

Abstract
T4 polynucleotide kinase/phosphatase (Pnkp) exemplifies a family of bifunctional enzymes with 5′-kinase and 3′-phosphatase activities that function in nucleic acid repair. T4 Pnkp is a homotetramer of a 301-aa polypeptide, which consists of an N-terminal kinase domain of the P-loop phosphotransferase superfamily and a C-terminal phosphatase domain of the DxD acylphosphatase superfamily. The homotetramer is formed via pairs of phosphatase-phosphatase and kinase-kinase homodimer interfaces. Here we identify four side chains – Asp187, Ser211, Lys258, and Asp277 – that are required for 3′ phosphatase activity. Alanine mutations at these positions abolished phosphatase activity without affecting kinase function or tetramerization. Conservative substitutions of asparagine or glutamate for Asp187 did not revive the 3′ phosphatase, nor did arginine or glutamine substitutions for Lys258. Threonine in lieu of Ser211 and glutamate in lieu of Asp277 restored full activity, whereas asparagine at position 277 had no salutary effect. We report a 3.0 Å crystal structure of the Pnkp tetramer, in which a sulfate ion is coordinated between Arg246 and Arg279 in a position that we propose mimics one of the penultimate phosphodiesters (5′NpNpNp-3′) of the polynucleotide 3′-PO4 substrate. The amalgam of mutational and structural data engenders a plausible catalytic mechanism for the phosphatase that includes covalent catalysis (via Asp165), general-acid base catalysis (via Asp167), metal coordination (by Asp165, Asp277 and Asp278), and transition state-transition stabilization (via Lys258, Ser211, backbone amides, and the divalent cation). Other critical side chains play architectural roles (Arg176, Asp187, Arg213, Asp254). To probe the role of oligomerization in phosphatase function, we introduced six double-alanine cluster mutations at the phosphatase-phosphatase domain interface, two of which (R297A-Q295A and E292A-D300A) converted Pnkp from a tetramer to a dimer and ablated phosphatase activity.
Keywords: RNA repair, end-healing, polynucleotide 3′ phosphatase
INTRODUCTION
T4 polynucleotide kinase/phosphatase (Pnkp) is the prototype of a family of repair enzymes that heal broken termini in RNA or DNA by converting 3′PO4/5′OH ends into 3′OH/5′PO4 ends, which are then suitable for sealing by RNA or DNA ligases. During T4 infection, Pnkp participates in an RNA-based immune response whereby the bacterium attempts to thwart T4 protein synthesis by inducing site-specific breakage of host-cell tRNAs, to which the phage responds by repairing the broken tRNAs using Pnkp and a phage-encoded RNA ligase (Amitsur et al., 1987). T4 Pnkp catalyzes two reactions in this pathway: (i) the transfer of the γ phosphate from ATP to the 5′OH terminus of RNA and (ii) the hydrolytic removal of a 3′PO4 terminus from RNA (Richardson, 1965; Novogrodsky and Hurwitz, 1966; Novogrodsky et al., 1966; Becker and Hurwitz, 1967; Cameron and Uhlenbeck, 1977).
T4 Pnkp is a homotetramer of a 301-aa polypeptide (Panet et al., 1973; Midgley and Murray, 1985; Wang and Shuman, 2001, 2002; Wang et al., 2002; Galburt et al., 2002). Essential constituents of the separate active sites for the 5′ kinase and 3′ phosphatase activities have been identified by alanine-scanning mutagenesis (Wang and Shuman, 2001, 2002). Amino acids required for catalysis of the 5′ kinase reaction map to the N-terminal half, whereas residues essential for the 3′ phosphatase function cluster in the C-terminal half. The C-terminal half of the Pnkp protomer, from residues 148-301, comprises an autonomous phosphatase domain with a homodimeric quaternary structure (Wang et al., 2002).
The crystal structure of the isolated kinase domain (aa 1-153) revealed that the kinase is also a homodimer (Wang et al., 2002). The kinase domain protomer consists of a central four-stranded parallel β-sheet flanked by three α helices on each side. The kinase active site is composed of (i) a classical P-loop motif (9GxxGxGKS16) that coordinates the β phosphate of the NTP donor; (ii) essential side chain Arg126, which also coordinates the NTP β phosphate; (iii) essential side chain Arg38, which coordinates the 3′ phosphate of the 5′OH acceptor, plus (iv) essential side chain Asp35, a putative general acid that activates the 5′OH for direct nucleophilic attack on the NTP γ phosphate. The active site is located within a tunnel through the heart of the enzyme. The tunnel entrance on the P-loop side admits the NTP phosphate donor and controls release of the NDP reaction product. The tunnel opening on the opposite face allows ingress of the 5′ end of a single-stranded polynucleotide to the kinase active site (Wang et al., 2002; Galburt et al., 2002; Eastberg et al., 2004). The homodimeric kinase crystal structure and the biochemical evidence for a homodimeric phosphatase domain prompted a model for assembly of the native Pnkp tetramer via separate kinase-kinase and phosphatase-phosphatase dimer interfaces (Wang et al., 2002) that was confirmed immediately by crystal structures of the Pnkp tetramer from the Stoddard lab (Galburt et al., 2002; Eastberg et al., 2004).
Insights to the architecture of the phosphatase active site of Pnkp emerged from a mutational analysis that identified 8 amino acids required for 3′ phosphatase activity: Asp165, Asp167, Arg176, Arg213, Arg246, Asp254, Asp278, and Arg279 (Wang and Shuman, 2002) (Fig. 1). Two of the essential phosphatase residues, Asp165 and Asp167, are located within a 165DxDxT169 motif that defines a superfamily of phosphotransferases that act through a covalent aspartyl phosphate intermediate (Collet et al., 1998; Burroughs et al., 2006). A comparison of the crystal structure from the Stoddard group with a structurally characterized DxDxT protein established that the Pnkp phosphatase domain is a bona fide member of this superfamily (Galburt et al., 2002; Cho et al., 2001). We have proposed that Pnkp belongs to a subgroup of acylphosphatases that includes the eukaryal Fcp1 protein serine phosphatases that dephosphorylate the carboxyl-terminal domain of RNA polymerase II (Hausmann and Shuman, 2003; Hausmann et al., 2004).
Fig. 1. Primary and secondary structure of the phosphatase domain of T4 Pnkp.

The amino acid sequence of T4 Pnkp from amino acids 159 to 301 is aligned with that of the phosphatase domain of the ORF86 polypeptide encoded by Autographa californica nucleopolyhedrovirus (AcNPV). Positions of side chain identity/similarity are denoted by ^ below the aligned sequences. The secondary structure elements of T4 Pnkp are shown above the amino acid sequence with β strands as arrows and α helices as bars. The amino acids of Pnkp that were mutated previously to alanine are denoted by + (nonessential for activity) or ● (essential or important for 3′ phosphatase activity). The amino acids in Pnkp that were mutated singly to alanine in the present study are indicated by !.
