

Abstract
Enantioenriched 2,3,4-trisubstituted thiochromanes have been synthesized by using a cupreine-catalyzed tandem Michael addition–Henry reaction between 2-mercaptobenzaldehydes and β-nitrostyrenes. Good diastereoselectivities and enantioselectivities were obtained for the title compounds, which may be further improved through a single recrystallization (up to 98% de and> 99% ee).
Keywords: Henry reaction, 2-mercaptobenzaldehyde, Michael addition, β-nitrostyrene, organocatalysis, tandem reaction
Chromanes are an important class of compounds that are found in many biologically active natural products.[1] Thiochromanes, the sulfur analogues of chromanes, have also been reported to possess important biological activities.[2] There are also reports that the replacement of the oxygen atom in chromanes with a sulfur atom results in enhanced bioactivities.[2d,f] Some representative examples are collected in Figure 1. Tertatolol (1) has been shown to have affinity to 5HTA1 receptors in human and rat brains.[2a,b] Thiochromane derivative 2 also displays a mixed binding affinity to dopamine D2, D3 and 5HTA1 receptors,[2d] whereas thiochromane derivative 3 shows potent anti-HIV activity.[2e]
Figure 1.
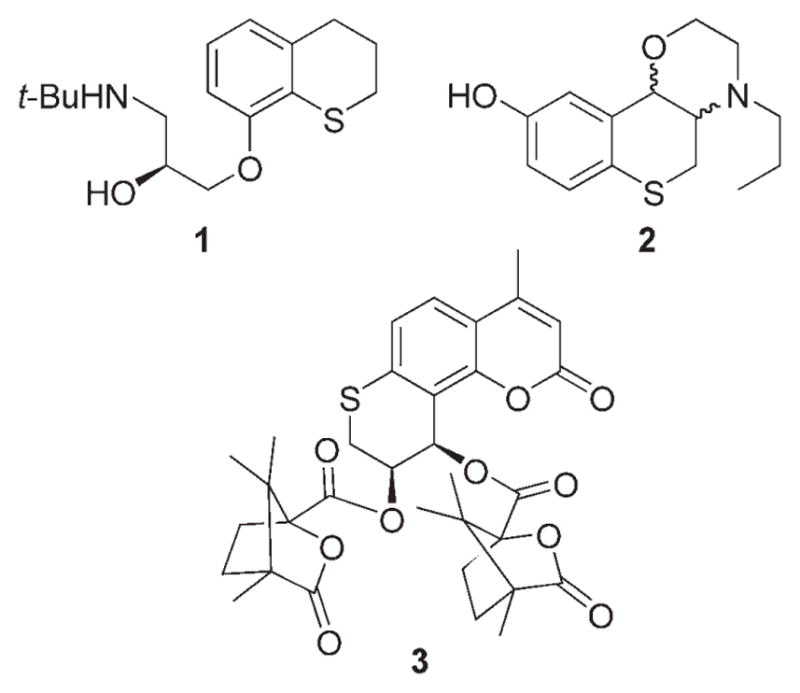
Some biologically active thiochromane derivatives.
Owing to their biological activities, several asymmetric methods have been developed for the synthesis of these compounds, which include asymmetric reduction of thiochroman-4-ones,[3] enzymatic resolution of racemic 4-hydroxythiochromanes or 4-acyloxythiochromanes,[4] and enantioselective alkylation of thiochromanes via conjugate addition.[5] Last year Wang and co-workers reported an enantioselective tandem Michael–aldol reaction for the synthesis of thiochromenes catalyzed by a proline derivative.[6] Most recently, the same group also reported a quinine thiourea-catalyzed tandem Michael–aldol reaction for synthesis of thiochromanes.[7] Nonetheless, even with these progresses, an enantioselective method for the synthesis of 3-amino-4-hydroxythiochromanes (such as 2) is still lacking.
Recently, as part of our continued interest in enantioselective organocatalysis,[8] we reported a highly enantioselective Henry reaction[9] for the synthesis of α-hydroxyphosphonates by using cupreine derivatives as the catalysts.[10] During this study, we envisioned that a tandem[11] Michael–Henry reaction may be achieved if the starting material has both a nucleophilic and an electrophilic site, such as 2-mercaptobenzaldehyde. Although tandem Michael–Henry reactions are known in the literature,[12] only very recently have Hayashi and co-workers reported a catalytic enantioselective method.[13] Moreover, the synthesis of thiochromanes by using this tandem reaction has never been studied.[14] Herein, we report an organocatalytic, enantioselective tandem Michael–Henry reaction for the direct synthesis of 2-aryl-3-nitro-4-hydroxythiochromanes.
