

Abstract
Global changes in the phosphorylation state of human H1 isoforms isolated from UL3 cells have been investigated using mass spectrometry. Relative changes in H1 phosphorylation between untreated cells and cells treated with dexamethasone or various CDK inhibitors were determined. The specific cyclin-dependent kinase consensus sites of phosphorylation on the histone H1 isoforms that show changes in phosphorylation were also investigated. Three sites of phosphorylation on histone H1.4 isoforms have been identified.
Keywords: H1 histones, chromatin, phosphorylation, mass spectrometry, CDK inhibitors, tandem mass spectrometry
Introduction
Histones, assembled with DNA to form nucleosomes, are the major structural proteins associated with chromatin.1–3 The histone proteins have been classified into two categories, core histones (e.g., histone H2A, H2B, H3, and H4) and linker histones (e.g., histone H1 and H5).2 The nucleosome core consists of a histone octamer consisting of an H3–H4 tetramer and two H2A–H2B dimers around which DNA wraps. A variable length of DNA connects the resulting nucleosome structures. The linker histones, H1 histones, associate noncovalently to the linker DNA and the core histones.4,5
The H1 histones consist of a globular domain flanked by two less structured lysine-rich N- and C-terminal domains. The globular domain binds noncovalently to one linker DNA strand to stabilize the compaction of chromatin.3–5 Histones are subjected to various signal transduction and cell cycle dependent post-translational modifications that can alter the interactions of the histones with DNA.5–10 These modifications include methylation, acetylation, phosphorylation, ubiquitination, glycosylation, and ADP-ribosylation. These modifications have been associated with a variety of biological processes such as gene expression, DNA replication, and chromatin assembly.8–14
Glucocorticoids are important endocrine modulators of metabolism and immunity. Synthetic glucocorticoids, such as dexamethasone (dex) and prednisone, are used clinically for long-term treatments of many autoimmune diseases and are critical tools in transplantation medicine. For a number of years, the Archer laboratory has been investigating the mechanisms of glucocorticoid-mediated transcription activation.15–18 In these studies, the hormone inducible mouse mammary tumor virus (MMTV) promoter has been utilized. It was reported that when mouse cells were exposed to prolonged treatment with dexamethasone, a significant decrease in the level of histone H1 phosphorylation was observed. This decreased level of phosphorylation could be correlated with the MMTV promoter becoming refractory to further glucocorticoid stimulation. For this work, Western analyses were performed with anti-histone H1 and phospho-specific H1 antibodies. Unfortunately, these analyses cannot differentiate between the specific isoforms (H1.0, H1.1, H1.2, H1.3, H1.4, and H1.5) of histone H1 proteins.
Therefore, we have applied ESI/MS to the analysis of mouse histone H1 isoforms.19,20 Utilizing this approach, we investigated the phosphorylation state of the specific H1 isoforms before and after prolonged treatment with dexamethasone and after exposure of cells to specific kinase inhibitors. Specifically, our previous experiments showed that the relative phosphorylation levels of the histone H1.3, H1.4, and H1.5 isoforms decrease after prolonged hormone exposure. For example, when the total additive phosphorylation for H1.3, H1.4, and H1.5 in control cells is normalized to 1, the corresponding level of phosphorylation for these isoforms in cells treated for 48 h with dexamethasone is 0.39, 0.2, and 0.2, respectively. The other histone H1 isoforms (i.e., H1.0, H1.1, and H1.2) showed no significant change in the level of phosphorylation.
In an effort to identify specific phosphorylation sites in H1 and delineate roles of cyclin-dependent kinases (CDK) in these events, we undertook the identification of the specific CDK consensus sites [(S/T)PX(R/K)] of phosphorylation on the mouse histone H1 isoforms that showed changes in phosphorylation after prolonged treatment with dexamethasone (e.g., H1.3 and H1.4). For this work, we are using a combination of enzymatic proteolysis and peptide sequencing by mass spectrometry. We have previously reported16 that the CDK2 inhibitor, CVT313, blocked H1 phosphorylation in mouse 1471.1 cells and so are most interested in determining those sites of phosphorylation. After these phosphorylation sites were determined, we generated an antibody that is phospho-site specific for H1.3/H1.4 isoforms.
Concurrent with these studies, we were interested in investigating the global phosphorylation status of human histone H1 isoforms to determine whether similar changes are observed for the human H1 isoforms as for the mouse H1 isoforms upon dexamethasone treatment. In the cell line used for this work (UL3 cells derived from the osteosarcoma cell line U2OS21), we found that only three isoforms of H1 were expressed, H1.0, H1.2, and H1.4. Using mass spectrometry, we show that both the H1.2 and H1.4 isoforms in UL3 cells are phosphorylated, and that phosphorylation is reduced in the presence of dexamethasone. We are also interested in determining whether a similar decrease in phosphorylation of the H1 proteins upon treatment with a CDK2 inhibitor will be observed for the human H1 proteins as was observed for the mouse H1 proteins. To determine the effects of CDK inhibitors on global phosphorylation of the human H1 isoforms, intact H1 proteins following treatment with CDK inhibitors were analyzed by electrospray ionization mass spectrometry. Because only three isoforms were observed in the UL3 cells, this also allowed us the opportunity to identify specific sites of phosphorylation within the human H1.4 isoform and compare those to the sites of phosphorylation determined in the mouse H1.4 isoform.
Experimental Section
Chemicals and Antibodies
Dexamethasone and DMSO were purchased from Sigma-Aldrich (St. Louis, MO). CVT313 (e.g., CDK2 inhibitor) and CGP74514 (CDK1 inhibitor) were purchased from Calbiochem/EMD Biosciences and dissolved in DMSO (Sigma). Monoclonal antibody to H1 (clone AE4) was purchased from Upstate USA, Inc. (Chicago, IL).
Cell Culture
Mouse 1471.1 cells are derived from a C127 parental cell line and the UL3 cells are derived from U2OS cells.15,21 Cells were grown at 37 °C in Dulbecco's Modified Eagle's medium (DMEM) with 10% fetal bovine serum (Hyclone), penicillin-streptomycin (Invitrogen), and 6% CO2. Drug treatments were initiated when cells were growing exponentially. Cells were plated in 10 × 100 mm tissue culture dishes in regular media at 60% confluence. The following day, cells were treated with either 0.01% DMSO, 10 μM CVT313, 25 μM CVT313, or 2.5 μM CGP74514 for 24 h. The reported IC50 for CVT313 is 0.5 μM for CDK2/cylin A22 and the IC50 for CGP74514 for CDK1 is 25 nM.23 The doses used in these experiments are in the effective concentration range reported for these drugs.16,22,24,25 In addition, cells were subjected to prolonged treatment (48 h) with 10 nM dexamethasone. Cells were harvested by trypsinization, collected into PBS, and fixed in cold 70% ethanol overnight at 4 °C.
Polyclonal Antibody Production to H1.4pT18
A custom peptide antigen was made corresponding to mouse H1.3 amino acids 15–27 (VEKTPVKKKSKKT) containing a phosphate on the threonine at position 18 (Princeton Biomolecules). Polyclonal antibodies were generated by injecting peptide into rabbits housed at Covance Laboratories and collecting serum at intervals using standard methods. Serum was tested by Western blot for detection of H1. Antiserum was purified using Nab Protein A Spin purification Kit (Pierce). Protein concentration of resulting fractions was determined by absorbance at 280 nm using a standard curve of purified rabbit IgG (Santa Cruz) and pooled. Specificity was determined using dot blot of serial 5× dilutions of peptides corresponding to H1.3 and H1.4 with and without a phosphorylated threonine at position 18, respectively, onto nitrocellulose membrane and probed with antisera.
Histone H1 Extraction
Total H1 was extracted from whole cells by resuspending cell pellets into 5% perchloric acid for 2 h on ice. Acid soluble proteins in the supernatant were precipitated by adjusting the volume to 30% trichloroacetate and incubating at −20 °C overnight. Pellets were washed several times with acetone and then resuspended in water. Protein concentration was determined via a UV-spectrophotometer (Molecular Devices) with detection at 214 nm after 1:50 dilutions in 1 mM Tris in a quartz cuvette using a standard curve of calf histone H1 (Upstate).
Western Blot Analysis
Isolated H1 proteins were separated by SDS-PAGE on 16% Tris-glycine gels and transferred to a PVDF membrane for 3 h at 300 mA at 4 °C in a cold room. Membranes were blocked using 5% fat-free powdered skim milk and 2% horse serum in Tris-buffered saline plus Tween (TTBS) overnight. After a brief wash in TTBS, membranes were incubated with protein-A purified antibody at a concentration of 1–2 μg/mL for 1–3 h at room temperature and then detected using horseradish peroxidase coupled goat anti-rabbit IgG (Amersham) and enhanced chemiluminescence (ECL, Amersham).
