other substrates. Our approach involving the in-situ polymerization of a liquid layer of monomers and porogens can be applied to different substrates provided the final polymer can adhere to the substrate. For example, in addition to glass surfaces, we prepared superhydrophobic polymer layers on stainless steel plates (Fig. 2a) and aluminum foil (Fig. 2b). A portion of the superhydrophobic layer can also be readily transferred to a plastic adhesive tape by attaching its sticky side to the polymer layer prepared on a glass plate and peeling off a superficial portion of the porous layer from the glass surface (Fig. 2c). This procedure leaves most of the porous material on the glass surface and increases the micro-scale roughness of the remaining surface as evidenced by an increase in the receding WCA of the porous polymer layer that remains on the glass plate.
Figure 2.

Water droplets residing on superhydrophobic surfaces prepared by one-step polymerization on different substrates. (a) Stainless steel plate. (b) Aluminum foil. (c) Plastic tape. A color version of this figure can be found in the Supporting Information.
Rough superhydrophobic surfaces are usually composed of “forests” of micro-protrusions. Because of the mechanical weakness of such microscopic features, superhydrophobicity is usually very easily destroyed when the surface is scratched or even wiped. Such mechanical weakness represents one of the main problems for the practical application of artificial superhydrophobic surfaces. However since our superhydrophobic layers are constituted of a porous structure that encompasses their entire thickness, they are able to display significant durability as wiping their surface only removes the most superficial layer of the structure. As the top layer becomes damaged, the underlying structure becomes exposed and superhydrophobicity remains unaffected.
The long-term preservation of superhydrophobicity is an important criterion for real outdoor applications of superhydrophobic materials. In order to examine this parameter, a glass plate coated with the superhydrophobic layer of porous poly(butyl methacrylate-co-ethylene dimethacrylate) was kept in an urban outdoor environment for eight weeks. The receding WCA decreased only by 10°, while the advancing WCA did not change at all and the surface remained superhydrophobic. Any dust particle that accumulate over the layer exposed to the elements is readily removed by spraying with water.
The low surface energy of apolar liquids makes superhydrophobic surfaces oleophilic and enables them to be wetted with most non-aqueous solvents but not with water. This property enables the use of superhydrophobic coatings prepared on membranes or meshes[13] for the separation of water from oil. Therefore, a microporous layer of poly(butyl methacrylate-co-ethylene dimethacrylate) was prepared on an aluminum mesh, rendering it both superhydrophobic and superoleophilic. When a mixture of diesel fuel and water is dropped onto such a membrane only diesel fuel passes through it by gravity and it can be collected separately from water that rests on top of the mesh (Video S2). Such superhydrophobic membranes could be useful, for example, in microextraction or in processes involving the separation of oil from water.
Poly(styrene-co-divinylbenzene) is a common material that exhibits an intrinsic θst of 97°, and is therefore slightly more hydrophobic than poly(butyl methacrylate-co-ethylene dimethacrylate). To demonstrate the generality of our approach to superhydrophobic polymers, we prepared a porous polymer layer from a mixture consisting of styrene (monomer), divinylbenzene (crosslinker), 1-decanol and tetrahydrofuran (porogens) and 2,2′-azobisisobutyronitrile (initiator). Thermally initiated polymerization was used in this case since the aromatic monomers are not UV transparent. The θst on the surface of the porous layer of poly(styrene-co-divinylbenzene) increased to 170° (Fig. 1b). The values of θadv and θrec increased from 102° and 83°, respectively, as measured for the nonporous polymer layer, to 171° and 162° for the porous polymer with the same composition. SEM micrographs again revealed a network of interconnected microglobules, but with sizes significantly smaller than those of the superhydrophobic poly(butyl methacrylate-co-ethylene dimethacrylate) (Fig. 1b). The dual scale roughness of superhydrophobic poly(styrene-co-divinylbenzene) is again readily observed in the SEM micrographs in Fig. 1b.