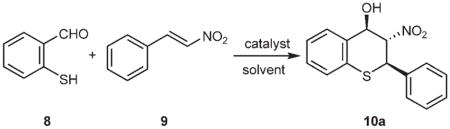
By using 2-mercaptobenzaldehyde (8) and β-nitrostyrene (9) as the model compounds, we initially studied the tandem Michael–Henry reaction using 5 mol% of DABCO as the catalyst at room temperature (entry 1, Table 1). We were pleased to find that the reaction was completed in just 5 min and only two diastereomers of the expected 2-phenyl-3-nitro-4-hydroxythiochromane (10a) were obtained in a ratio of 70:30, according to the 1H NMR analysis of the crude product. Encouraged by this result, we further screened some readily available alkaloid catalysts 4–7 (Figure 2) in an effort to develop an enantioselective reaction, and the results are summarized in Table 1.
Table 1.
Catalyst screening and reaction condition optimizations.a
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Conversion [%]b | drb | ee [%]c |
| 1 | DABCO | Et2O | 100 | 70:30 | - |
| 2 | 4 | Et2O | 100 | 75:25 | 8d |
| 3 | 5 | Et2O | 100 | 67:33 | 29 |
| 4 | 6 | Et2O | 100 | 70:30 | 75 |
| 5 | 7 | Et2O | 100 | 70:30 | 75d |
| 6 | 6 | toluene | 100 | 65:35 | 56 |
| 7 | 6 | CHCl3 | 100 | 60:40 | 42 |
| 8 | 6 | EtOAc | 100 | 56:44 | 72 |
| 9e | 6 | Et2O | 100 | 70:30 | 80 |
| 10f | 6 | Et2O | 100 | 70:30 | 86 |
| 11g | 6 | Et2O | 100 | 68:32 | 82 |
| 12f,h.i | 6 | Et2O | 100(50) | 70:30(94:6) | 86(> 99) |
Unless otherwise specified, all reactions were conducted at room temperature with 0.22 mmol of 8, 0.20 mmol of 9 and the catalyst (5 mol%) in anhydrous diethyl ether (10 mL) for 5 min.
Determined by 1H NMR spectroscopy on the crude product; yield of isolated product after flash column chromatography was over 95% based on conversion.
Enantiomeric excess of major isomer as determined by HPLC analysis on a Chiralpak AD-H column.
The other enantiomer was obtained as the major product in this case.
Reaction was performed at 0°C.
Reaction was performed at −10°C.
Reaction was performed at −25°C.
With 2 mol% of catalyst loading.
Values in parantheses are the results of yield, dr, and ee of the major product after a single recrystallization.
Figure 2.
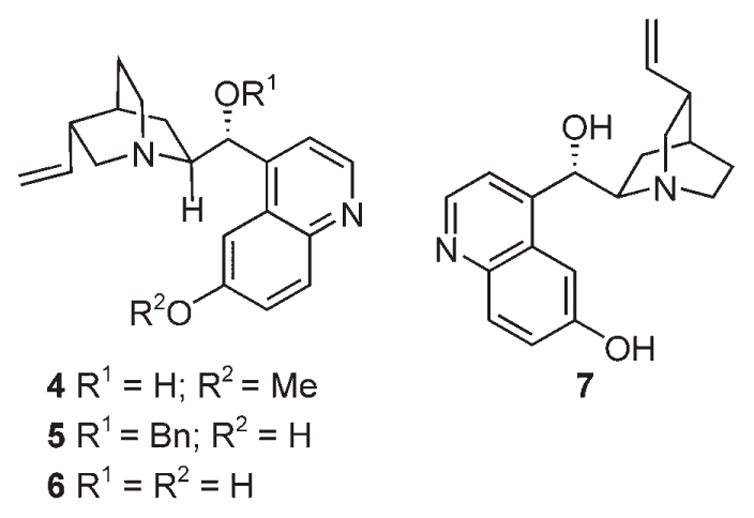
Catalysts screened for the tandem Michael-Henry reaction.