To further delineate the phosphatase active site and catalytic mechanism of Pnkp, we conducted two lines of investigation: (i) additional mutational analysis of the C-terminal phosphatase domain, guided by structure and sequence comparisons to a eukaryotic viral homolog (Galburt et al., 2002; Martins and Shuman, 2004); and (ii) crystallization of the native Pnkp homotetramer in the presence of sulfate, a phosphate mimetic. We report the effects of 19 mutations at 12 conserved positions of Pnkp, which identify four residues (Asp187, Ser211, Lys258, and Asp277) as essential for activity. A new Pnkp crystal structure identifies a sulfate-binding site near the phosphatase active site that likely mimics an interaction with the phosphodiester backbone of the polynucleotide substrate.
The rationale for the tetrameric quaternary structure of T4 Pnkp is not clear given that: (i) the kinase and phosphatase active sites appear to be contained within single protomer domains; and (ii) eukaryal cellular Pnkp is composed of structurally similar catalytic modules but has a monomeric structure (Bernstein et al., 2005). To explore the relevance of cross protomer interactions for enzyme activity, we introduced alanine cluster mutations at pairs of amino acids that comprise the crystallographic phosphatase-phosphatase domain interface. We thereby identified changes that converted Pnkp to a homodimer. The Pnkp dimer mutants retained kinase function, but were defective for 3′ phosphatase activity. We surmise that dimerization of the C-terminal domain of Pnkp is important for phosphatase function.
RESULTS AND DISCUSSION
Defining the Active Site of the Pnkp Phosphatase Domain
A goal of the present study was to extend the functional map of the phosphatase active site of T4 Pnkp. To do this, we introduced single alanine changes at amino acids within the phosphatase domain that are conserved in its baculovirus homologue (Fig. 1). This conservation guided a previous alanine-scan of T4 Pnkp, in which we identified eight residues – Asp165, Asp167, Arg176, Arg213, Arg246, Asp254, Asp278, Arg279 – as essential or important for 3′ phosphatase activity (these are denoted by • in Fig. 1; unimportant residues are denoted by +). In classifying mutational effects on Pnkp function, we adopted the following criteria of significance: (i) functional groups were deemed essential when their replacement by alanine reduced specific activity to ≤5% of wild-type Pnk; (ii) residues were deemed important when alanine substitution lowered activity to between 6 and 25% of wild-type; (iii) residues were deemed unimportant when alanine mutation elicited less than a 4-fold activity decrement. Here we targeted 12 new conserved positions for mutational analysis (indicated by ! in Fig. 1). We focused on: (i) conserved basic residues Lys198, Arg229, Lys253, and Lys258; (ii) conserved acidic residues Asp187, Glu219, Glu233, Asp277; (iii) conserved hydrophilic side chains Ser211, Thr222, and Thr251; and (iv) Gly212 flanking the essential residue Arg213.
Full-length wild-type Pnkp and the 12 Pnk-Ala mutants were produced in bacteria as His10-tagged fusions and purified from soluble bacterial extracts by Ni-agarose chromatography (Fig. 2A). Phosphatase activity was measured by the release of inorganic phosphate from deoxythymidine 3′ monophosphate (Becker and Hurwitz, 1967; Cameron and Uhlenbeck, 1977). Wild-type Pnkp released 0.66 nmol of Pi from 3′ dTMP per ng of protein in the linear range of enzyme-dependence, which translates into a turnover number of ~20 s−1. The specific activities of the 12 Ala-mutants were determined by enzyme titration and normalized to that of the wild-type Pnkp (Table I). Eight of the mutants had activity similar to wild-type Pnkp: K198A (82%), G212A (37%), E219A (51%), T222A (94%), R229A (94%), E233A (45%), T251A (120%), and K253A (95%). We surmise that the Lys198, Glu219, Thr222, Arg229, Glu233, Thr251, and Lys253 side chains are unimportant for catalysis of the 3′ phosphatase reaction. Four Ala-mutants were severely defective: D187A (<1%), S211A (<1%), K258A (<1%), and D277A (<1%). Thus, we conclude that the Asp187, Ser211, Lys258, and Asp277 side chains are essential for 3′ phosphatase function.
Fig. 2. Pnkp mutants.

Aliquots (5 μg) of the nickel-agarose preparations of full-length wild-type (WT) Pnkp and the indicated mutants were analyzed by SDS-PAGE. Polypeptides were visualized by staining with Coomassie Blue dye. The positions and sizes (in kDa) of marker proteins are indicated on the left.
Table I.
Effect of Alanine Mutations on 3′ Phosphatase Activity
| Pnkp Mutant | 3′ phosphatase (% of WT) |
5′ kinase (% of WT) |
|---|---|---|
| D187A | <1 | 110 |
| K198A | 82 | 80 |
| S211A | <1 | 83 |
| G212A | 37 | 90 |
| E219A | 51 | 64 |
| T222A | 94 | 87 |
| R229A | 94 | 87 |
| E233A | 45 | 66 |
| T251A | 120 | 95 |
| K253A | 95 | 83 |
| K258A | <1 | 59 |
| D277A | <1 | 53 |
Each of the Ala-mutant enzymes was also assayed for 5′ kinase activity, which was measured as the transfer of 32P from [γ-32P]ATP to 3′CMP to form [5′-32P]pCp. All of the mutant proteins retained 5′ kinase activity (within a factor of two of the specific activity of wild-type Pnkp) (Table I). We conclude that the severe loss of 3′ phosphatase activity elicited by alanine mutations at Asp187, Ser211, Lys258, and Asp277 reflects a specific requirement for these side chains in the folding or catalytic function of the phosphatase active site, rather than a general effect on Pnkp structure or activity.
The native size of the four phosphatase-defective mutant proteins was investigated by glycerol gradient sedimentation. Marker proteins catalase (248 kDa), BSA (66 kDa), and cytochrome c (13 kDa) were included as internal standards. After centrifugation, the polypeptide compositions of the odd-numbered gradient fractions were analyzed by SDS-PAGE (Fig. 3). The wild-type Pnkp polypeptide sedimented between catalase (248 kDa) and BSA (66 kDa), consistent with a tetrameric quaternary structure. By interpolation to a linear standard curve of the marker sedimentation coefficients versus gradient fraction, we calculated a sedimentation coefficient of 7.5 S for wild-type Pnkp. The D187A, S221A, K258A, and D277A mutant polypeptides also sedimented as single discrete peaks between catalase and BSA. Thus, none of the mutations that abrogated 3′ phosphatase activity affected the tetrameric quaternary structure of Pnkp.