HPLC Purification
HPLC purifications were performed as previously described,19,20 except gradients were performed using an Agilent (Palo Alto, CA) HP Series 1100 HPLC system consisting of an autoinjector with a 400 μL sample loop, a Series 1100 binary pump, a Series 1100 diode array detector, and a BioRad Model 2110 fraction collector (Hercules, CA). The H1 isoform purifications were performed at a flow rate of 1 mL/min using a Vydac C4 column and a gradient of 5% B for 10 min, 5–33% B for 10 min, then 33–45% B over 65 min. For purification of the endoproteinase Arg-C digestions, a Vydac C18 reverse phase column was used with a gradient of 1% B for 10 min, 1–40% B for 60 min, then 40–95% B over 10 min. Fractions were collected at 10 s to 1 min intervals, dried, and stored at −80 °C until use. Just prior to MALDI/MS analysis, fractions were reconstituted in 20 μL of water. Following MALDI/MS analysis, the HPLC fractions were pooled by individual isoforms and stored at −80 °C.
Enzymatic Digestion Conditions
For digestion with Arg-C, selected HPLC fractions were pooled and diluted to a total volume of 76 μL in 5 mM DTT/5 mM sodium phosphate buffer (pH 7.6). Sigma endoproteinase Arg-C (1 μg/μL in H2O) was diluted 1:1000 in 5 mM DTT. A total of 80 pg of the enzyme solution was added to the pooled HPLC fractions and the reactions were allowed to proceed at room temperature for approximately 1.5–2.5 h. Reactions were monitored by MALDI/MS analysis, and if necessary, additional enzyme (80 pg) was added to complete the digestions. For the tryptic digestions, an aliquot of the pooled HPLC purified H1 isoform samples were further diluted with 10–50 mM ammonium bicarbonate buffer (pH 8) and subjected to tryptic digestion by adding 1–2 μg of porcine trypsin (Promega Corporation, Madison, WI) and incubating the reaction overnight at 37 °C, then storing at −80 °C until analyzed.
Matrix-Assisted Laser Desorption Ionization Mass Spectrometry
Positive ion matrix-assisted laser desorption ionization (MALDI) MS analyses were performed using a Voyager Super De-STR (Applied Biosystems, Framingham, MA) delayed-extraction time-of-flight mass spectrometer. The instrument is equipped with a nitrogen laser (337 nm) to desorb and ionize the samples. A close external calibration, using two points to bracket the mass range of interest, was used. A 0.5 μL aliquot of the reconstituted fraction was spotted with 0.5 μL of MALDI matrix on a stainless steel sample target and allowed to dry at room temperature. A saturated solution of α-cyano-4-hydroxycinnamic acid in 45:45:10 ethanol/water/formic acid (v/v) was used as the MALDI matrix. Following MALDI/MS analysis, the HPLC fractions were pooled by individual isoforms and stored at −80 °C.
Electrospray Mass Spectrometry
A Micromass Q-Tof Ultima Global (Waters/Micromass, Milford, MA) hybrid tandem mass spectrometer was used for the acquisition of the electrospray ionization (ESI) mass spectra and tandem mass spectra (MS/MS). This instrument was operated at a mass resolution (fwhm) of 8000 and a mass accuracy of better than 0.01%. All data were acquired following an external calibration performed once daily. The Ultima Global is equipped with a nanoflow electrospray source and consists of a quadrupole mass filter and an orthogonal acceleration time-of-flight mass spectrometer. The needle voltage was ∼3200 V and the collision energy was 10 eV for the MS analyses.
For the ESI/MS analyses, 2 μL of the pooled histone isoform fraction was added to 4 μL of 50:50 acetonitrile/water (0.1% formic acid). The resulting solutions were analyzed by flow injection analysis at ∼300 nL/min using a pressure injection vessel.26 Measurements were taken from at least three separate ESI/MS analyses from each of two independent biological experiments (n ≥ 6).
For the LC/MS and LC/MS/MS analyses of the tryptic digests, an Agilent (Palo Alto, CA) Cap1100 HPLC system consisting of binary pumps and a micro autosampler was used to deliver the gradients. Injections of 1–8 μL were made and a linear water/acetonitrile (0.1% formic acid) gradient of 5% acetonitrile for 10 min, 5–50% acetonitrile over 45 min, then 50–95% acetonitrile over 10 min or other similar gradient was used for the chromatographic separations. The column used was a 15 cm × 75 μm i.d. Hypersil C18 (“pepmap”) column (LC Packings, San Francisco, CA) at a flow rate of 500 nL/min. To verify the sites of phosphorylation, the instrument was set to cycle through a series of spectra throughout the entire LC gradient. The cycle typically consisted of the acquisition of one mass spectrum followed by one to four MS/MS spectra of specific ions of interest. The ions of interest for the MS/MS analyses were determined from a prior LC/MS analysis. The collision energy used for the MS/MS experiments was 25 eV. Data analysis was accomplished with a MassLynx data system, MaxEnt deconvolution software, and ProteinLynx software supplied by the manufacturer.
Results and Discussion
Previously, the relative levels of phosphorylation on specific H1 isoforms in asynchronously dividing mouse tumor cells following prolonged treatment with dexamethasone and after exposure of cells to staurosporine, a potent kinase inhibitor, were investigated using mass spectrometry.19,20 From these studies, it was determined that specific mouse histone H1 isoforms showed differential changes in phosphorylation based on treatment. These data pointed specifically to the mouse H1.3, H1.4, and H1.5 isoforms as targets of CDK-specific phosphorylation. This information suggests that the H1 isoforms may have differing functions. Thus, identification of the sites of phosphorylation of the specific isoforms may be important in defining their roles in such activities as transcription.
Identification of H1.4 Phosphorylation Sites in Mouse 1471.1 Cells
Initially, we have investigated the sites of phosphorylation on the mouse histone H1 isoforms that showed changes in phosphorylation after prolonged treatment with dexamethasone (e.g., H1.3 and H1.4). Because these isoforms co-purify by HPLC and because of the high sequence homology between these two isoforms (nearly 87% identical), endoproteinase Arg-C is the only enzyme readily available that will yield unique peptides with CDK2 consensus phosphorylation sites upon proteolysis. Figure 1A shows the amino acid sequences for mouse H1.3 and H1.4 with the CDK2 consensus sequences outlined in boxes, and the arrows indicate Arg-C cleavage sites. The Arg-C peptides which would yield unique sequences containing CDK2 consensus phosphorylation sites are Arg-C fragments one and six from H1.4 and Arg-C fragments one and four from H1.3. Unfortunately, the mass of some of these peptides are too large (greater than 5000 Da) for good MS/MS sequencing using traditional mass spectrometric instrumentation. Therefore, to determine the sites of phosphorylation unique to these peptides, the H1.3/H1.4 HPLC fractions were pooled and subjected to Arg-C digestion and the resulting peptides were separated by HPLC (data not shown). All of the HPLC fractions were lyophilized, reconstituted in water, and analyzed by MALDI/MS to determine which fractions contain the unique H1.3 or H1.4 peptides with the CDK2 consensus phosphorylation sites. Figure 1B shows the linear-mode MALDI mass spectrum of one of the HPLC fractions. The major ion observed has an average m/z of 9702 and corresponds in mass to Arg-C fragment 6 of mouse histone H1.4 (average theoretical m/z = 9703). Also observed are ions corresponding in mass to the addition of one and two phosphate groups (80 Da) to the Arg-C fragment 6 peptide (ions of m/z 9783 and 9862, respectively). Under the chromatographic conditions used, the phosphorylated H1.4 peptides were separated from the H1.3 peptides. All of the phosphorylated H1.3 peptide fractions, however, also contained some phosphorylated H1.4 peptides.
Figure 1.