2.2. Superhydrophilic porous polymer layers
To demonstrate the importance of the chemistry of the material itself as it impinges on hydrophobicity, we replaced butyl methacrylate with the more hydrophilic 2-hydroxyethyl methacrylate. The nonporous layer of poly(2-hydroxyethyl methacrylate-co-ethylene dimethacrylate) exhibited an intrinsic θst of 47° while its porous counterpart, possessing morphological features similar to those of porous poly(butyl methacrylate-co-ethylene dimethacrylate) (Fig. S1), afforded a superhydrophilic material with θst, θadv and θrec all equal to zero. In this case, roughness magnifies the hydrophilicity of the surface.[8,14]
2.3. Superhydrophobic powder
Because the superhydrophobicity of the porous polymers is a bulk property of the material and not just of its surface, it should be preserved upon grinding the polymer into a powder. Such a powder could then prove useful in conferring superhydrophobicity to any surface to which it is applied. To test this concept we prepared a bulk microporous poly(butyl methacrylate-co-ethylene dimethacrylate) by thermal polymerization of a mixture of monomers and porogens on a larger (10 mL) scale. The material was then powdered by conventional grinding means and sprayed onto pressure sensitive adhesive tape (Fig. 3a). The measured θst, θadv and θrec values for this superhydrophobic polymer tape were as high as 172, 178, and 170°, respectively (see inset in Fig. 3a). In other demonstrations a rubber glove and a tissue paper were coated with the powder rendering them superhydrophobic (Fig. 3b,c and videos S3,S4) and seemingly impervious to concentrated aqueous acid or base.
Figure 3.



(a) SEM micrographs of an adhesive tape coated with the superhydrophobic powder. Inset: water droplet on this surface. (b) Water droplets on an exam glove coated with the superhydrophobic powder. (c) Droplets of concentrated aqueous solutions of sodium hydroxide (left) and hydrochloric acid (right) residing on a paper tissue coated with the superhydrophobic powder. A color version of this figure can be found in the Supporting Information.
2.4. Increasing transparency of porous polymers by tuning the morphology of the porous structure
Transparency and superhydrophobicity are properties that are not readily combined as the roughness required to achieve superhydrophobicity is detrimental to transparency because of the scattering of light. The porous polymethacrylate and polystyrene described above are completely opaque due to the extensive light scattering of the micrometer sized polymer globules. To avoid Mie scattering of visible light (ca. 380–760 nm), feature sizes within the rough surface should be significantly smaller than the wavelength of light, preferably less than 80 nm. Since it is difficult to control surface roughness on the nanometer scale, only a few groups have attempted the preparation of transparent superhydrophobic coatings.[15–21] For example, transparent superhydrophobic films have been obtained by coating TiO2 with fluoroalkylsilanes[17, 18]; while evaporation of solvent from a solution of fluoropolymer covered by a layer of water droplets was used to generate a transparent superhydrophobic nanoporous polymer film.[19] Bravo et al. created transparent superhydrophobic films by a layer-by-layer deposition of differently sized silica nanoparticles on a glass substrate.[20]
In order to improve transparency of our polymers we have to significantly reduce the feature sizes of the porous structure. An advantage of our approach is the flexibility with which it is possible to control the morphology of the porous structure.[22] We exploited this possibility in an attempt to improve transparency of the porous poly(butyl methacrylate-co-ethylene dimethacrylate) by decreasing the globule and pore size of the polymer.
Five polymerization mixtures with varying proportions of cyclohexanol and 1-decanol were prepared. All mixtures contained 30% (vol.) butyl methacrylate, 20% (vol.) ethylene dimethacrylate, 1% (w/w, with respect to the monomers) 2,2′-dimethoxy-2-phenylacetophenone photoinitiator and 50% (vol.) porogens (cyclohexanol and 1-dodecanol). The volume percentage of cyclohexanol in the polymerization mixtures was 0% in mixture 1 (pure 1-dodecanol as the sole porogen), 10% in mixture 2, 25% in mixture 3, 40% in mixture 4, and finally 50% in mixture 5 (pure cyclohexanol as the sole porogen). These polymerization mixtures were then used to prepare 50 μm thick porous polymer layers on glass substrate using UV-initiated free-radical polymerization.
SEM of the porous layers shown in Fig. 4 confirm the gradual decrease in the size of microglobules and pores as the cyclohexanol content of the polymerization mixture is increased. The average globule size for the porous polymers obtained from mixtures 1–5 is 486, 105, 82, 48, and 45 nm, respectively (see supporting information). Thus, by increasing the volume ratio of cyclohexanol to 1-decanol in the polymerization mixture and keeping the amounts of monomer and cross-linker unchanged we were able to achieve a more than ten-fold decrease of both the pore and globule size of the polymer. This morphological change resulted in a significant improvement of the transparency of the polymer layers as demonstrated in Fig. 4 (bottom). UV-NIR transmittance spectra also corroborate the increased transparency of the layers upon reducing the feature size of porous structure. The transparency increases first in the IR range and finally in the visible range (Fig. 5a), which agrees with the theory of light scattering. In addition to the control of microstructure of the layer, the transmittance of visible light was further improved by reducing the thickness of the porous layer to 7.5 μm (Fig. 5b).