Fig. 3. Velocity sedimentation of phosphatase-defective Pnkp-Ala mutants.

Sedimentation analysis was performed as described under Methods. The distributions of Pnkp (either WT, D187A, S211A, K258A or D277A as indicated) and the marker proteins catalase, BSA, and cytochrome c in each gradient were analyzed by SDS-PAGE. Scans of the Coomassie-blue stained gels are shown.
Structure-Activity Relationships at Essential 3′-Phosphatase Residues
To further evaluate the contributions of Asp187, Ser211, Lys258 and Asp277 to the 3′ phosphatase reaction, we tested the effects of conservative substitutions. Aspartate was replaced by asparagine and glutamine, lysine by glutamine and arginine, and serine by threonine. The D187N, D187E, S211T, K258Q, K258R, D277N and D277E proteins were purified from soluble bacterial extracts by Ni-agarose chromatography (Fig. 2B). The 3′ phosphatase specific activities of the D187N, D187E, and D277N mutants were <1% of the activity of wild-type Pnk, whereas the D277E mutant was 93% as active as wild-type (Table II). These data establish a requirement for a carboxylate at positions 187 and 277 of T4 Pnkp and they indicate a tight steric constraint on the distance from the main-chain to the carboxylate at position 187, such that the active site is perturbed by the additional methylene group of glutamate. A longer glutamate side chain can be accommodated at position 277 with no detrimental effect. The K258Q and K258R mutants had <1% of wild-type phosphatase activity (Table II), indicating a requirement for a positively charged residue, and lysine specifically, at this critical position. 3′ phosphatase activity was restored fully when Ser211 was replaced by threonine, signifying that the hydroxyl moiety is essential at position 211.
Table II.
Effect of Conservative Mutations on 3′ Phosphatase Activity
| Pnkp Mutant | 3′ phosphatase (% of WT) |
|---|---|
| D187E | <1 |
| D187N | <1 |
| S211T | 130 |
| K258Q | <1 |
| K258R | <1 |
| D277E | 93 |
| D277N | <1 |
New Crystal Structure of the Pnkp Tetramer
The Pnkp homotetramer was first crystallized by Galburt et al. (2002) in space group I222 with a single Pnkp protomer in the asymmetric unit. Here, we determined the structure of full-length Pnkp crystallized in space group P21212 with a much larger unit cell (124 × 128 × 357 Å) and an asymmetric unit composed of 12 Pnkp protomers organized as three homotetramers. We solved the structure by molecular replacement using the Stoddard group’s structure as a search model. The refined model at 3.0 Å resolution had a crystallographic R factor of 23.7% and an Rfree of 28.1% (Table I). For the purpose of illustrating the relevant structural features, we focus on tetramer 1 composed of Pnkp protomers A, B, C and D. These protomers were punctuated by gaps (aa 176-177 and 182-183 in A; aa 176 in B; aa 176-177 in C) in a conformationally flexible surface loop of the phosphatase domain. Gaps in this region (aa 174-185) were noted previously by Eastberg et al. (2004). Protomer D has a disordered gap from aa 216-220 in another surface loop of the phosphatase domain. The electron density maps revealed extra density appended to the Cys184 sulfur of protomers A, B, C and D, consistent with a DTT-mediated reaction of cysteine with the arsenic center of the cacodylate (dimethyl arsinic acid) present in the crystallization buffer (Jacobson et al., 1972; Tsao and Maki, 1991; Maignan et al., 1998; Tete-Favier et al., 2000; Junop et al., 2000). The arsenic adduct is located 3 to 3.5 Å above the planar aromatic Trp213 side chain; a similar reaction of a tryptophan-proximal cysteine with cacodylate has been reported for EcoRI methyltransferase and E. coli peptide methionine sulphoxide reductase (Tsao and Maki, 1991; Tete-Favier et al., 2000).
The Pnkp homotetramer adopted a flat rhomboid architecture with a central cavity, as reported by Galburt et al. (2002). Pairwise kinase-kinase and phosphatase-phosphatase dimer interfaces are located at the oblique and acute corners of the rhombus, respectively. The structures of the four kinase domains of the homotetramer were virtually identical to one another and were essentially identical to that of the kinase homodimer reported previously (Wang et al., 2002). Two sulfate ions (derived from the ammonium sulfate included in the crystallization buffer) were bound by the kinase active site in positions corresponding to the β phosphate of the NTP donor and the phosphate of the 5′-terminal HONp-N dinucleotide of the polynucleotide phosphate acceptor (not shown). Superposition of the kinase domains from our three homotetramers with the kinase domains of the several homotetramer structures from the Stoddard lab (Galburt et al., 2002; Eastberg et al., 2004) showed coincidence of all secondary structure elements; slight variations were confined to a surface loop from aa 45-50 that lines the central cavity of the rhomboid (not shown).
Phosphatase Domain and Active Site
The tertiary structure of the phosphatase domain comprises a central five-stranded parallel β sheet flanked by two α helices on either side (see Fig. 4A). Whereas a superposition of the phosphatase domains from all 12 protomers in the asymmetric unit reveals coincidence of all secondary structure elements, there was protomer-to-protomer variability in the positions of the surface loops from aa 217-222 and 173-188. The latter loop, which is located on the outer edge of the Pnkp rhomboid, was also the main source of variability of the main-chain trace between the present structures and those from the Stoddard lab. Fig. 4A shows the superimposed phosphatase active sites of protomers A, B, C and D (with the carbon atoms colored cyan for chain A, beige for chain B, yellow for chain C, and magenta for chain D) and the active site from the symmetrical Pnkp homotetramer of Galburt et al. (carbon atoms in green). There is little or no variability in the side chain locations on rotamer conformations of Asp165 (the nucleophile in the acylphosphate reaction mechanism), Ser211, Arg246, Asp254, Lys258, Asp277, Asp278, or Arg279. In contrast, the main chain of the loop that includes Asp167 (an essential residue and a putative general acid-base catalyst) varies in position from one protomer to the next, implying some conformational mobility. The Asp167 carboxylate forms an ion pair with Arg213 in every protomer; Arg213 is strictly essential for 3′ phosphatase activity (Wang and Shuman, 2002). Although the Cα position of Arg213 does not change, the arginine adopts different rotamer conformations to accommodate the subtle backbone movements and side chain rotations of Asp167.
Fig. 4. Pnkp phosphatase active site.

(A) Stereo view of the superimposed phosphatase active sites from Pnkp protomers A, B, C, and D (with the carbon atoms colored cyan for chain A, beige for chain B, yellow for chain C, and magenta for chain D) and the active site from the symmetrical Pnkp homotetramer structure reported previously (with carbon atoms in green). The Mg2+ of protomer B is depicted as a magenta sphere. The sulfate of protomer B is shown poised between Arg279 and Arg246. (B) Refined 2Fo-Fc electron density map of protomer D contoured at 1σ. The Mg2+ (cyan sphere) and sulfate ligands are shown. Waters are depicted as red spheres. The images were prepared in Pymol (DeLano, 2002).