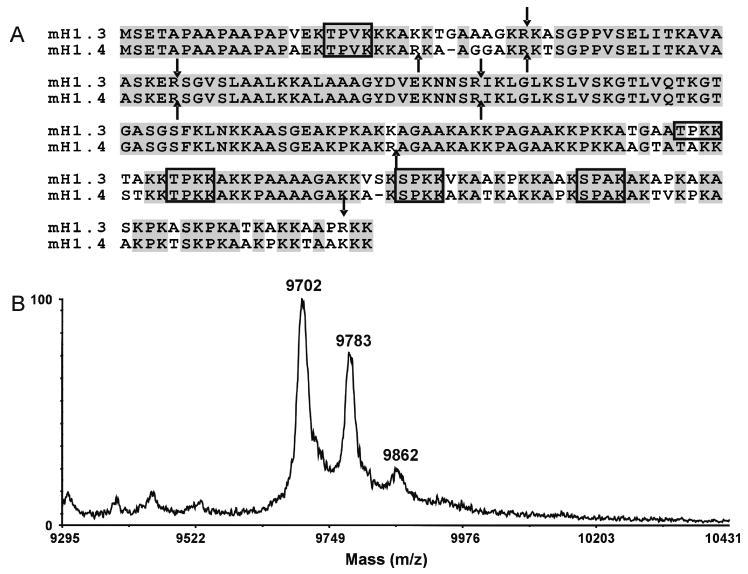
(A) Sequence alignment for mouse histone H1.3 and H1.4 isoforms. CDK consensus sites ((S/T)PX(R/K)) are outlined with a box. These histones are nearly 87% identical (shaded in gray). The arrows indicate endoproteinase Arg-C cleavage sites. (B) MALDI mass spectrum of an HPLC fraction which contains a unique mouse H1.4 peptide with a CDK2 consensus site. The MALDI mass spectrum shows ions which correspond in mass to Arg-C peptide fragment 6 of H1.4. In addition, ions are observed which correspond in mass to the addition of one and two phosphate groups to this peptide fragment.
The HPLC fractions containing the unique H1.4 Arg-C fragments were further subjected to tryptic digestion followed by LC/MS and LC/MS/MS analyses. The MS/MS spectra of three tryptic phosphopeptides containing CDK2 consensus sites are shown in Figure 2. The MS/MS spectrum of the ion of m/z 411.21 which corresponds in mass to monophosphorylated tryptic peptide SPK of Arg-C fragment 6 of histone H1.4 is shown in Figure 2A. A nearly complete series of both y and b ions and the loss of HPO3 (80 Da) from the a2 and b2 ions and the loss of H3PO4 (98 Da) from the a2 ion are observed. Abundant ions corresponding to the loss of H3PO4, HPO3, and H3PO4 + H2O from the molecular ion are observed. From these data, phosphorylation is verified and the site of phosphorylation in this peptide can be assigned to Ser-172 of histone H1.4.
Figure 2.
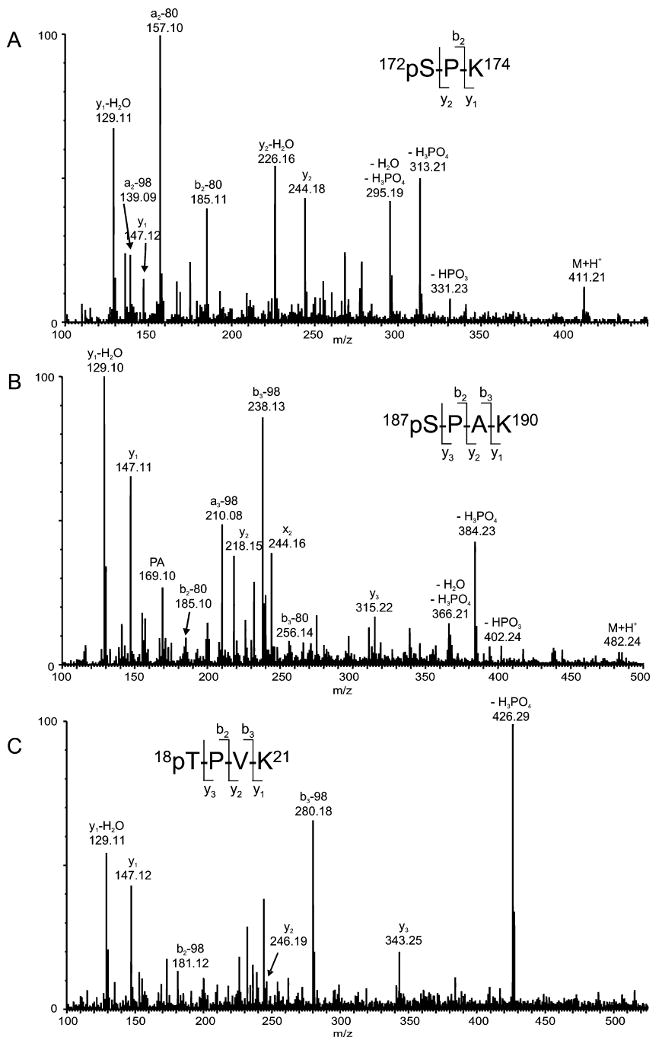
Tandem mass spectra of phosphorylated tryptic peptides derived from Arg-C fragment six of mouse histone H1.4. (A) MS/MS data of the ion of m/z 411.21 corresponding in mass to monophosphorylated SPK; (B) MS/MS data of the ion of m/z 482.24 corresponding in mass to monophosphorylated SPAK; and (C) MS/MS data of the ion of m/z 524.27 corresponding in mass to monophosphorylated TPVK.
The MS/MS spectrum of the ion of m/z 482.24 (Figure 2B) which corresponds in mass to monophosphorylated tryptic peptide SPAK of Arg-C fragment 6 of histone H1.4 was acquired. Again, both y and b ions as well as the loss of HPO3 and H3PO4 from y and b ions are observed. In addition, the loss of H3PO4, HPO3, and H3PO4 + H2O from the molecular ion, verifying phosphorylation is observed. These structurally informative fragment ions allow the assignment of the site of phosphorylation in this peptide to Ser-187 of histone H1.4.
Figure 2C shows the MS/MS spectrum of the (M + H)+ ion of m/z 524.27 which corresponds in mass to monophophorylated tryptic peptide TPVK from Arg-C fragment 1 of histone H1.4. The most abundant fragment ion observed in this spectrum corresponds to the loss of H3PO4 from the molecular ion. Other structurally informative ions observed include y1, y2, and y3 as well as the loss of H3PO4 from the b2 and b3 ions. These data allow the assignment of the phosphorylation site in this peptide to Thr-18 of histone H1.4.
Characterization of Histone H1 Isoform Phosphorylation in Human UL3 Cells
Concurrent with LC/MS/MS analyses of the mouse H1 isoforms, we also investigated the post-translational modification status of human H1 isoforms in osteosarcoma UL3 cells. H1 proteins were acid-extracted, purified by reverse phase HPLC, and screened by MALDI/MS analyses. From the mass of the ions observed from these data, it was determined that two chromatographic peaks contained H1 isoforms; the first peak contained the H1.0 isoform, whereas the H1.2 and H1.4 isoforms co-eluted in the second HPLC peak (data not shown). Interestingly, we did not find ions corresponding in mass to the H1.1, H1.3, or H1.5 isoforms in the UL3 cells in any of the fractions analyzed. This is not unexpected, as it had been previously reported that in cells in culture and in mammalian tissue, the H1.2 and H1.4 isoforms are more broadly expressed in all cell types, whereas expression of other H1 isoforms are often down-regulated in differentiated cells.27,28
To determine the phosphorylation status of these human H1 isoforms, the two HPLC fractions were analyzed by electrospray mass spectrometry and the data are summarized in Table 1. The ESI/MS analyses of the first HPLC peak show ions which correspond in mass to the unmodified H1.0 isoform. The deconvoluted ESI mass spectrum of the second HPLC peak (Figure 3A) shows ions which correspond in mass to acetylated H1.2 as well as the mono- and diphosphorylated forms of acetylated H1.2. In addition, ions corresponding in mass to the acetylated H1.4 isoform were observed as well as the mono-, di-, and triphosphorylated forms of this protein.
Table 1.
Observed Average Masses from the Deconvoluted ESI Mass Spectra for the Human Histone H1 Isoforms Isolated from UL3 Cells
| isoform | modificationa | calc. Mrb | observed Mrc |
|---|---|---|---|
| H1.0 | None | 20733 | 20732 |
| H1.2 | 1A | 21277 | 21276 |
| H1.2 | 1A, 1P | 21357 | 21356 |
| H1.2 | 1A, 2P | 21437 | 21436 |
| H1.4 | 1A | 21777 | 21777 |
| H1.4 | 1A, 1P | 21857 | 21857 |
| H1.4 | 1A, 2P | 21937 | 21937 |
| H1.4 | 1A, 3P | 22017 | 22017 |
A = acetyl (42 Da), P = phosphate (80 Da).
Calculated average protonated mass including loss of the N-terminal methionine.
Observed average protonated mass.
Figure 3.