Figure 4.

(top) Scanning electron micrographs and (bottom) photographs of 50 μm thick porous poly(butyl methacrylate-co-ethylene dimethacrylate) layers prepared using mixtures 1–5 showing the relative transparency of the layers. Scale bar – 1 μm. A color version of this figure can be found in the Supporting Information.
Figure 5.
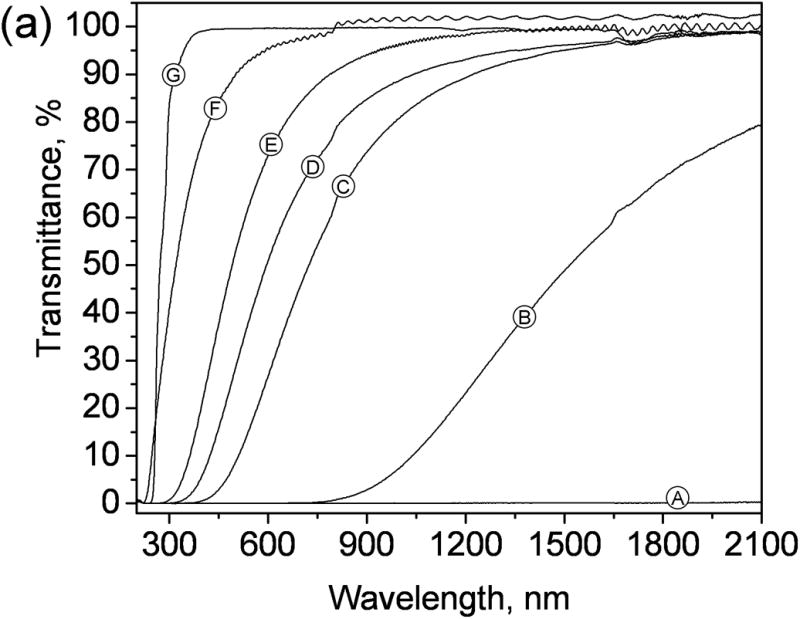

(a) UV-NIR transmittance spectra of poly(butyl methacrylate-co-ethylene dimethacrylate) layers prepared on glass support. Layers A – E are 50μm in thickness. Globules and pore size of the polymers are reduced from A to E by changing the composition of the polymerization mixtures, with A to E corresponding to mixtures 1 to 5 respectively. Layer F made from mixture 5 is 7.5 μm in thickness. Layer G is nonporous, 50 μm in thickness. (b) Photograph showing the relative transparency of layer F.
2.5. Increasing hydrophobicity of the transparent porous polymers by surface modification
Superhydrophobicity is attained through a combination of suitable material chemistry and suitable morphology of the surface. Therefore the change of the morphology of the porous polymers that improves transparency may also influence the superhydrophobic properties of the surface. Fig. 6a and Table 1S show the θst, θadv and θrec WCAs for the porous layers obtained from the five polymerization mixtures 1–5. Clearly, a decrease in the size of microglobules from 486 to 45 nm is accompanied by a large drop in θrec from 140° to 0°, indicating a significant increase in water adhesion to the surface (Video S5). Since the chemical nature of the polymers produced using mixture 1–5 is the same, the observed decrease of θrec is the result of surface morphology. The loss of the superhydrophobic property can be explained by the loss of microscale roughness associated with the 10-fold decrease in the size of the structural features of the polymer layer (Fig. 4). The importance of the bimodal micro/nano scale roughness for achieving superhydrophobicity has been noted several times in the literature.[23–25]
Figure 6.


Water contact angles measured for the nanoporous poly(butyl methacrylate-co-ethylene dimethacrylate) layers before (a) and after (b) surface modification with 2,2,3,3,3-pentafluoropropyl methacrylate. Advancing (triangles), static (circles) and receding (squares) water contact angles are plotted as a function of the cyclohexanol content in the polymerization mixtures and hence as a function of the globule size in the porous polymer structure.