The mobility of the Asp167-Arg213 pair affects the access of the phosphoryl substrate to the active site nucleophile. Although none of the Pnkp structures has a phosphate or phosphate analog vicinal to Asp165, reference to the structures of other DxD phosphotransferases captured as a pentavalent transition state analog (Lahiri et al., 2993; Baxter et al., 2006) or as covalent aspartyl-BeF3 adducts (a mimetic of the acylphosphate intermediate) (Cho et al., 2001; Kamenski et al., 2004) points toward essential residues Lys258 and Ser211 as the likely contacts to the phosphate oxygens (see Fig. 5). Remarkably, in protomer B (colored beige in Fig. 4A), the Asp167 carboxylate has swung into the putative oxyanion hole normally reserved for phosphate, such that it makes a hydrogen bond from Oδ1 to the Ser211 hydroxyl and an ionic interaction of Oδ2 with Lys258. This “closed” conformation is incompatible with catalysis. However, in the three other protomers of the homotetramer, Asp167 is oriented away from the phosphate-binding site and makes no contacts to either Ser211 or Lys258, leaving these functional groups available to bind the 3′ phosphate substrate (Fig. 4A). Moreover, the movement of Asp167 opens a path to Asp165 Oδ1 (the nucleophilic atom), which is occluded by Asp167 in protomer B.
Fig. 5. Active site similarity between Pnkp phosphatase and CTD serine phosphatase.

Stereo view of the phosphatase active site from Pnkp protomer D (green) superimposed on the BeF3-modified active site of CTD phosphatase Scp1 (beige). The aspartyl-BeF3 adduct is depicted with the beryllium in yellow and the fluorines colored red (reflecting their mimicry of the phosphate of the aspartyl-phosphate intermediate). The Mg2+ of Scp1 is depicted as a cyan sphere and the associated waters as red spheres. Atomic interactions in the Scp1 active site are indicated by dashed lines. Pnkp side chains are labeled.
Protomers A, B, C and D have density consistent with a magnesium ion adjacent to Asp165 and a sulfate ion coordinated by Arg246 and Arg249. Fig. 4A depicts the Mg2+ and sulfate ligands from protomer B. The refined 2Fo-Fc electron density map highlighting the Mg2+ and sulfate of protomer D is shown in Fig. 4B. The divalent cation is surrounded with octahedral geometry by the backbone carbonyl of Asp167, Asp165 Oδ, Thr169 Oγ, Asp277 Oδ, Asp278 Oδ, and a water (Figs. 4A and B). We attribute the essentiality of the Asp277 side chain, established in the present study (Table I), to its involvement in metal coordination. Apparently, the active site can accommodate the longer glutamate side chain (Table II), but the amide carbonyl oxygen of asparagine does not suffice for metal coordination. The imputed role of Asp278 carboxylate in metal binding (Figs. 4A and B) is consistent with previous findings that D278A and D278N mutations abolished 3′ phosphatase activity, which was restored partially by a conservative D278E change (Wang and Shuman, 2002).
Fig. 5 shows a superposition of the “open” active site of the Pnkp D protomer (green carbon atoms) on the active site of human Scp1, a DxD-family protein serine phosphatase that dephosphorylates the carboxyl-terminal domain of RNA polymerase II (Kamenski et al., 2004). The Scp1 structure (depicted with beige carbon atoms) captures the stable covalent apspartyl-BeF3 adduct as a mimetic of the labile aspartyl-phosphate intermediate. Lys258 and Ser211 of Pnkp correspond to lysine and threonine side chains of Scp1 that contact the aspartyl-phosphate oxygens. Mutational analysis of the homologous S. pombe CTD phosphatase Fcp1 has established that the lysine and threonine residues are essential for catalysis (Hausmann and Shuman, 2003; Hausmann et al., 2004). The structural alignment suggests that the Val166, Asp167, and Gly212 main chain amides of Pnkp also coordinate the phosphate oxygens (Fig. 5, see orange dashed lines), thereby comprising (with the lysine and serine side chains) the oxyanion hole of the Pnkp phosphatase.
The Scp1 structure reveals at higher resolution the occupancy of the octahedral magnesium coordination complex, which is composed of the counterparts of essential Pnkp residues Asp165, Asp167, Asp277, and Asp278 (Fig. 5). Three of the six magnesium coordination sites entail direct contacts to protein side-chain or main-chain oxygens; two sites are occupied by waters coordinated by protein side chains, and the sixth site entails metal coordination to a phosphate oxygen. The metal organizes constituents of the active site contributed by adjacent strands of the β sheet, in addition to participating directly in phosphoryl transfer chemistry by stabilizing the developing negative charge on the presumptive pentacoordinate transition state. We presume that Pnkp forms an analogous metal complex in the presence of substrate.
The sulfate ion near the phosphatase active site is situated between Arg279 and Arg246 (Figs. 4A and B). Prior studies showed that R246A and R279A mutations reduced phosphatase activity by an order or magnitude compared to wild-type Pnkp (Wang and Shuman, 2002). Both arginines engage in ionic interactions with the Asp254 carboxylate, which is itself essential for 3′-phosphatase activity (Wang and Shuman, 2002). The Asp254 backbone amide also donates a hydrogen bond to the sulfate. We speculate that the interactions of this sulfate might mimic those to one of the internucleotide phosphates of a polynucleotide 3′-phosphate substrate. Based on the distance from the aspartate nucleophile and the putative 3′-phosphate binding site, the sulfate is most likely to one of the two terminal phosphodiesters in a 5′-NpNpNp oligomer.
In sum, the Pnkp structures now suggest plausible roles (in catalysis, substrate binding, or active site organization) for ten of the twelve key residues identified by mutagenesis: Asp165, Asp167, Ser211, Arg213, Arg246, Asp254, Lys258, Asp277, Asp278 and Arg279. The remaining two essential residues – Arg176 and Asp187 – are located on the protein surface in our homotetramer structure within what appear to be mobile loops. Indeed, Arg176 is disordered in three of the protomers in tetramer 1 in our structure and is also disordered in the tetramer structures of Eastberg et al. (2004). However, the surface loop is ordered in the structure of Galburt et al. (2002), which reveals that Asp187 Oδ1 and Oδ2 form salt bridges to the Arg176 and Arg213 side chains, respectively. Thus, Arg176 and Asp187 play architectural roles, by interacting with each other to organize the surface loop and to help tether it to Arg213.