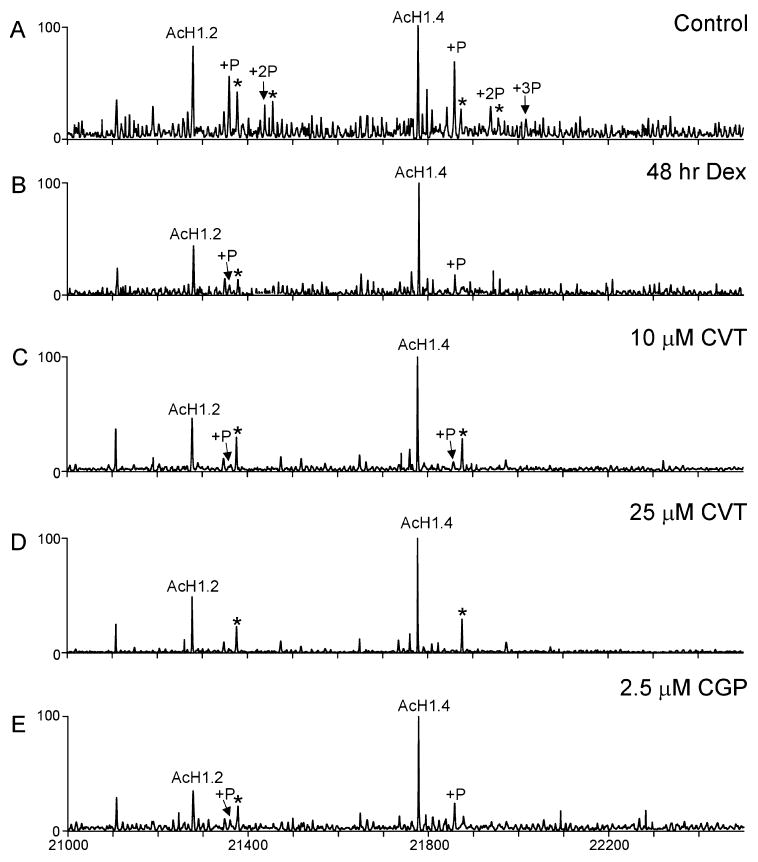
Deconvoluted electrospray mass spectra of intact H1 proteins detected in UL3 cells following treatments with dexamethasone and two CDK inhibitors. Mass spectra from (A) control cells and cells following treatment with (B) dexamethasone for 48 h, (C) 10 μM CVT313, (D) 25 μM CVT, and (E) 2.5 μM CGP74514. The ions labeled with an asterisk (*) correspond in mass to the addition of a noncovalent phosphate (+98 Da).
CDK Inhibitor Studies of Human H1 Isoform Phosphorylation
Using Western blot analyses, we have previously reported16 that the CDK2 inhibitor, CVT313, blocked H1 phosphorylation in mouse 1471.1 cells and are interested in determining whether the same phenomenon is observed for human H1 isoforms. Additionally, the recent development of a CDK1 inhibitor, CGP74514, allows one to determine the role of CDK1 in H1 phosphorylation. To determine the effects of CDK inhibitors on global phosphorylation of the human H1 isoforms, we used electrospray ionization mass spectrometry to analyze the intact H1 proteins following treatment of the cells with these CDK inhibitors. For these treatments, cells were treated for 24 h with either 0.01% DMSO (as a control), 10 μM CVT313, 25 μM CVT313, or 2.5 μM CGP74514. Additionally, cells were treated for 48 h with 10 nM dexamethasone. Following treatment, the cells were harvested and the H1 proteins were isolated, purified by HPLC, and analyzed by ESI/MS. From the resulting ESI mass spectra, the relative phosphorylation of the human H1 isoforms following 24 h treatments with each inhibitor was determined. Comparison of these data show that, when cells are pretreated with dexamethasone, the CDK2 inhibitor CVT313, or the CDK1 inhibitor CGP74514, the global levels of phosphorylation of both the H1.2 and H1.4 isoform were significantly decreased by all treatments (Figure 3B–E).
To determine the relative quantitative levels of the individual phosphorylated isoforms present, the ESI/MS analyses were repeated in triplicate from two independent biological experiments. Although the MS analysis of proteins, in general, is not very quantitative, relative changes in phosphorylation on a specific protein can be determined. Generally speaking, the addition of negatively charged modifications on a protein (i.e., phosphorylation) would reduce its ionization potential in the mass spectrometer. In the case of the histone H1 proteins, however, these proteins have large overall positive charges (e.g., over 50 positively charged amino acid residues per isoform). The addition, therefore, of one or two negatively charged phosphate groups per molecule is not expected to significantly affect the ionization potential of the protein, and this is not expected to significantly affect the relative sensitivity of the differentially phosphorylated protein ions.
The relative quantitation of each isoform of the individual human H1.2 and H1.4 proteins was computed by first determining the relative height abundance of the ions corresponding in mass to each specific isoform. These heights were then normalized to the total amount of the H1 isoform observed, thereby, reflecting the relative percentage of each species (Figure 4A and B; Supplementary Tables 1 and 2). For example, the relative amounts of each of the H1.4 isoforms present in control cells was determined to be 39%, 34%, 18%, and 8% for acetylated H1.4, acetylated monophosphorylated H1.4, acetylated diphosphorylated H1.4, and acetylated triphosphorylated H1.4, respectively. For the H1.2 isoform in control cells, the relative amounts of each species observed is 58% acetylated, 34% acetylated monophosphorylated, and 8% acetylated diphosphorylated.
Figure 4.
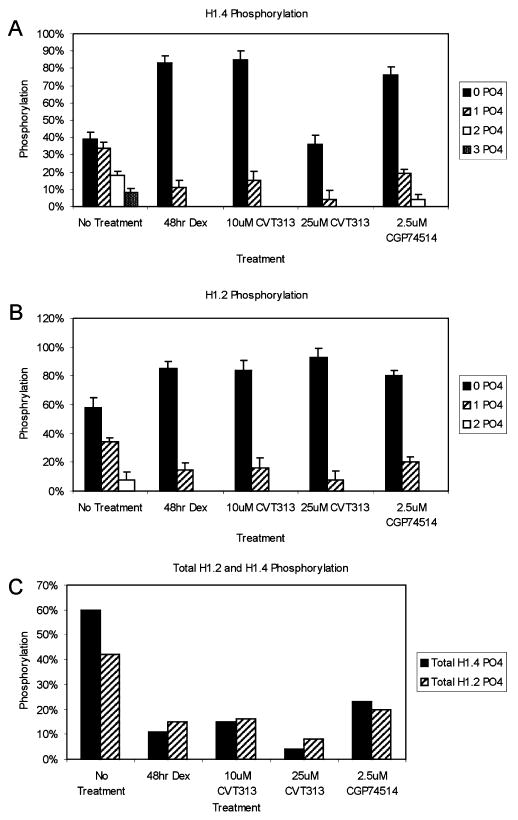
Relative quantitation of phosphorylation in each human H1 isoform following treatment with dexamethasone or CDK inhibitor. (A) Relative amounts of each isoform detected for human H1.4 protein; (B) relative amounts of each individual isoform detected for human H1.2 protein; (C) total overall level of phosphorylation in human H1.2 and H1.4 protein based on treatment.
Similarly, the relative quantitation of each species was determined after treatment of the cells with the various drugs (Figure 3 and Figure 4; Supplementary Tables). For the H1.4 isoform, only the nonphosphorylated isoform and a relative small amount (15% or less) of the monophosphorylated isoform were observed from cells treated with dexamethasone and CVT313, whereas both the monophosphorylated (19%) and the diphosphorylated (4%) forms were observed after the CGP745145 treatment. For the H1.2 isoform, for all drug treatments studied, only relative small amounts (20% or less) of the monophosphorylated species were observed in addition to the acetylated form. Overall, these data suggest that CDK kinase activity is responsible for nearly all of the phosphorylation of the human histone H1 isoforms in these cells. Collectively, the overall level of phosphorylation can be determined for the H1.2 and H1.4 proteins based on treatment (Figure 4C; Supplementary Table 3). For the H1.4 protein in the control cells, 60% of the H1.4 total protein detected was observed in a phosphorylated form. The total phosphorylation level in H1.4 decreased to 11%, 15%, 4%, and 23% after treatment with dexamethasone, 10 μM CVT, 25 μM CVT, and 2.5 μM CGP74514, respectively. Similar results were observed for the H1.2 isoform; 42% of the total protein was observed in a phosphorylated form in control cells. Following the same treatments as with H1.4, these total phosphorylation levels dropped to 15%, 16%, 8%, and 20%.