Thus, while transparency was achieved by reducing the feature size within the porous polymers, superhydrophobicity was lost. Since superhydrophobicity results from the combination of morphology and chemistry, we explored a change of surface chemistry as a means to improve hydrophobicity of the transparent polymers. UV initiated photografting[26,27] of 2,2,3,3,3-pentafluoropropyl methacrylate onto the nanoporous poly(butyl methacrylate-co-ethylene dimethacrylate) films prepared using mixture 3, 4 and 5 led to a significant improvement of the hydrophobicity of the transparent polymers (Figs. 6b, S2, Table S1 and Video S6). However, about 5–10% decrease in the visible light transmittance accompanied the grafting process.
These experiments demonstrate that despite the lack of micro scale roughness, films of the nanoporous polymers obtained from mixtures 3–5 could be rendered superhydrophobic by reducing the surface energy of the material.
3. Conclusions
We demonstrated a new approach to superhydrophobic polymer coatings involving the formation of microporous layers through a simple polymerization in the presence of porogens. The materials obtained by this process can be ground into a superhydrophobic powder, which is readily applied to surfaces to render them superhydrophobic.
This study demonstrates that a dual scale (micro/nano) roughness is important to confer superhydrophobicity to materials with inherently low hydrophobicity characterized by advancing WCA on smooth surfaces in the range of 70–100°. When microscale roughness is absent and only nanoscale roughness prevails on surfaces consisting of such materials a significant decrease of the receding WCA leads to strong adhesion of water to such surface. In contrast, highly hydrophobic materials (WCA ~ 120°) possessing solely nanoscale roughness can still be superhydrophobic.
4. Experimental
Materials and Instrumentation
An OAI Model 30 deep UV collimated light source (San Jose, CA, USA) fitted with a 500-W HgXe lamp was used for UV exposures. The irradiation power was calibrated to 12.0 mW/cm2 (4.4 mW/cm2 after the cover glass plate) using an OAI Model 306 UV power meter with a 260-nm probe head. Scanning electron micrographs were obtained using the Zeiss Gemini Ultra-55 Analytical Scanning Electron Microscope. The samples were gold-sputtered using the BAL-TEC SCD 050 sputter coater. UV-3000 Shimadzu Spectrophotometer was used for acquiring UV-NIR spectra. “Easy Drop” Kruss GmbH (Germany) instrument was used to take pictures and videos of water droplets and to measure static water contact angles. ImageJ software with a DropSnake plugin was used to measure the dynamic contact angles. 12 cm × 3.3 cm, 1.1 mm thick, Borofloat glass plates were purchased from S. I. Howard Glass Co. Inc., Worcester, MA.
All chemicals were purchased from Sigma-Aldrich. Monomers were purified by passing them through a short column packed with inhibitor remover (Aldrich).
Polymerization mixtures
– Porous poly(butyl methacrylate-co-ethylene dimethacrylate) (BMA-EDMA) via photoinitiation: butyl methacrylate (BMA) (24% wt.), ethylene dimethacrylate (EDMA) (16% wt.), 1-decanol (40% wt.), cyclohexanol (20% wt.) and 2,2-dimethoxy-2-phenylacetophenone (DMPAP) (1% wt. with respect to monomers).
– Porous BMA-EDMA via thermal initiation: BMA (24% wt.), EDMA (16% wt.), 1-decanol (40% wt.), cyclohexanol (20% wt.) and AIBN (1% wt. with respect to monomers).
– Nonporous BMA-EDMA via photoinitiation: BMA (60% wt.), EDMA(40% wt.) and DMPAP (1% wt. with respect to monomers).
– Porous poly(2-hydroxyethyl methacrylate-co-ethylene dimethacrylate) via photoinitiation: 2-hydroxyethyl methacrylate (24% wt.), EDMA (16% wt.), 1-decanol (40% wt.), cyclohexanol (20% wt.) and DMPAP (1% wt. with respect to monomers).
– Nonporous poly(2-hydroxyethyl methacrylate-co-ethylene dimethacrylate) via photoinitiation: 2-hydroxyethyl methacrylate (60% wt.), EDMA (40% wt.) and DMPAP (1% wt. with respect to monomers).