As shown in Table 1, with 5 mol% loading of quinine (4) in diethyl ether, the reaction of 8 and 9 proceeds smoothly, and complete conversion was achieved in just 5 min (entry 2). The same two diastereomers were formed in a ratio of 75:25. However, the ee value of the major diastereomer was only 8% for the enantiomer of 10a [i.e., the (2S,3R,4S)-stereo-isomer]. Catalyst 5 (9-O-benzylcupreine) also produces a similar diastereoselectivity (67:33) with a modest ee value of 29%, but with 10a as the major product of the major diastereomer (entry 3). In contrast, cupreine (6) proved to be a good catalyst for this reaction, and an ee value of 75% for 10a was obtained under similar conditions (entry 4). As expected, catalyst 7, the diastereomer of 6, yielded 10a in similar diasteroselectivity and enantioselectivity as 6, except that the enantiomer of 10a was obtained (entry 5). The reaction conditions were further optimized for 6. Screening of some common organic solvents (toluene, CHCl3, and EtOAc, entries 6–8) revealed that ether (entry 4) is the best one in terms of both diastereoselectivity and enantioselectivity.
It was also found that the reaction temperature has subtle effects on the reaction (entries 9–11): the highest enantioselectivity of 86% was obtained by carrying out the reaction at −10°C (entry 10), whereas higher (entries 5 and 9) or lower temperatures (entry 11) all led to inferior ee values. In contrast, the diastereoselectivity of this reaction is essentially not affected by the reaction temperature (entries 5, 9, and 11). Since the reaction proceeds very quickly under the optimized conditions (−10°C in ether), 5 mol% of catalyst loading seems unnecessary. Indeed, the reaction with only 2 mol% loading gave similar results (entries 12 and 10). It should be pointed out that the major diastereomer is a crystalline compound and the diastereoselectivity and enantioselectivity may be easily improved by recrystallization from hexane/EtOAc. For example, the diastereoselectivity and enantioselectivity of compound 10a were improved to 94:6 and > 99% ee, respectively, after a single recrystallization (entry 12, values in parentheses).
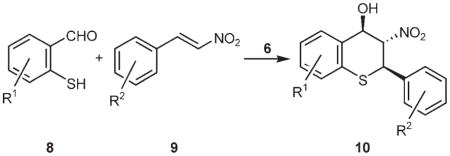
To test the substrate scope of this new tandem reaction, the reaction of various 2-mercaptobenzaldehydes[15] and β-nitrostyrenes was studied under the optimized conditions using 2 mol% of cupreine (6) as the catalyst. The results are collected in Table 2.
Table 2.
Enantioselective tandem Michael–Henry reaction of 2-mercaptobenzaldehydes (8) with various β-nitrostyrenes (9).a
 | |||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | 10, Yield [%]b | drc,d | ee [%]d,e |
| 1 | H | H | a, 95 (50) | 70:30 (94:6) | 86 (>99) |
| 2 | H | 2-Br | b, 97 (60) | 77:23 (93:7) | 85 (>99) |
| 3 | H | 4-Br | c, 95 (50) | 74:26 (92:8) | 76 (99) |
| 4 | H | 3-Cl | d, 96 (42) | 65:35 (92:8) | 78 (>99) |
| 5 | H | 4-Cl | e, 97 (46) | 73:27 (96:4) | 72 (97) |
| 6 | H | 2-NO2 | f, 95 (51) | 75:25 (99:1) | 78 (90) |
| 7 | H | 2-OMe | g, 94 (52) | 78:22 (93:7) | 80 (>99) |
| 8 | H | 4-OMe | h, 96 (51) | 68:32 (93:7) | 85 (>99) |
| 9 | H | 4-Me | i, 94 (48) | 71:29 (92:8) | 80 (>99) |
| 10 | 4-OMe | H | j, 95 (50) | 70:30 (91:9) | 82 (98) |
| 11 | 4-OMe | 4-Br | k, 96 (62) | 72:28 (94:6) | 80 (85) |
| 12 | 4-Me | H | l, 84 (29) | 70:30 (87:13) | 72 (95) |
| 13 | 4-Cl | H | m, 85 (32) | 70:30 (93:7) | 75 (97) |
Unless otherwise specified, all reactions were conducted at −10°C with the substituted β-nitrostyrene (0.2 mmol), the substituted 2-mercaptobenzaldehyde (0.22 mmol) and cupreine (6, 2 mol%) in anhydrous diethyl ether (10 mL) for 5 min. The absolute configuration of the major isomer 10b was determined by X-ray crystallography and the rest were assigned on the basis of the reaction mechanism.