These data clearly demonstrate that the in vivo levels of phosphorylation of both the H1.2 and H1.4 isoforms are greatly reduced upon treatment for 48 h with dexamethasone. To determine whether these changes could be correlated with CDK activity, the levels of phosphorylation of H1.2 and H1.4 from control and dexamethasone-treated cells were compared to cells treated with either a CDK1 or a CDK2 inhibitor. Following treatment with 10 μM CVT313 or 2.5 μM CGP74514, the level of phosphorylation of H1.2 and H1.4 was reduced to a comparable level observed following dexamethasone treatment. Treatment with 25 μM CVT313 decreased the level of phosphorylation to an even greater extent, although a small amount of phosphorylation was still observed following all treatment conditions. These results suggest that the phosphorylation events of histone H1 proteins are complex and are a dynamic process which occurs throughout the cell cycle, with possibly a variety of factors influencing these modifications.
Identification of H1.4 Phosphorylation Sites in Human UL3 Cells
For the mouse H1.4 isoforms in 1471.1 cells, three CDK consensus sites of phosphorylation were identified; therefore, we were interested in determining the CDK-dependent sites of phosphorylation in the human H1.4 isoform. In the UL3 cells, both H1.2 and H1.4 were observed as phosphorylated. Figure 5 shows the amino acid sequences for human H1.2 and H1.4 with the CDK consensus sequences outlined in boxes. The consensus target residues correspond to threonine-18, -146, -154, and serine-174 and -187. To identify sites of phosphorylation in the human H1.4 protein, the HPLC purification of the intact histone H1 proteins from control cells was repeated, collecting 10 s fractions. By doing so, we could collect a fraction that contained mostly the H1.4 isoform with little or no H1.2 isoform. From this fraction, we performed LC/MS analyses of the tryptic digest of the H1.4 fraction. From a theoretical digest of the H1.4 isoform, those masses which contained the consensus CDK sites were searched in the LC/MS analyses. The potential consensus CDK-containing tryptic peptides and the theoretical nonphosphorylated protonated masses and theoretical phosphorylated protonated masses are as follows: TPVK (threonine-18; 444.2822, 524.2485), ATGAATPK (threonine-146; 716.3943, 796.3606), TPK (threonine-154; 345.2138, 425.1801), SPK (serine-174; 331.1981, 411.1645), and SPAK (serine-187; 402.2353, 482.2016). Of these, ions were observed which correspond in mass to all of the nonphosphorylated form of the peptides, whereas only masses corresponding to phosphorylated TPVK (m/z obs = 524.28), SPK (m/z obs= 411.19), and SPAK (m/z obs = 482.19) were observed. No ions were observed which would correspond in mass to phosphorylated ATGAATPK or TPK. Additional LC/MS analyses were performed where the instrument was set to acquire MS/MS spectra of specific masses throughout the entire LC/MS run. Figure 6 shows the MS/MS spectra of three tryptic peptides that were found to be phosphorylated in H1.4. In Figure 6A and B, fragment ions are observed which correspond to the loss of HPO3, H3PO4, and H3PO4 + H2O from the protonated molecule. The observation of these fragment ions indicates the presence of a phosphate group within these peptides. In addition, a series of b and y ions and the loss of phosphate (either as the loss of 80 Da or the loss of 98 Da) from the b ions are observed. These structurally informative fragment ions allow the assignment of the site of phosphorylation to Ser-172 and Ser-187 in these two peptides, respectively. Figure 6C shows the MS/MS spectrum of the (M + H)+ ion of m/z 524.28 which corresponds in mass to monophosphorylated tryptic peptide 2 (T2), TPVK (aa 18–21). Although the S/N ratio in this spectrum is low, structurally diagnostic ions, especially y3 and b3-98, are observed. In addition, several of these same structural ions are observed in the MS/MS spectrum of mouse monophosphorylated T2:18–21 (Figure 3C). These data allow confirmation of a phosphate group on the threonine residue within this tryptic peptide. From these data, we can confirm that T18, S172, and S187 are phosphorylated in the histone H1.4 isoform of UL3 cells.
Figure 5.

Sequence alignment for human H1.2 and H1.4 histones. CDK consensus sites ((S/T)PX(R/K)) are outlined with a box. These histones are more than 86% identical (shaded in gray).
Figure 6.
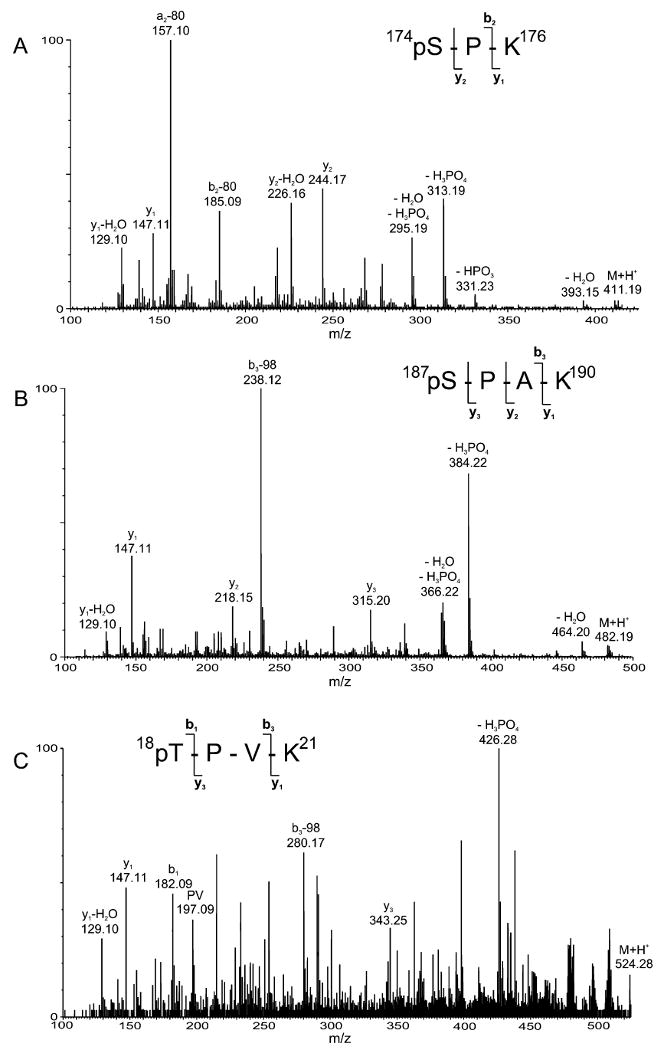
Tandem mass spectra of phosphorylated tryptic peptides derived from human histone H1.4. (A) MS/MS data of the ion of m/z 411.19 corresponding in mass to monophosphorylated SPK; (B) MS/MS data of the ion of m/z 482.19 corresponding in mass to monophosphorylated SPAK; and (C) MS/MS data of the ion of m/z 524.28 corresponding in mass to monophosphorylated TPVK.
Tryptic digests of both the mouse and human isolated H1.4 isoforms resulted in the identification of phosphorylated peptides corresponding to three CDK consensus sites in both the N- and C-terminal tails of H1.4. These sites of phosphorylation were determined to be located at Ser-174, Ser-187, and Thr-18. Sites of post-translational modifications of H1 have been previously addressed using similar MS/MS techniques on the H1 isoforms.29–31 With this approach, Hunt et al.29 identified seven serine or threonine residues (including Ser-174, Ser-187, and Thr-18) that were phosphorylated in human H1.4 in asynchronous HeLa cells. Four of the identified H1.4 phosphorylation sites contain a putative cyclin-dependent kinase motif; however, three of the sites were identified from non-CDK consensus motifs. Conversely, Mann et al.31 identified two (possibly three) sites of phosphorylation in human H1.4 in both human MCF7 and HeLa cells. In addition to the two cell lines, Mann et al.31 also analyzed and compared the post-translational modifications of histone H1 in nine mouse tissues. From this work, these authors showed that differences in the post-translational modifications of histone H1 proteins could be determined between different tissues and different cell types. Several of the serine and threonine phosphorylation sites identified by these groups29,31 were found within peptides which did not contain CDK-consensus sites. This, therefore, implies that other kinases can influence H1 phosphorylation. Indeed, previous research has implicated that H1 phosphorylation is influenced by MAP kinases as well.32,33 Although this study has focused on the phosphorylation of CDK consensus sites because of the correlation between CDK2 inhibition and loss of MMTV activation found in our previous work,15,16 the results from the H1.4 analyses from the cell lines reported by Mann et al.31 are similar to the number of sites of phosphorylation found in this work, that is, three sites. Therefore, we hypothesize that different phosphorylation sites are utilized differentially between cells and are unique to particular isoforms with functional consequences.