Effects of Mutations at the Phosphatase Dimer Interface of the Pnkp Tetramer
The rationale for the homodimeric quaternary structure of T4 Pnkp is not clear. It has been proposed that the tetramer facilitates simultaneous processing of both termini of the broken tRNA substrate in vivo (Galburt et al., 2002), but there is no evidence that the two reactions are coupled during a single substrate binding event. To probe the contributions of quaternary structure to the phosphatase function of Pnkp, we introduced six double-alanine cluster mutations at amino acids located at or near the phosphatase dimer interface of the Pnkp crystal structure. Figure 6 highlights the polar and hydrophobic interactions at that interface. The amino acid pairs subjected to cluster mutations were: Asn190-Met192, Met199-Tyr200, Glu195-Tyr205, Arg287-Gln295, Glu292-Trp294, and Ser298-Asp300. The six full-length Pnkp-Ala/Ala mutants were produced in bacteria as His10-tagged fusions and purified from soluble bacterial extracts by Ni-agarose chromatography in parallel with wild-type Pnkp; SDS-PAGE analysis confirmed that the preparations were of equivalent purity (not shown).
Fig. 6. The phosphatase homodimer interface.

A stereo view is shown with one phosphatase protomer colored beige and the other colored gray. Amino acids that line the homodimer interface are shown in stick representation; residues from the gray promoter are labeled in italics and those from the beige protomer are in regular font style. Hydrogen-bonding and ionic interactions of these side chains are indicated by dashed lines. Landmark active site residues and the Mg and sulfate ligands are shown for the beige protomer only.
The native sizes of the six Ala-cluster mutants were gauged by glycerol gradient sedimentation with internal standards (Fig. 7). The N190A-M192A, E195A-Y205A, M199A-Y200A, and S298A-D300A proteins sedimented as discrete peaks between catalase (248 kDa) and BSA (66 kDa), consistent with a tetrameric quaternary structure. These results tells us that 8 of the side chains located at the intersubunit surface are not required per se to form a stable phosphatase homodimer. Ser298-Asp300 engage in a polar cross-subunit interaction network that embraces Arg137 (located in the last α helix of the kinase domain of the neighboring subunit) plus Glu284 and Arg287 of the opposing phosphatase domain (Fig. 6). Glu195-Tyr205 form a cross-subunit hydrogen bond. Asn190-Met192 and Met199-Tyr200 make van der Waals contacts at and across the dimer interface (Fig. 6).
Fig. 7. Sedimentation analysis of phosphatase dimer interface mutants.
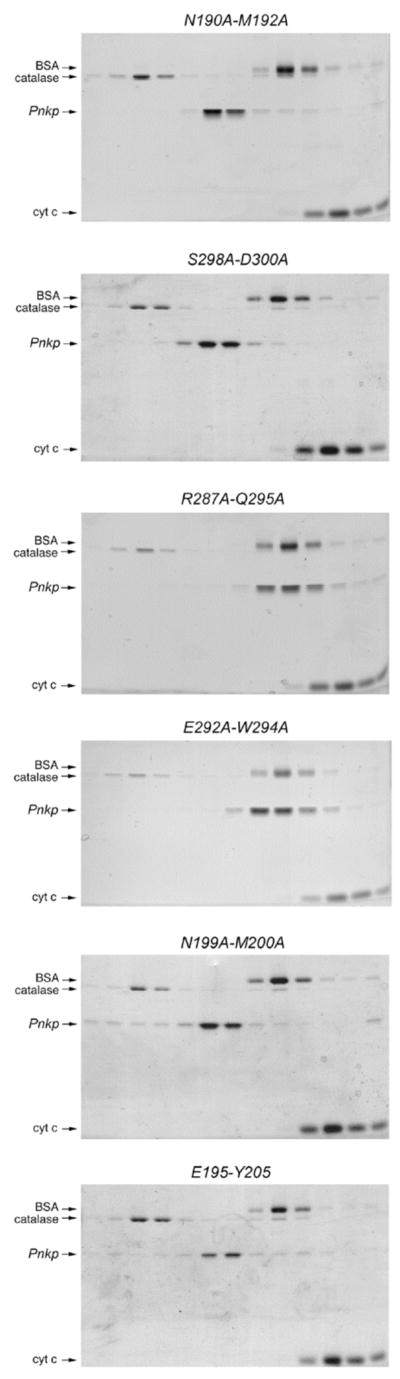
Sedimentation analysis was performed as described under Methods. The distributions of Pnkp Ala-Ala mutants and the marker proteins catalase, BSA, and cytochrome c in each gradient were analyzed by SDS-PAGE. Scans of the Coomassie-blue stained gels are shown.
The R287A-Q295A and E292A-W294A proteins also sedimented as discrete peaks, but were shifted toward lighter positions in the gradients such that they sedimented with BSA (Fig. 7). We surmise that the R287A-Q295A and E292A-W294A proteins are homodimers rather than tetramers. The simplest explanation for the change in Pnkp quaternary structure is that the cluster mutations disrupted the phosphatase-phosphatase interface, while sparing the kinase-kinase interface. Trp294, while making a hydrogen bond from Nε to Glu292 in the same subunit, engages in multiple cross-subunit van der Waals contacts to other hydrophobic side chains, including Val193, Leu196, and the Trp294 in the opposing subunit (Fig. 6). Arg287 and Gln295 are neighbors on the subunit interaction surface but they don’t interact with each other. Gln295 forms an intrasubunit hydrogen bond to Thr280, whereas Arg287 coordinates the backbone carbonyl of Trp294 of the same subunit (Fig. 6). Trp294 and Arg287 are adjacent to each other on the central surface of the dimer interface.
The kinase and phosphatase specific activities of the Ala-cluster mutants were determined by enzyme titration and normalized to that of the wild-type Pnkp (Table IV). Whereas the kinase activities ranged from 30–95% of wild-type and thus did not meet our criteria for a significant mutational effect, two of the cluster mutants (R287A-Q295A and E292A-W294A) reduced phosphatase activity to 2–3% of the wild-type level. The other four Ala-cluster mutations elicited less drastic reduction in phosphatase activity, to between 18 and 27% of the wild-type level (Table IV). These results suggest that homodimerization of the phosphatase domain is important for phosphatase activity. Because the phosphatase active site is composed of functional groups from a single protomer, with no apparent cross-subunit contributions (Fig. 6), we surmise that the homodimer interface is likely to contribute to the proper folding or stability of the active site, so that changes at the dimer interface exert long-range effects on catalysis.
Table IV.