– Porous poly(styrene-co-1,4-divinylbenzene) via thermal initiation: styrene (24% wt.), 1,4-divinylbenzene (16% wt.), 1-decanol (50% wt.), tetrahydrofuran (10% wt.) and 2,2′-azobisisobutyronitrile (1% wt. with respect to monomers).
– Nonporous poly(styrene-co-1,4-divinylbenzene) via thermal initiation: styrene (60% wt.), 1,4-divinylbenzene (40% wt.) and 2,2′-azobisisobutyronitrile (1% wt. with respect to monomers).
All mixtures used in the subsections 2.4 and 2.5 comprised BMA (30% vol.), EDMA (20% vol.), DMPAP (1% wt. with respect to the monomers) and porogens (50% vol.). The porogens composition in the mixtures 1–5: mix 1 - 1-decanol (50% vol.), mix 2 - 1-decanol (40% vol.) and cyclohexanol (10% vol.), mix 3 - 1-decanol (25% vol.) and cyclohexanol (25% vol.), mix 4 - 1-decanol (10% vol.) and cyclohexanol (40% vol.), mix 5 - cyclohexanol (50% vol.).
Glass surface modification
To achieve the covalent attachment of the porous polymer layers to the glass surface, the glass plates were first functionalized. The plates were washed with water, dried, and immersed in aqueous sodium hydroxide (1 mol/L) for 1h. Then the plates were rinsed with water and immersed in hydrochloric acid solution (0.2 mol/L) for 30 min followed by washing with water and drying with a nitrogen gun. The activated glass surface was functionalized with 3-(trimethoxysilyl)propyl methacrylate. Typically, a few drops of 20% vol. ethanol solution of 3-(trimethoxysilyl)propyl methacrylate with apparent pH adjusted to 5 using acetic acid were placed on the surface of the glass plate and covered with another plate. The solution was reapplied after 30 min and left to react for another 30 min. The pair of functionalized plates was washed with acetone and dried under a flow of nitrogen.
Polymerization procedures
The polymerization mixture was injected into a mold assembled from two silanized glass plates separated by two 50 μm thick Teflon strips (American Durafilm Co.) defining the thickness of the polymer layer. To prepare the 7.5 μm thick layer, polyimide film strips (American Durafilm Co.) with that thickness were used. Photopolymerizations were initiated by irradiating the filled mold with UV light for 15 min. Thermally initiated polymerizations were carried out at a temperature of 70°C for 24 h. The mold was then carefully opened using a razor blade. The porous polymer layer usually adheres to the top glass plate. After completion of the polymerization, the layer was washed with methanol for 2 min and dried in air. If the layer was used for photografting it was immersed in methanol for 1 h, and then dried under vacuum. Both top and bottom glass plates of the mold must be silanized to avoid formation of a glossy nonporous surface on the polymer layer, which is especially important for the nanoporous polymers possessing much higher mechanical strength than their microporous counterparts.
Bulk samples of porous BMA-EDMA were prepared in a glass vial using the thermally initiated polymerization of 10 mL of the standard polymerization mixture. The resulting solid polymer was then ground with mortar and pestle and sieved through a 106 μm mesh metal sieve (USA standard testing sieve, Gilson, Worthington, Ohio, USA).
Photografting
The 50 µm thick porous layers prepared using mixtures 1–5 were wetted with the solution of 15% wt. 2,2,3,3,3-pentafluoropropyl methacrylate in 1/3 (v/v.) of water - tert-butanol mixture containing 0.25% wt. benzophenone. The layer was covered with a quartz plate and exposed to the UV light (10.8 mW/cm2 after the quartz plate) for 5 min followed by washing with methanol and acetone, and drying in a stream of nitrogen.
Supplementary Material
Footnotes
Support of this research by a grant of the National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health (EB-006133) is gratefully acknowledged. Characterization work at the Molecular Foundry was supported by the Director, Office of Science, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering, of the US Department of Energy under Contract No. DE-AC02-05CH11231. (Supporting Information is available online from Wiley InterScience).