Quantitative Changes in Specific Sites of Phosphorylation in H1.4
LC/MS data from one each of the tryptic digests of mouse H1.4 isoforms from 1471.1 control cells versus cells treated for 48 h with dexamethasone were compared to see if changes in the relative abundance of the phosphorylated peptides could be determined. Because the absolute amount of protein in each experiment is not known, only relative changes in the abundances can be stated. First, the ratio of ion counts of the extracted ion chromatograms of five different peptides, which do not contain a CDK2 consensus site, from the tryptic digest of Arg-C fragment 6 of H1.4 were compared. The change in the counts between the control cells versus cells treated for 48 h with dexamethasone for each of these peptides was relatively the same (average ratio of counts of dexamethasone-treated cells/control cells = 0.9). Figure 7A shows the extracted ion chromatograms of the protonated ion of m/z 784.5 (amino acids KPAAAAGAK) from both the control cells and the 48 h dexamethasone-treated cells. The ion counts for each of these masses is 329 and 308 for the control cells and the dexamethose-treated cells, respectively. Conversely, for the tryptic peptides containing phosphorylated Ser-172 (pSPK) and phosphorylated Ser-187 (pSPAK) of Arg-C fragment 6 of H1.4, an overall decrease in the counts of the extracted ion chromatograms of these ions was observed in the 48 h dexamethasone-treated cells versus the control cells (average ratio of counts of dexamethasone-treated cells/control cells = 0.5). The extracted ion chromatograms of the protonated ion of m/z 411.2 (pSPK) from the control cells (ion count of 124) versus dexamethasone-treated cells (ion count of 71) are shown in Figure 7B. Additionally, the ion counts from the extracted ion chromatograms of ions corresponding in mass to the nonphosphorylated tryptic peptides SPK and SPAK containing the CDK consensus sites showed an overall increase in the 48 h dexamethasone-treated cells treatment versus control cells (average ratio of counts of dexamethasone-treated cells/control cells/dexamethasone-treated cells = 1.1), albeit a slight increase (data not shown). Because of the small size of some of these tryptic peptides (3 or 4 amino acids in length), the peptides were not well-retained under the chromatographic conditions used. Therefore, quantitation of these ions is difficult. Nonetheless, these observations in the relative quantitation of these data are consistent with the observed changes in the relative phosphorylation levels of the full-length proteins reported previously.19
Figure 7.
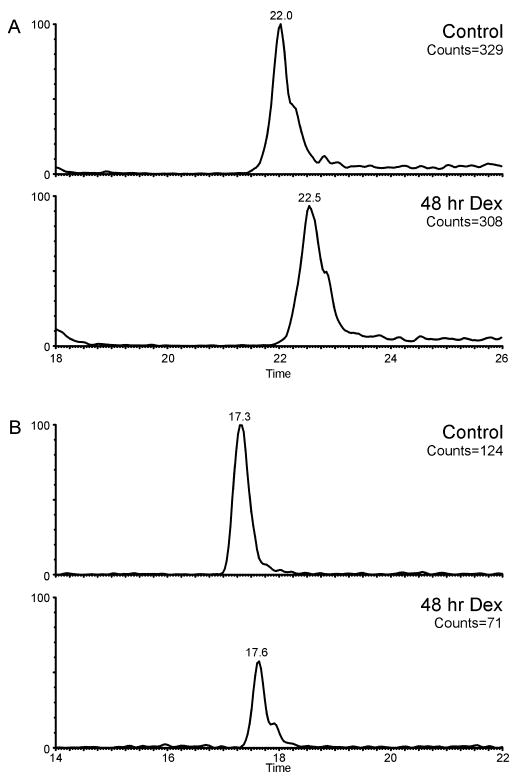
Extracted ion chromatograms of ions from the LC/MS/MS analyses of control cells and cells treated for 48 h with dexamethasone. (A) EIC of the protonated ion of m/z 784.5 (amino acids KPAAAAGAK); and (B) EIC of the protonated ion of m/z 411.2 (phosphorylated SPK). Within each panel, the extracted ion chromatograms were normalized to the ion counts from the control cells.
Phosphorylation Site-Specific Antibodies
To explore the dynamics of each H1 phosphorylation event in more detail, we generated phosphorylation site-specific antibodies to the serine and threonine residues found by MS to be phosphorylated in vivo. Up to now, the most widely used technique for assessing changes in the levels of histone modifications has been the use of Western blots with antibodies that specifically recognize the modification of interest. A significant advantage of this approach is that these antibodies can be highly sensitive. Potential disadvantages of this type of analysis are the lack of specificity of the antibody, the ability to only monitor a single modification at a time, and the modification of a residue near the site of interest may affect the specificity of the antibodies. Nonetheless, we generated polyclonal antisera to specific phosphorylation sites identified by MS/MS to accurately quantitate the changes in the levels of phosphorylation at specific residues.
Peptides corresponding to phosphorylated CDK consensus sites within both mouse H1.3 or H1.4 isoforms were used to immunize rabbits. One resulting antiserum (designated H1.4pT18 and/or 1754 antibody) recognized phosphorylated peptides corresponding to mouse H1.3 and H1.4 isoforms with phosphorylation on Thr-18, but did not recognize nonphosphorylated peptides of the same primary sequences (data not shown). The antibody was also tested by Western blot for recognition of four mutations of the mouse H1.3 isoforms (T18E, T18A, T18Q, and T18D) that were transiently expressed in UL3 cells. Each of these mutations abrogated recognition by the H1.4pT18 antibody (Figure 8A). This antiserum was then used to probe immunoblots of acid extracted H1 proteins from UL3 cells following various time hormone treatments. As shown in Figure 8B, the anti-H1.4pT18 antibody strongly recognizes a band corresponding to the H1.4 isoform in asynchronous UL3 cells not treated with hormone. Following 24 and 48 h of hormone treatment, band intensity diminished significantly to nearly undetectable levels by 48 h. After the 48 h treatment, hormone was washed away and cells were allowed to recover for 24 h, at which time band intensity returned to basal levels.
Figure 8.
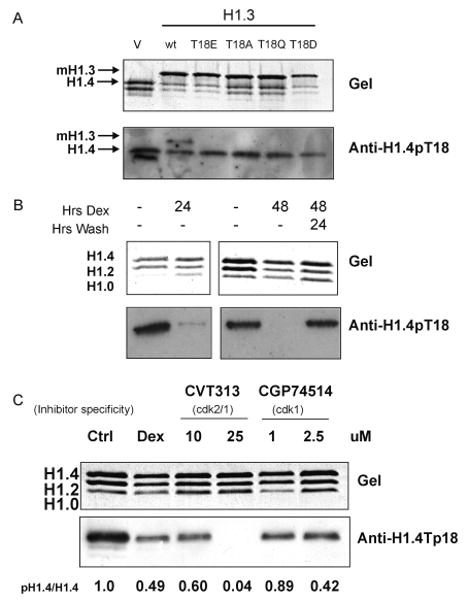
Anti-H1.4pT18 antibody used to detect hormone-associated changes in H1 phosphorylation in UL3 cells. Two micrograms of acid-extracted proteins was separated by SDS-PAGE and analyzed by Western blot using the anti-H1.4pT18 (1754) antibody. Coomassie stained gel is shown as a loading control. (A) Specificity of anti-H1.4pT18 antibody for mouse H1.4 and not for mouse H1.3 or four different mouse H1.3 mutants (T18E, T18A, T18Q, and T18D). V refers to the empty vector control. (B) UL3 cells were treated with dexamethasone as indicated. (C) Phosphorylation of H1.4T18 is reduced by inhibitors for either CDK1 or CDK2 in UL3 cells. Cells were treated with dexamethasone, CVT313, or CGP74514 at indicated concentrations for 24 h. The ratios of phosphorylated H1.4 to total H1.4 based on densitometry are shown as pH1.4/H1.4.
To determine if threonine-18 in human H1.4 is a target for either CDK1 or CDK2 in vivo, UL3 cells were treated with either CVT313 or CGP74514 at two concentrations each for 24 h, after which, Western blot analyses with the H1.4pT18 antibody were performed. Treatment of UL3 cells with dexamethasone or CVT313 showed a dose-dependent decrease in H1.4pT18 phosphorylation after 24 h relative to the phosphorylation observed in untreated cells. Treatment with the inhibitor CGP74514 also showed a decrease in H1.4pT18 phosphorylation levels relative to that observed in untreated cells, but what appears to be a lesser extent than with CVT313 treatment (Figure 8C). To quantitate the amount of total H1.4 that is phosphorylated, densitometry was used to compare each band detected by the polyclonal H1.4pT18 antibody to the corresponding H1.4 band on the coomassie-stained gel. The average relative quantitation of two independent experiments of phosphorylated H1.4 to total H1.4 (pH1.4/H1.4) was determined and is shown in Figure 8C. On the basis of densitometry, treatment of the cells with dexamethasone reduced levels of H1.4pT18 by 51% compared to untreated cells. The CDK2 inhibitor CVT313 reduced H1.4pT18 by 40% and 96% at 10 and 25 μM doses, respectively. The CDK1 inhibitor CGP74514 did not significantly reduce H1.4pT18 at 1 μM (11%), but reduced levels by 58% at the 2.5 μM concentration. For comparison, the ratio of phosphorylated isoforms relative to the total H1 isoform (normalized to untreated cells) was calculated from the MS analyses (Supplementary Table 4). Although these numbers arise from different biological samples on different days, similar results were obtained from the MS analyses and the densitometry analyses, that is, a decrease in the ratio of phosphorylated H1.4 to total H1.4 was observed following treatment with the cdk inhibitors.