Yields of nonseparable diastereomeric mixture after chromatography; values in parentheses are those after a single recrystallization.
Determined by 1H NMR analyses on the crude products or the recrystallized products.
Values in parantheses are those of the recrystallized products.
Values of enantiomeric excess of the major isomers as determined by HPLC analyses on a Chiralpak AD-H column (entries 1–12) or a Chiralcel OD-H column (entry 13).
As shown in Table 2, the reaction of 2-mercaptobenzaldehyde with various β-nitrostyrenes all give excellent yields of the desired products (≥ 94%, entries 2–9). According to NMR analyses of the crude products, only two diastereomers were formed, even though three stereogenic centers were generated. The diastereoselectivities were in the range of 68:32 to 77:23, and the ee values of the major enantiomers were from 72 to 86%. β-Nitrostyrenes with electron-withdrawing substituents on the ortho, meta or para positions afford thiochromanes with slightly inferior enantioselectivities (entries 3–6), except for the 2-bromo substrate (entry 2), for which the product was obtained in 85% ee. Besides the prototype 2-mercaptobenzaldehyde, substituted 2-mercaptobenzaldehydes also react with β-nitrostyrenes. For example, the reaction of 2-mercapto-4-methoxybenzaldehyde and β-nitrostyrene (entry 10) and 4-bromo-β-nitrostyrene (entry 11) both give similar diastereoselectivities (ca. 70:30) and enantioselectivities (ca. 80% ee). These results are comparable with those of 2-mercaptobenzaldehyde (entry 1). Similar results were also observed when 4-methyl-2-mercaptobenzaldehyde (entry 12) and 4-chloro-2-mercaptobenzaldehyde (entry 13) and β-nitrostyrene were used; except that the yields (ca. 85%) and ee values (ca. 75%) were slightly lower. As with compound 10a (entry 1), all these diastereomeric mixtures cannot be separated by column chromatography; however, they may be readily separated by recrystallization. As shown in Table 2 (data in parentheses), after a single recrystallization using hexane/EtOAc, the major diastereomers of thiochromanes 10a–m may be obtained in very good diastereomeric purity (dr ≥ 87:13) and high optical purity (>99% ee in several cases).
The absolute configuration of major diastereomer was assigned on the basis of the X-ray crystallographic analysis on compound 10b (CCDC deposition number: 647961). The stereochemistry of the major trans,trans-isomer is 2R,3S,4R.[16] A mechanism as shown in Scheme 1 is proposed to explain the observed results. The mercapto group of the mercaptoaldehyde is deprotonated by the catalyst and the newly formed anion is associated with the catalyst through charge interactions. When β-nitrostyrene approaches this complex, the nitro group will form hydrogen bonds with the two hydroxy groups of the curpreine. Among the two possible orientations of the alkene substrate (Scheme 1), the re face approach is favored (left structure), as the unfavourable steric interaction between the phenyl group and the catalyst backbone is avoided. The attack of the sulfide anion onto the re face of the β-nitrostyrene leads to the observed major enantiomer of the product.
Scheme 1.