Effect of Double-Alanine Mutations on 3′-Phosphatase Activity
| Pnkp Mutant | 3′ phosphatase (% of WT) |
5′ kinase (% of WT) |
|---|---|---|
| N190A-M192A | 18 | 67 |
| M199A-Y200A | 19 | 76 |
| E195A-Y205A | 22 | 39 |
| R287A-Q295A | 2 | 34 |
| E292A-W294A | 3 | 30 |
| S298A-D300A | 27 | 95 |
MATERIALS AND METHODS
Pnkp Purification and Crystallization
The Pnkp ORF was amplified by PCR with primers that introduced BamHI restriction sites over the start codon and 3′ of the stop codon, thereby facilitating in-frame insertion into vector pET28-His10-Smt3. A 2-liter culture of E. coli BL21(DE3)/pET28-His10-Smt3-Pnkp was grown in Luria-Bertani medium containing 0.06 mg/ml kanamycin until the A600 reached 0.6; the culture was then adjusted to 0.3 mM isopropyl β-D-thiogalactoside and incubated for 3.5 h at 30°C. Cells were harvested by centrifugation and the pellet was stored at −80°C. All subsequent procedures were performed at 4°C. Thawed bacteria were resuspended in 125 ml of lysis buffer (50 mM Tris-HCl, pH 7.5, 1.2 M NaCl, 15 mM imidazole, 10% glycerol). Lysozyme, PMSF, benzamidine, and Triton X-100 were added to final concentrations of 1 mg/ml, 0.2 mM, 1 mM and 0.2% respectively. The lysate was sonicated to reduce viscosity and insoluble material was removed by centrifugation for 45 min at 17,000 rpm in a Sorvall SS34 rotor. The soluble extract (900 mg protein) was applied to a 12-ml column of Ni2+-nitrilotriacetic acid-agarose (Qiagen). The column was washed sequentially with 50 ml of buffer A (50 mM Tris-HCl, pH 7.5, 200 mM NaCl, 10% glycerol), 50 ml of 50 mM Imidazole in buffer A, and 50 ml of 100 mM imidazole in buffer A. The bound His10-Smt3-Pnkp protein was then eluted with 200 mM imidazole in buffer A. The elution profile was monitored by SDS-PAGE. The peak Pnkp-containing fractions were pooled and dialyzed overnight against 50 mM Tris-HCl, pH 7.5, 200 mM NaCl, 1 mM DTT, 10% glycerol. A faint cloudy precipitate was removed from the dialysate by centrifugation. The clarified supernatant (81 mg protein) was treated for 1 h at room temperature with 81 μg of the Smt3-specific protease Ulp1 (Mossessova and Lima, 2000). The digest was applied to a 4.5-ml column of Ni-agarose and the tag-free Pnkp protein was recovered in the flow-through fractions.
Crystals of Pnkp were grown at 23°C by the hanging drop vapor diffusion method by mixing aliquots of Pnkp (3 μl; 2.9 mg/ml in buffer containing 10 mM MgCl2) with an equal volume of reservoir buffer containing 100 mM sodium cacodylate (pH 6.6), 12% PEG-8000, 0.2 M ammonium sulfate, 20 mM urea, 5 mM DTT. Prior to diffraction, the crystals were cryo-preserved in reservoir buffer containing 30% glycerol and then flash-frozen in liquid nitrogen. X-ray diffraction data for a native crystal were collected at the National Synchrotron Light Source (Brookhaven, NY) at beamline X9A using a 165-mm MAR CCD detector. Data were integrated, scaled, and merged using DENZO and SCALEPACK (Otwinowski and Minor, 1997). Pnkp crystallized in space group P21212 (a=124.63 Å, b= 128.05 Å, c=357.12 Å; α,β,γ = 90°). Due to the large unit cell, non-ideal crystal-to-detector orientation, and limited detector size, diffraction data were collected in 0.5° oscillations.
Structure Determination and Refinement
The structure of Pnkp was solved via molecular replacement using a combination of the “fastdirect” cross-rotation and “general” translation functions in combination with Patterson correlation refinement in CNS (Brunger et al., 1998). Six rotation-translation solutions with correlation coefficients from 6.4% to 11.6% were combined to give a complete initial model, which yielded an R-factor of 39.2% after rigid-body refinement. Subsequent refinement was carried out using a combination of positional, grouped and individual B-factor, and simulated annealing refinement procedures in CNS, cross-validated by an Rfree dataset consisting of 2.5% of the reflection data. Non-crystallographic symmetry restraints were applied to 217 residues of each monomer using a separate 12-fold operator for the kinase and phosphatase domains. Model building was done in O (Jones et al., 1991).
The final model consists of 12 Pnkp protomers organized as three homotetramers, with protomers A–D, E–H, and I–L comprising tetramers 1, 2, and 3, respectively. The structure of the each protomer is unique in light of variable conformations of loop regions connecting secondary structure elements of the phosphatase domain. In particular, the loop from residues 173 to 185 is the most variable element from protomer to protomer. One or more residues or side chains within this loop were disordered and therefore omitted from the final model in each of the protomers. Aside from the flexible loops, each protomer shows an average root mean square deviation of ~0.5 Å for 280 Cα atoms.
In 11 out of 12 protomers, Cys184 appears to be modified by covalent attachment to an arsenic atom derived from the cacodylate (dimethyl arsinic acid) buffer. This covalent modification of cysteine by arsenic is well described and depends on the presence of both cacodylate and DTT (Jacobson et al., 1972; Tsao and Maki, 1991; Maignan et al., 1998). The canonical product of such covalent modification is dimethyl-arsenic cysteine, RS-As(CH3)2, and the modified cysteine is modeled as such in several protein crystal structures (Maignan et al., 1998; Tete-Favier et al., 2000; Junop et al., 2000). An alternate product, dimethyl arsinoyl cysteine, RS-As(O)(CH3)2, has also been modeled in several proteins that were crystallized in the presence of cacodylate and DTT (Shi et al., 2006; Elkins et al., 2006). Because it was not possible to differentiate between these two products at 3.0 Å resolution, we modeled only a single non-bonded arsenic atom for this adduct.
Three sulfate ions were bound per Pnkp protomer. Two sulfates were located within the kinase active site, as observed previously in the crystal structure of the isolated kinase homodimer (Wang et al., 2002). A novel sulfate-binding site was seen in the phosphatase domain. The occupancy of the sulfate atoms in the phosphatase active site was manually adjusted to eliminate corresponding fo-fc electron density. Refinement statistics are listed in Table I. The coordinates are deposited in the Protein Data Bank under ID code 2IA5.
Pnkp mutants
Missense mutations were introduced into the Pnkp ORF by using the two-stage PCR-based overlap extension method as described previously (Wang and Shuman, 2001, 2002). The PCR products were digested with NdeI and BamHI and then inserted into pET16b. The inserts were sequenced completely to confirm the desired mutations and exclude the acquisition of unwanted changes. The pET-Pnkp plasmids were introduced into E. coli BL21(DE3). Recombinant protein production was induced by adjusting exponentially growing cultures (100 ml) to 0.3 mM IPTG and incubating them at 17°C for 15 h with continuous shaking. The wild-type and mutant His10-Pnkp proteins were purified from soluble bacterial lysates by Ni-agarose chromatography as described previously (Wang and Shuman, 2001, 2002). Protein concentrations were determined by using the BioRad dye reagent with bovine serum albumin as the standard.