The H1.4pT18 polyclonal antibody was shown to be specific for phosphorylated Thr-18 of H1.4, and because UL3 cells only express the H1.4 isoform, these antisera allowed us to track phosphorylation of H1.4 in vivo in control cells and cells treated with hormone and/or CDK inhibitors. This antibody strongly recognizes a band corresponding to the H1.4 isoform in control cells, and this band is greatly reduced following treatment of UL3 cells with dexamethasone for 24 or 48 h. The intensity of the band returns to control levels if, after the 48 h treatment, the cells are washed of the hormone and allowed to recover for 24 h.
Similarly, the antibody was useful for determining if this specific site of phosphorylation (threonine-18) in human H1.4 is a target for either CDK1 or CDK2 in vivo. Western blot analyses with the H1.4pT18 antibody on cells treated with either CVT313 or CGP74514 at two concentrations each for 24 h showed a dose-dependent decrease in H1pT18 phosphorylation after 24 h. Treatment with the inhibitor CGP74514 also showed a decrease in H1pT18 levels, but to a lesser extent than was seen with CVT313 treatment. Densitometry comparing each band detected in the Western analyses was used to quantitate the amount of total H1.4 that is phosphorylated. From this, an average relative quantitation was determined and compared to the relative quantitation levels obtained using mass spectrometry. A decrease in the relative quantitation levels of phosphorylation following CDK inhibitor treatments were detected by both the MS analyses and the Westerns with the H1.4pT18 antibody, thereby suggesting that the remaining phosphorylation seen by MS is perhaps, at least in part, due to residual T18 phosphorylation. The ability to accurately quantitate changes in the levels of specific phosphorylation sites in response to different hormone or CDK treatments is essential to fully understand the functional role of histones and/or their modifications. Although mass spectrometry is a useful tool for the quantitation of post-translational modifications of peptides, the presence of the modification can affect the ionization potential of the peptide. Recently, the use of stable isotope labeling in conjunction with mass spectrometric analyses was reported for the quantitation of changes in specific histone modifications.34 Perhaps this approach may prove to be the most useful technique for the quantitation of localized changes in histone post-translational modifications.
Conclusions
Several studies have alluded to the important influence of linker histone phosphorylation in chromatin regulation. Phosphorylation of CDK consensus S/T residues in mammalian H1s have been shown to alter mobility of H1 in chromatin, suggesting phosphorylation assists in release of H1 from chromatin.35–37 Phosphorylation of the Tetrahymena H1 on S/T residues has been shown to influence transcriptional activity by creating an electrostatic charge patch that repels contact with DNA.38,39 Purified H1 from the human HeLa cells and H5 from chicken erythrocytes have been shown to inhibit ATP-dependent chromatin remodeling activities in vitro.37,38 In the case of H5, inhibition of remodeling was abrogated by saturating phosphorylation of H5 using the cdc2 kinase.41 Furthermore, using Xenopus extract model systems, only nonphosphorylated recombinant mammalian H1 represses chromatin remodeling during DNA replication and repair.42,43 These experiments highlight the importance of phosphorylation of H1 in inhibiting the repressive function of H1 in chromatin remodeling and DNA metabolism across species. Further functional studies on these sites of phosphorylation should increase our understanding of their impact on chromatin structure and the role these specific isoforms play in MMTV activation. Moreover, the continued developments in the area of mass spectrometry should provide new insights into not only the function of proteins, but also the basic regulatory mechanisms that control cellular functions.
In summary, we have characterized global changes in the phosphorylation state of human H1 isoforms isolated from UL3 cells using mass spectrometry. In untreated cells, the H1.2 isoform is observed as both mono- and diphosphorylated, whereas the H1.4 isoform is observed as mono-, di-, and triphosphorylated. Following treatment of the cells with dexamethasone or the CDK2 inhibitor, CVT313, low levels of only the monophosphorylated forms of these proteins were observed. Treatment of the cells with the CDK1 inhibitor, CGP74514, led to a decrease in the levels of phosphorylation of both H1.2 and H1.4, albeit to a lesser extent.
Using a combination of proteolysis enzymes, peptide mapping, and MS sequencing methodologies, three sites of phosphorylation on both the mouse and human histone H1.4 isoforms have been identified. The three sites identified, Ser-187, Ser-172, and Thr-18, are all CDK2 consensus sites. Because CDK2 is often overactive in tumor cells, it is possible that resulting changes in phosphorylation of H1 could alter the way tumors respond to hormones.
Supplementary Material
Acknowledgments
The authors thank Dr. Allison Schorzman and Dr. Harriet Kinyamu for critical review of this manuscript. This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Footnotes
Supporting Information Available: Tables listing the relative percentage of each H1.2 and H1.4 isoform, the total H1.2 and H1.4 phosphorylation, and ratio of phosphorylated isoforms to total isoforms (normalized to untreated cells). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wolffe AP, Hayes JJ. Chromatin disruption and modification. Nucleic Acids Res. 1999;27:711–720. doi: 10.1093/nar/27.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 3.Hansen JC. Conformational dynamics of the chromatin fiber in solution: determinants, mechanisms, and functions. Annu Rev Biophys Biomol Struct. 2002;31:361–392. doi: 10.1146/annurev.biophys.31.101101.140858. [DOI] [PubMed] [Google Scholar]
- 4.Khochbin S. Histone H1 diversity: bridging regulatory signals to linker histone function. Gene. 2001;271:1–12. doi: 10.1016/s0378-1119(01)00495-4. [DOI] [PubMed] [Google Scholar]
- 5.Parseghian MH, Hamkalo BA. A compendium of the histone H1 family of somatic subtypes: an elusive cast of characters and their characteristics. Biochem Cell Biol. 2001;79:289–304. [PubMed] [Google Scholar]
- 6.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 7.Horn PJ, Carruthers LJ, Logie C, Hill DA, Solomon MJ, Wade PA, Imbalzano AN, Hansen JC, Peterson CL. Phosphorylation of linker histones regulates ATP-dependent chromatin remodeling enzymes. Nat Struct Biol. 2002;9:263–267. doi: 10.1038/nsb776. [DOI] [PubMed] [Google Scholar]
- 8.Ito T. Role of histone modification in chromatin dynamics. J Biochem. 2007;141:609–614. doi: 10.1093/jb/mvm091. [DOI] [PubMed] [Google Scholar]
- 9.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 10.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Meijer M, Smerdon MJ. Accessing DNA damage in chromatin: insights from transcription. BioEssays. 1999;21:596–603. doi: 10.1002/(SICI)1521-1878(199907)21:7<596::AID-BIES8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 12.Demeret C, Vassetzky Y, Mechali M. Chromatin remodeling and DNA replication: from nucleosomes to loop domains. Oncogene. 2001;20:3086–3093. doi: 10.1038/sj.onc.1204333. [DOI] [PubMed] [Google Scholar]
- 13.Urnov FD, Wolffe AP. A necessary good: nuclear hormone receptors and their chromatin templates. Mol Endocrinol. 2001;15:1–16. doi: 10.1210/mend.15.1.0589. [DOI] [PubMed] [Google Scholar]