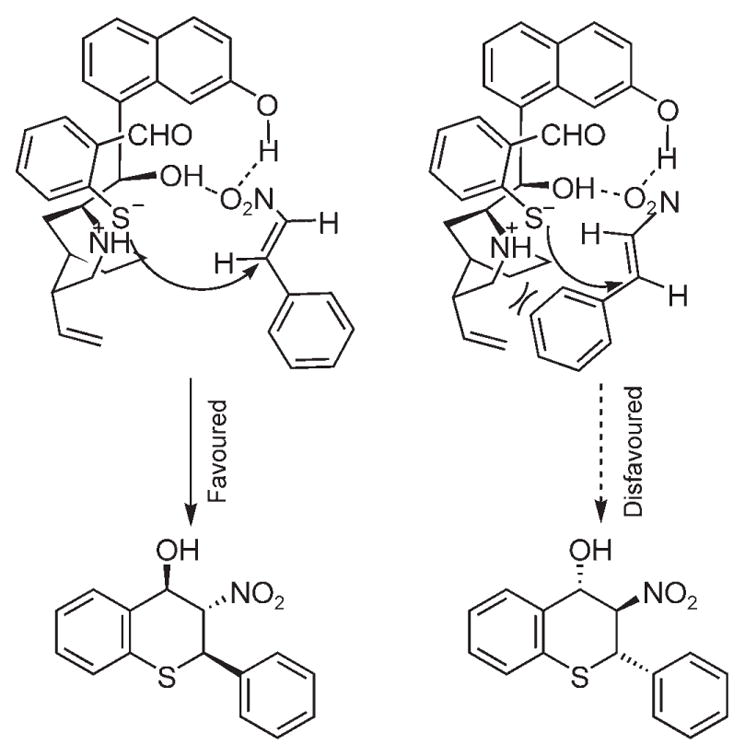
Proposed transition states.
To show the utility of this method in the preparation of enantioenriched 3-amino-4-hydroxythiochromanes,[2d] the nitro compound 10a obtained in this study was reduced with hydrogen gas under the catalysis of palladium/carbon. As shown in Eq. (1), the reaction leads to the desired amino derivative 11 in good yield and with complete retention of the stereochemistry.
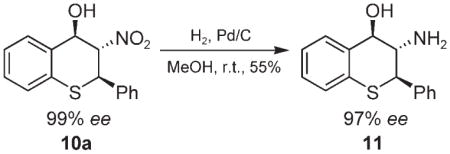 |
(1) |
In summary, we have developed a tandem Michael–Henry reaction of 2-mercaptobenzaldehydes and β-nitrostyrenes by using cupreine (6) as the catalyst. Chiral 2-aryl-3-nitrothiochroman-4-ols have been synthesized with enantioselectivities up to 86% and diastereomeric ratios up to 78:22. A single recrystallization of the diastereomeric mixture from hexane/EtOAc enhances both the enantioselectivities (up to >99% ee) and diastereoselectivities (up to 98% de). These products are useful for the synthesis of biologically active thiochromane derivatives, such as compound 2.
Experimental Section
General Experimental Procedure
A solution of β-nitrostyrene (9, 0.2 mmol) and cupreine (6, 1.25 mg, 0.004 mmol) in anhydrous diethyl ether (5.0 mL) was stirred at −10°C for 2 min. After which, a precooled solution (−10°C) of 2-mercaptobenzaldehyde (8, 0.22 mmol) in anhydrous diethyl ether (5.0 mL) was added over a period of 1 min. The reaction mixture was then stirred at the same temperature for 5 min. The solvent was removed at 0°C under reduced pressure. The crude mixture was subjected to flash column chromatography over silica gel (eluted with 9:1 hexane/EtOAc and then EtOAc) to furnish a mixture of two diastereomers, which was further recrystallized using a mixture of hexane and ethyl acetate to afford 2,3,4-trisubstituted thiochromanes 10a–m as colorless crystals. The compounds were fully characterized by 1H, 13C NMR, microanalysis and optical rotation data.
Supplementary Material
Acknowledgments
Generous financial support of this project from the Welch foundation (grant no. AX-1593) and the NIH-MBRS program (grant no. S06 08194) is gratefully acknowledged.
Footnotes
Dedicated to Professor Rong-Yu Zhang on the occasion of his 73th birthday.
Supporting information for this article is available on the WWW under http://asc.wiley-vch.de/home/.
Supporting Information
Detailed experimental procedure, NMR spectra for new compounds, HPLC analysis and X-ray data are available as Supporting Information
References
- 1.For a review on chromanes and tocopherols, see: Parkhurst RM, Skinner WA. In: Chromanes and Tocopherols in Chemistry of Hererocyclic Compounds. Ellis GP, Lockhardt IM, editors. Vol. 36. Wiley; New York: 1981. pp. 1–469.