Contributor Information
Dr. Pavel A. Levkin, College of Chemistry, University of California at Berkeley, Berkeley, CA 94720-1460, USA
Dr. Frantisek Svec, The Molecular Foundry, E.O. Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
Prof. Jean M.J. Frechet, College of Chemistry, University of California at Berkeley, Berkeley, CA 94720-1460, USA, frechet@berkeley.edu
References
- 1.Quere D. Rep Prog Phys. 2005;68:2495–2532. [Google Scholar]
- 2.Callies M, Quere D. Soft Matter. 2005;1:55–61. [Google Scholar]
- 3.Oner D, McCarthy T. Langmuir. 2000;16:7777–7782. [Google Scholar]
- 4.Li XM, Reinhoudt D, Crego-Calama M. Chem Soc Rev. 2007;36:1350–1368. doi: 10.1039/b602486f. [DOI] [PubMed] [Google Scholar]
- 5.Zhang X, Shi F, Niu J, Jiang Y, Wang Z. J Mater Chem. 2008;18:621–633. [Google Scholar]
- 6.Genzer J, Efimenko K. Biofouling. 2006;22:339–360. doi: 10.1080/08927010600980223. [DOI] [PubMed] [Google Scholar]
- 7.Tuteja A, Choi W, McKinley GH, Cohen RE, Rubner MF. Mater Res Bull. 2008;33:752–758. [Google Scholar]
- 8.Dorrer C, Ruhe J. Adv Mater. 2008;20:159–163. [Google Scholar]
- 9.Pastine S, Okawa D, Kessler B, Rolandi M, Llorente M, Zettl A, Fréchet JMJ. J Am Chem Soc. 2008;130:4238–4239. doi: 10.1021/ja8003446. [DOI] [PubMed] [Google Scholar]
- 10.Svec F, Frechet JMJ. Chem Mater. 1995;7:707–715. [Google Scholar]
- 11.Nosonovsky M, Bhushan B. Microelectron Eng. 2007;84:382–386. [Google Scholar]
- 12.Barthlott W, Neinhuis C. Planta. 1997;202:1–8. [Google Scholar]
- 13.Feng L, Zhang ZY, Mai ZH, Ma YM, Liu BQ, Jiang L, Zhu DB. Angew Chem, Int Ed. 2004;43:2012–2014. doi: 10.1002/anie.200353381. [DOI] [PubMed] [Google Scholar]
- 14.Feng XJ, Jiang L. Adv Mater. 2006;18:3063–3078. [Google Scholar]
- 15.Nakajima A. J Ceram Soc Jpn. 2004;112:533–540. [Google Scholar]
- 16.Tadanaga K, Katata N, Minami T. J Am Ceram Soc. 1997;80:1040–1042. [Google Scholar]
- 17.Zhang X, Kono H, Liu Z, Nishimoto S, Tryk DA, Murakami T, Sakai H, Abe M, Fujishima A. Chem Commun. 2007:4949–4951. doi: 10.1039/b713432k. [DOI] [PubMed] [Google Scholar]
- 18.Nakajima A, Hashimoto K, Watanabe T, Takai K, Yamauchi G, Fujishima A. Langmuir. 2000;16:7044–7047. [Google Scholar]
- 19.Yabu H, Shimomura M. Chem Mater. 2005;17:5231–5234. [Google Scholar]
- 20.Bravo J, Zhai L, Wu ZZ, Cohen RE, Rubner MF. Langmuir. 2007;23:7293–7298. doi: 10.1021/la070159q. [DOI] [PubMed] [Google Scholar]
- 21.Artus GRJ, Jung S, Zimmermann J, Gautschi HP, Marquardt K, Seeger S. Adv Mater. 2006;18:2758–2762. [Google Scholar]
- 22.Eeltink S, Hilder EF, Geiser L, Svec F, Fréchet JMJ, Rozing GP, Schoenmakers PJ, Kok WT. J Sep Sci. 2007;30:407–413. doi: 10.1002/jssc.200600316. [DOI] [PubMed] [Google Scholar]
- 23.Gao LC, McCarthy TJ. Langmuir. 2006;22:2966–2967. doi: 10.1021/la0532149. [DOI] [PubMed] [Google Scholar]
- 24.Patankar NA. Langmuir. 2004;20:8209–8213. doi: 10.1021/la048629t. [DOI] [PubMed] [Google Scholar]
- 25.Zhao N, Xu J, Xie QD, Weng LH, Guo XL, Zhang XL, Shi LH. Macromol Rapid Commun. 2005;26:1075–1080. [Google Scholar]
- 26.Ranby B. Macromol Symp. 1992;63:55–67. [Google Scholar]
- 27.Rohr T, Hilder EF, Donovan JJ, Svec F, Frechet JMJ. Macromolecules. 2003;36:1677–1684. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.