5′ Kinase Assay
Reactions mixtures (10 μl) containing 70 mM Tris-HCl (pH 7.6), 10 mM MgCl2, 5 mM DTT, 25 μM [γ32P]ATP, 1 mM 3′ CMP (Sigma) and 0, 4.7, 9.4, 18.8, 37.5, or 75 ng of wild-type or mutant Pnkp were incubated for 20 min at 37°C. The reactions were quenched by adding 5 μl of 5M formic acid. Aliquots of the mixtures were applied to a polyethyleneimine-cellulose TLC plate, which was developed with 1 M formic acid, 0.5 M LiCl. The [γ32P]ATP substrate and [α32P]pCp product were visualized and quantified by scanning the gel with a Fujix BAS2500 phosphorimager. The kinase specific activity of each protein was determined from the slope of the titration curve and then normalized to the specific activity of wild-type Pnkp (defined as 100%). The results are shown in Tables I and IV.
3′ Phosphatase Assay
Reaction mixtures (25 μl) containing 100 mM imidazole (pH 6.0), 10 mM MgCl2, 10 mM β-mercaptoethanol, 0.1 mg/ml BSA, 1.6 mM 3′ dTMP (Sigma), and 0, 4.7, 9.4, 18.8, 37.5, or 75 ng of wild-type or mutant Pnkp as specified were incubated for 20 min at 37°C. The reactions were quenched by adding 75 μl of cold water and 1 ml of malachite green reagent (BIOMOL Research Laboratories). Phosphate release was determined by measuring A620 and interpolating the value to a phosphate standard curve. The phosphatase specific activity of each protein was determined from the slope of the titration curve and then normalized to the specific activity of wild-type Pnkp (defined as 100%). The results are shown in Tables I, II, and IV.
Velocity Sedimentation
Aliquots (40 μg) of the Pnkp preparations were mixed with catalase (30 μg), BSA (30 μg), and cytochrome c (30 μg) and the mixtures were applied to 4.8-ml 15–30% glycerol gradients containing 50 mM Tris-HCl (pH 8.0), 0.2 M NaCl, 1 mM EDTA, 2.5 mM DTT, 0.1% Triton X-100. The gradients were centrifuged in a SW50 rotor at 50,000 rpm for 16 h at 4°C. Fractions were collected from the bottoms of the tubes. Aliquots (15 μl) of the odd-numbered gradient fractions were analyzed by SDS-PAGE.
Table III.
T4 polynucleotide kinase/phosphatase crystallographic data.
| Space group P21212 a=124.63 Å, b= 128.05 Å, c= 357.12 Å; α, β, γ = 90° at 125°K | |
| Crystallographic Data Quality: (370 0.5° oscillations) | |
| Resolution | 40.0 – 3.0 Å (3.05 – 3.0 Å) |
| Rsym | 9.8% (38.4%) for I ≥ -σI for individual observations |
| Mean Redundancy | 4.38 (3.5) for 112,850 unique reflections |
| Completeness | 98.8% (98.4%) All measured reflections |
| 88.3% (66.9%) I ≥ σI for merged reflections | |
| Mean I/σI | 13.25 (2.34) |
| Refinement Residuals: 20.0 – 3.0 Å (3.11 – 3.0 Å), I ≥ σI: | |
| Rfree | 28.1% (41.9%) for 2.5% of data |
| Rwork | 23.7% (35.4%) |
| Model Quality: | |
| Bond deviations lengths / angles (rms) | 0.0126Å / 1.503° (Engh and Huber parameters) |
| Ramachandran distribution | 89.0% core / 11.0% allowed/0.0% other |
| B-factors | All atoms: 60.60 Å2 |
| Protein: 61.26 ± 10.88 Å2 (s.d. by monomer) | |
| Heteroatoms: 44.69 Å2 | |
| Model Contents: 12 Pnkp monomers containing 3542 protein residues. | |
| Heteroatoms: 36 sulfate, 7 Mg++, 11 arsenic, 976 water molecules. | |
Standard definitions are used for all of the parameters. Figures in parentheses refer to data in the highest resolution bin. The data collection statistics come from SCALEPACK, with anomalous pairs merged throughout. The refinement / geometric statistics come from CNS, and the Ramachandran analysis was performed with PROCHECK.
Acknowledgments
This work was supported by NIH Grant GM42498. SS is an American Cancer Society Research Professor.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amitsur M, Levitz R, Kaufman G. Bacteriophage T4 anticodon nuclease, polynucleotide kinase, and RNA ligase reprocess the host lysine tRNA. EMBO J. 1987;6:2499–2503. doi: 10.1002/j.1460-2075.1987.tb02532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter NJ, Olguin LF, Golicnik M, Feng G, Hounslow AM, Bermel W, Blackburn GM, Hollfelder F, Waltho JP, Williams NH. A Trojan horse transition state analogue generated by MgF3− formation in an enzyme active site. Proc Natl Acad Sci USA. 2006;103:14732–14737. doi: 10.1073/pnas.0604448103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker A, Hurwitz J. The enzymatic cleavage of phosphate termini from polynucleotides. J Biol Chem. 1967;242:936–950. [PubMed] [Google Scholar]
- Bernstein NK, Williams RS, Rakovszky ML, Cui D, Green R, Galicia S, Koch CA, Cass CE, Durocher D, Weinfeld M, Glover JNM. The molecular architecture of the mammalian DNA repair enzyme, polynucleotide kinase. Mol Cell. 2005;17:657–670. doi: 10.1016/j.molcel.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Burroughs AM, Allen KN, Dunaway-Mariano D, Aravind L. Evolutionary genomics of the HAD superfamily: understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J Mol Biol. 2006;361:1003–1034. doi: 10.1016/j.jmb.2006.06.049. [DOI] [PubMed] [Google Scholar]
- Cameron V, Uhlenbeck OC. 3′-Phosphatase activity in T4 polynucleotide kinase. Biochemistry. 1977;16:5120–5126. doi: 10.1021/bi00642a027. [DOI] [PubMed] [Google Scholar]
- Cho H, Wang W, Kim R, Yokota H, Damo S, Kin SH, Wemmer D, Kustu S, Yan D. BeF3− acts as a phosphate analog in proteins phosphorylated on aspartate: structure of a BeF3− complex with phosphoserine phosphatase. Proc Natl Acad Sci USA. 2001;98:8525–8530. doi: 10.1073/pnas.131213698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collet JF, Stroobant V, Pirard M, Delpierre G, Van Schaftingen E. A new class of phosphotransferases phosphorylated on an aspartate residue in an amino-terminal DXDX(T/V) motif. J Biol Chem. 1998;273:14107–14112. doi: 10.1074/jbc.273.23.14107. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL molecular graphics system. DeLano Scientific; San Carlos, CA: 2002. http://www.pymol.org. [Google Scholar]