- 2.a) Castro ME, Harrison PJ, Pazos A, Sharp T. J Neurochem. 2000;75:755–762. doi: 10.1046/j.1471-4159.2000.0750755.x. [DOI] [PubMed] [Google Scholar]; b) Lejuene F, Rivet JM, Gobert A, Canton H, Millan MJ. Eur J Pharmacol. 1993;240:307–310. doi: 10.1016/0014-2999(93)90915-5. [DOI] [PubMed] [Google Scholar]; c) Bolognesi ML, Bortolini M, Cavalli A, Andrisano V, Rosini M, Minarini A, Melchiorre CJ. J Med Chem. 2004;47:5945–5952. doi: 10.1021/jm049782n. [DOI] [PubMed] [Google Scholar]; d) Vliet LAV, Rodenhuis N, Dijkstra D, Wikstrom H, Pugsley TA, Serpa KA, Meltzer LT, Heffner TG, Wise LD, Lajiness ME, Huff RM, Svensson K, Sundell S, Lundmark M. J Med Chem. 2000;43:2871–2882. doi: 10.1021/jm0000113. [DOI] [PubMed] [Google Scholar]; e) Chen Y, Zhang Q, Zhang B, Xia P, Xia Y, Yang ZY, Kilgore N, Wild C, Morris-Natschka SL, Lee KH. Bioorg Med Chem. 2004;12:6383–6387. doi: 10.1016/j.bmc.2004.09.038. [DOI] [PubMed] [Google Scholar]; f) Spruce LW, Rajadhyaksha SN, Berlin KD, Gale JB, Miranda ET, Ford WT, Blossey EC, Verma AK, Hossain MB, Helm DVD, Breitman TR. J Med Chem. 1987;30:1474–1482. doi: 10.1021/jm00391a033. [DOI] [PubMed] [Google Scholar]
- 3.a) Ursini CV, Dias GHM, Rodrigues JAR. J Organomet Chem. 2005;690:3176–3186. [Google Scholar]; b) Micskei K, Hajdu C, Wessjohann LA, Mercs L, Kiss-Szikszai A, Patonay T. Tetrahedron: Asymmetry. 2004;15:1735–1744. [Google Scholar]; c) Evans DA, Michael FE, Tedrow JS, Campos KR. J Am Chem Soc. 2003;125:3534–3543. doi: 10.1021/ja012639o. [DOI] [PubMed] [Google Scholar]; d) Zhang X, Taketomi T, Yoshizumi T, Kumobayashi H, Akutagawa S, Mashima K, Takaya H. J Am Chem Soc. 1993;115:3318–3319. [Google Scholar]; e) Quallich GJ, Woodall TM. Tetrahedron Lett. 1993;34:785–788. [Google Scholar]
- 4.a) Holland HL, Manoharan TS, Schweizer F. Tetrahedron: Asymmetry. 1991;2:335–338. [Google Scholar]; b) Honig H, Shi N, Polanz G. Biocatalysis. 1994;9:61–69. [Google Scholar]
- 5.a) Xie J-W, Chen W, Li R, Zheng M, Du W, Yue L, Chen Y-C, Wu Y, Zhu J, Deng J-G. Angew Chem. 2007;119:393–396. [Google Scholar]; Angew Chem Int Ed. 2007;46:389–392. [Google Scholar]; b) Xue D, Chen YC, Wang QW, Cun LF, Zhu J, Deng JG. Org Lett. 2005;7:5293–5296. doi: 10.1021/ol052283b. [DOI] [PubMed] [Google Scholar]
- 6.Wang W, Li H, Wang J, Zu L. J Am Chem Soc. 2006;128:10354–10355. doi: 10.1021/ja063328m.See also: Rios R, Sunden H, Ibrahem I, Zhao GL, Cordova A. Tetrahedron Lett. 2006;47:8679–8682.