- 14.Swedlow JR, Hirano T. The making of the mitotic chromosome: modern insights into classical questions. Mol Cell. 2003;11:557–569. doi: 10.1016/s1097-2765(03)00103-5. [DOI] [PubMed] [Google Scholar]
- 15.Lee HL, Archer TK. Prolonged glucocorticoid exposure dephosphorylates histone H1 and inactivates the MMTV promoter. EMBO J. 1998;17:1454–1466. doi: 10.1093/emboj/17.5.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhattacharjee RN, Banks GC, Trotter KW, Lee HL, Archer TK. Histone H1 phosphorylation by CDK2 selectively modulates mouse mammary tumor virus transcription through chromatin remodeling. Mol Cell Biol. 2001;21:5417–5425. doi: 10.1128/MCB.21.16.5417-5425.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Archer TK, Fryer CJ, Lee HL, Zaniewski E, Liang T, Mymryk JS. Steroid hormone receptor status defines the MMTV promoter chromatin structure in vivo. J Steroid Biochem Mol Biol. 1995;53:421–429. doi: 10.1016/0960-0760(95)00088-h. [DOI] [PubMed] [Google Scholar]
- 18.Kinyamu HK, Archer TK. Modifying chromatin to permit steroid hormone receptor-dependent transcription. Biochem Biophys Acta. 2004;1677:30–45. doi: 10.1016/j.bbaexp.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 19.Banks GC, Deterding LJ, Tomer KB, Archer TK. Hormone-mediated dephosphorylation of specific histone H1 isoforms. J Biol Chem. 2001;276:36467–36473. doi: 10.1074/jbc.M104641200. [DOI] [PubMed] [Google Scholar]
- 20.Deterding LJ, Banks GC, Tomer KB, Archer TK. Understanding global changes in histone H1 phosphorylation using mass spectrometry. Methods. 2004;33:53–58. doi: 10.1016/j.ymeth.2003.10.020. [DOI] [PubMed] [Google Scholar]
- 21.Fryer CJ, Kinyamu HK, Rogatsky I, Garabedian JJ, Archer TK. Selective activation of the glucocorticoid receptor by steroid antagonists in human breast cancer and osteosarcoma cells. J Biol Chem. 2000;275:17771–17777. doi: 10.1074/jbc.M908729199. [DOI] [PubMed] [Google Scholar]
- 22.Brooks EE, Gray NS, Joly A, Kerwar SS, Lum R, Mackman RL, Norman TC, Rosete J, Rowe M, Schow SR, Schultz PG, Wang X, Wick MM, Shiffman D. CVT-313, a specific and potent inhibitor of CDK2 that prevents neointimal proliferation. J Biol Chem. 1997;272:29207–29211. doi: 10.1074/jbc.272.46.29207. [DOI] [PubMed] [Google Scholar]
- 23.Imbach P, Capraro HG, Furet P, Mett H, Meyer T, Zimmermann J. 2,6,9,-Trisubstituted purines: optimization towards highly potent and selective CDK1 inhibitors. Bioorg Med Chem Lett. 1999;9:91–96. doi: 10.1016/s0960-894x(98)00691-x. [DOI] [PubMed] [Google Scholar]
- 24.Dai Y, Dent P, Grant S. Induction of apoptosis in human leukemia cells by the CDK1 inhibitor CGP74514A. Cell Cycle. 2002;1:143–152. [PubMed] [Google Scholar]
- 25.Yu C, Rahmani M, Dai Y, Conrad D, Krystal G, Dent P, Grant S. The lethal effects of pharmacological cyclin-dependent kinase inhibitors in human leukemia cells proceed through a phosphatidylinositol 3-kinase/akt-dependent process. Cancer Res. 2003;63:1822–1833. [PubMed] [Google Scholar]
- 26.Deterding LJ, Moseley MA, Tomer KB, Jorgenson JW. Coaxial continuous flow fast atom bombardment in conjunction with tandem mass spectrometry for the analysis of biomolecules. Anal Chem. 1989;61:2504–2511. doi: 10.1021/ac00197a011. [DOI] [PubMed] [Google Scholar]
- 27.Meergans T, Albig W, Doenecke D. Varied expression patterns of human H1 histone genes in different cell lines. DNA Cell Biol. 1997;16:1041–1049. doi: 10.1089/dna.1997.16.1041. [DOI] [PubMed] [Google Scholar]
- 28.Fu G, Ghadam P, Sirotkin A, Khochbin S, Skoultchi AI, Clarke HJ. Mouse oocytes and early embryos express multiple histone H1 subtypes. Biol Reprod. 2003;68:1569–1576. doi: 10.1095/biolreprod.102.012336. [DOI] [PubMed] [Google Scholar]
- 29.Garcia BA, Busby SA, Barber CM, Shabanowitz J, Allis CD, Hunt DF. Characterization of phosphorylation sites on histone H1 isoforms by tandem mass spectrometry. J Proteome Res. 2004;3:1219–1227. doi: 10.1021/pr0498887. [DOI] [PubMed] [Google Scholar]
- 30.Chagas da Cunha JP, Nakayasu ES, Elias MC, Pimenta DC, Tellez-Inon MT, Rojas F, Manuel M, Almeida IC, Schenkman S. Trypansoma cruzi histone H1 is phosphorylated in a typical cyclin dependent kinase site accordingly to the cell cycle. Mol Biochem Parasitol. 2005;140:75–86. doi: 10.1016/j.molbiopara.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 31.Wisniewski JR, Zougman A, Kruger S, Mann M. Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue. Mol Cell Proteomics. 2007;6:72–87. doi: 10.1074/mcp.M600255-MCP200. [DOI] [PubMed] [Google Scholar]
- 32.Chadee DN, Taylor WR, Hurta RAR, Allis CD, Wright JA, Davie JR. Increased phosphorylation of histone H1 in mouse fibroblasts transformed with oncogenes or constitutively active mitogen-activated protein kinase kinase. J Biol Chem. 1995;270:20098–20105. doi: 10.1074/jbc.270.34.20098. [DOI] [PubMed] [Google Scholar]
- 33.Sweet MT, Carlson G, Cook RG, Nelson D, Allis CD. Phosphorylation of linker histones by a protein kinase A-like activity in mitotic nuclei. J Biol Chem. 1997;272:916–923. doi: 10.1074/jbc.272.2.916. [DOI] [PubMed] [Google Scholar]
- 34.Knapp AR, Ren C, Su X, Lucas DM, Byrd JC, Freitas MA, Parthun MR. Quantitative profiling of histone post-translational modifications by stable isotope labeling. Methods. 2007;41:312–319. doi: 10.1016/j.ymeth.2006.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Contreras A, Hale TK, Stenoien DL, Rosen JM, Mancini MA, Herrera RE. The dynamic mobility of histone H1 is regulated by cyclin/CDK phosphorylation. Mol Cell Biol. 2003;23:8626–8636. doi: 10.1128/MCB.23.23.8626-8636.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dou Y, Bowen J, Liu Y, Gorovsky MA. Phosphorylation and an ATP-dependent process increase the dynamic exchange of H1 in chromatin. J Cell Biol. 2002;158:1161–1170. doi: 10.1083/jcb.200202131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hendzel MJ, Lever MA, Crawford E, Th'ng JP. The C-terminal domain is the primary determinant of histone H1 binding to chromatin in vivo. J Biol Chem. 2004;279:20028–20034. doi: 10.1074/jbc.M400070200. [DOI] [PubMed] [Google Scholar]
- 38.Dou Y, Mizzen CA, Abrams M, Allis CD, Gorovsky MA. Phosphorylation of linker histone H1 regulates gene expression in vivo by mimicking H1 removal. Mol Cell. 1999;4:641–647. doi: 10.1016/s1097-2765(00)80215-4. [DOI] [PubMed] [Google Scholar]
- 39.Dou Y, Gorovsky MA. Phosphorylation of linker histone H1 regulates gene expression in vivo by creating a charge patch. Mol Cell. 2000;6:225–231. doi: 10.1016/s1097-2765(00)00024-1. [DOI] [PubMed] [Google Scholar]
- 40.Hill DA, Imbalzano AN. Human SWI/SNF nucleosome remodeling activity is partially inhibited by linker histone H1. Biochemistry. 2000;39:11649–11656. doi: 10.1021/bi001330z. [DOI] [PubMed] [Google Scholar]
- 41.Horn PJ, Carruthers LM, Logie C, Hill DA, Solomon MJ, Wade PA, Imbalzano AN, Hansen JC, Peterson CL. Phosphorylation of linker histones regulates ATP-dependent chromatin remodeling enzymes. Nat Struct Biol. 2002;9:263–267. doi: 10.1038/nsb776. [DOI] [PubMed] [Google Scholar]
- 42.De S, Brown DT, Lu ZH, Leno GH, Wellman SE, Sittman DB. Histone H1 variants differentially inhibit DNA replication through an affinity for chromatin mediated by their carboxyl-terminal domains. Gene. 2002;292:173–181. doi: 10.1016/s0378-1119(02)00675-3. [DOI] [PubMed] [Google Scholar]
- 43.Lu ZH, Sittman DB, Brown DT, Munshi R, Leno GH. Histone H1 modulates DNA replication through multiple pathways in Xenopus egg extract. J Cell Sci. 1997;110:2745–2758. doi: 10.1242/jcs.110.21.2745. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.