- Eastberg JH, Pelletier J, Stoddard BL. Recognition of DNA substrates by bacteriophage T4 polynucleotide kinase. Nucleic Acids Res. 2004;32:653–660. doi: 10.1093/nar/gkh212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins JM, Soundararajan M, Yang X, Papagrigoriou E, Sundstrom M, Edwards A, Arrowsmith C, Weigelt J, Doyle DA. Structure of centaurin gamma 1. 2006 PBD ID 2IWR. [Google Scholar]
- Galburt EA, Pelletier J, Wilson G, Stoddard BL. Structure of a tRNA repair enzyme and molecular biology workhorse: T4 polynucleotide kinase. Structure. 2002;10:1249–1260. doi: 10.1016/s0969-2126(02)00835-3. [DOI] [PubMed] [Google Scholar]
- Hausmann S, Shuman S. Defining the active site of Schizosaccharomyces pombe CTD phosphatase Fcp1. J Biol Chem. 2003;278:13627–13632. doi: 10.1074/jbc.M213191200. [DOI] [PubMed] [Google Scholar]
- Hausmann S, Erdjument-Bromage H, Shuman S. Schizosaccharomyces pombe carboxyl-terminal domain (CTD) phosphatase Fcp1: distributive mechanism, minimal CTD substrate, and active site mapping. J Biol Chem. 2004;279:10892–10900. doi: 10.1074/jbc.M312513200. [DOI] [PubMed] [Google Scholar]
- Jacobson KB, Murphy JB, Das Sarma B. Reaction of cacodylate with organic thiols. FEBS Lett. 1972;22:80–82. doi: 10.1016/0014-5793(72)80224-2. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–118. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Junop MS, Modesti M, Guarné A, Ghirlando R, Gellert M, Yang W. Crystal structure of the Xrcc4 DNA repair protein and implications for end joining. EMBO J. 2000;19:5962–5970. doi: 10.1093/emboj/19.22.5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenski T, Heilmeier S, Meinhart A, Cramer P. Structure and mechanism of RNA polymerase CTD phosphatases. Mol Cell. 2004;15:399–407. doi: 10.1016/j.molcel.2004.06.035. [DOI] [PubMed] [Google Scholar]
- Lahiri SD, Zhang G, Dunaway-Mariano D, Allen KN. The pentacovalent phosphorus intermediate of a phosphoryl transfer reaction. Science. 2003;299:2067–2071. doi: 10.1126/science.1082710. [DOI] [PubMed] [Google Scholar]
- Maignan S, Guilloteau JP, Zhou-Lin Q, Clément-Mella C, Mikol V. Crystal structures of the catalytic domain of HIV-1 integrase free and complexed with its metal cofactor: high level of similarity of the active center with other viral integrases. J Mol Biol. 1998;282:359–368. doi: 10.1006/jmbi.1998.2002. [DOI] [PubMed] [Google Scholar]
- Martins A, Shuman S. Characterization of a baculovirus enzyme with RNA ligase, polynucleotide 5′ kinase and polynucleotide 3′ phosphatase activities. J Biol Chem. 2004;279:18220–18231. doi: 10.1074/jbc.M313386200. [DOI] [PubMed] [Google Scholar]
- Midgley CA, Murray NE. T4 polynucleotide kinase; cloning of the gene (pseT) and amplification of its product. EMBO J. 1985;4:2695–2703. doi: 10.1002/j.1460-2075.1985.tb03989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossessova E, Lima CD. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol Cell. 2000;5:865–876. doi: 10.1016/s1097-2765(00)80326-3. [DOI] [PubMed] [Google Scholar]
- Novogrodsky A, Hurwitz J. The enzymatic phosphorylation of ribonucleic acid and deoxyribonucleic acid: phosphorylation at 5′-hydroxyl termini. J Biol Chem. 1966;241:2923–2932. [PubMed] [Google Scholar]
- Novogrodsky A, Tal M, Traub A, Hurwitz J. The enzymatic phosphorylation of ribonucleic acid and deoxyribonucleic acid: further properties of the 5′-hydroxyl polynucleotide kinase. J Biol Chem. 1966;241:2933–2943. [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Meth Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Panet A, van de Sande JH, Loewen PC, Khorana HG, Raae AJ, Lillehaug JR, Kleppe K. Physical characterization and simultaneous purification of bacteriophage T4 induced polynucleotide kinase, polynucleotide ligase, and deoxyribonucleic acid polymerase. Biochemistry. 1973;12:5045–5050. doi: 10.1021/bi00749a003. [DOI] [PubMed] [Google Scholar]
- Richardson CC. Phosphorylation of nucleic acid by an enzyme from T4 bacteriophage-infected Escherichia coli. Proc Natl Acad Sci USA. 1965;54:158–165. doi: 10.1073/pnas.54.1.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Rojas R, Bonifacino JS, Hurley JH. The retromer subunit Vps26 has an arrestin fold and binds Vps35 through its C-terminal domain. Nat Struct Biol. 2006;13:540–548. doi: 10.1038/nsmb1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tete-Favier F, Cobessi D, Boschl-Muller S, Azza S, Branlant G, Aubry A. Crystal structure of the Escherichia coli peptide methionine sulphoxide reductase at 1.9 Å resolution. Structure. 2000;8:1167–1178. doi: 10.1016/s0969-2126(00)00526-8. [DOI] [PubMed] [Google Scholar]
- Tsao DHH, Maki AH. Optically detected magnetic resonance study of the interactions of an arsenic(III) derivative of cacodylic acid with EcoRI methyl transferase. Biochemistry. 1991;30:4565–4572. doi: 10.1021/bi00232a029. [DOI] [PubMed] [Google Scholar]
- Wang LK, Shuman S. Domain structure and mutational analysis of T4 polynucleotide kinase. J Biol Chem. 2001;276:26868–26874. doi: 10.1074/jbc.M103663200. [DOI] [PubMed] [Google Scholar]
- Wang LK, Shuman S. Mutational analysis defines the 5′-kinase and 3′-phosphatase active sites of T4 polynucleotide kinase. Nucleic Acids Res. 2002;30:1073–1080. doi: 10.1093/nar/30.4.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Lima CD, Shuman S. Structure and mechanism of T4 polynucleotide kinase – an RNA repair enzyme. EMBO J. 2002;21:3873–3880. doi: 10.1093/emboj/cdf397. [DOI] [PMC free article] [PubMed] [Google Scholar]