- 7.Zu L, Wang J, Li H, Xie H, Jiang W, Wang W. J Am Chem Soc. 2007;129:1036–1037. doi: 10.1021/ja067781+. [DOI] [PubMed] [Google Scholar]
- 8.a) Samata S, Liu J, Dodda R, Zhao CG. Org Lett. 2005;7:5321–5323. doi: 10.1021/ol052277f. [DOI] [PubMed] [Google Scholar]; b) Samata S, Zhao CG. J Am Chem Soc. 2006;128:7442–7443. doi: 10.1021/ja062091r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dodda R, Zhao CG. Org Lett. 2006;8:4911–4914. doi: 10.1021/ol062005s. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Samanta S, Krause J, Mandal T, Zhao CG. Org Lett. 2007;9:943–945. doi: 10.1021/ol070209i. [DOI] [PubMed] [Google Scholar]
- 9.For reviews, see: Ooi T, Maruoka K. Acc Chem Res. 2004;37:526–533. doi: 10.1021/ar030060k.Palomo C, Oiarbide M, Mielgo A. Angew Chem. 2004;116:5558–5560. doi: 10.1002/anie.200460506.Angew Chem Int Ed. 2004;43:5442–5444. doi: 10.1002/anie.200460506.Marcelli T, van Maarseveen JH, Hiemstra H. Angew Chem. 2006;118:7658–7666. doi: 10.1002/anie.200602318.Angew Chem Int Ed. 2006;45:7496–7504. doi: 10.1002/anie.200602318.
- 10.Mandal T, Samanta S, Zhao CG. Org Lett. 2007;9:943–945. doi: 10.1021/ol070209i. [DOI] [PubMed] [Google Scholar]
- 11.For selected examples on organocatalytic tandem reactions, see; Ramachary DB, Chowdari NS, Barbas CF., III Angew Chem. 2003;115:4365–4369. doi: 10.1002/anie.200351916.Angew Chem Int Ed. 2003;42:4233–4237. doi: 10.1002/anie.200351916.Yamamoto Y, Momiyama N, Yamamoto H. J Am Chem Soc. 2004;126:5962–5963. doi: 10.1021/ja049741g.Brandau S, Maerten E, Jorgensen KA. J Am Chem Soc. 2006;128:14986–14991. doi: 10.1021/ja065507+.Enders D, Huttl MRM, Grondal C, Raabe G. Nature. 2006;441:861–863. doi: 10.1038/nature04820.Maki BE, Chan A, Phillips EM, Scheidt KA. Org Lett. 2007;9:371–374. doi: 10.1021/ol062940f.Kotkar S, Chavan VB, Vilas B, Sudalai A. Org Lett. 2007;9:1001–1004. doi: 10.1021/ol063012j.Li H, Wang J, E-Nunu T, Zu L, Jiang W, Wei S, Wang W. Chem Commun. 2007:507–509. doi: 10.1039/b611502k.For reviews on tandem reactions, see: Nicolaou KC, Edmonds DJ, Bulger PG. Angew Chem. 2006;118:7292–7344. doi: 10.1002/anie.200601872.Angew Chem Int Ed. 2006;45:7134–7186. doi: 10.1002/anie.200601872.Guo HC, Ma JA. Angew Chem. 2006;118:362–375.Angew Chem Int Ed. 2006;45:354–366. doi: 10.1002/anie.200500195.Tietze LF. Chem Rev. 1996;96:115–136. doi: 10.1021/cr950027e.
- 12.a) Barco A, Baricordi N, Benetti S, Risi CD, Pollini GP. Tetrahedron Lett. 2006;47:8087–8090. [Google Scholar]; b) Ballini R, Barboni L, Fiorini D, Giarlo G, Palmieri A. Green Chem. 2005;7:828–829. doi: 10.1039/b500846h. [DOI] [PubMed] [Google Scholar]; c) Barco A, Benetti S, Risi CD, Pollini GP, Romagnoli R, Zanirato V. Tetrahedron Lett. 1996;37:7599–7602. [Google Scholar]; d) Barco A, Benetti S, Risi CD, Pollini GP, Romagnoli R, Zanirato V. Tetrahedron Lett. 1994;35:9293–9296. [Google Scholar]; e) Posner GH, Crouch RD. Tetrahedron. 1990;46:7509–7530. [Google Scholar]
- 13.Hayashi Y, Okano T, Arataka S, Hazelard D. Angew Chem. 2007;119:5010–5013. doi: 10.1002/anie.200700909. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:4922–4925. doi: 10.1002/anie.200700909. [DOI] [PubMed] [Google Scholar]
- 14.For a related example, see: Yao CF, Jang YJ, Yan MC. Tetrahedron Lett. 2003;44:3813–3816.
- 15.2-Mercaptobenzaldehydes were synthesized using a modified procedure originally reported by: Gallagher T, Pardoe DA, Porter RA. Tetrahedron Lett. 2000;41:5415–5418.
- 16.For details, see Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.