

Abstract
Patients with Down syndrome (DS) display a unique spectrum of malignancies, with a 10 to 20-fold increased risk of acute leukemias, and a markedly decreased incidence of solid tumors. This review discusses current understanding of the basis for this distinctive pattern of cancer incidence, and the clinical and biologic features of the malignant disorders most frequent in DS: transient myeloproliferative disease, acute megakaryoblastic leukemia, and acute lymphoblastic leukemia. We also review distinctive pharmacogenetic issues, highlighting the differential chemosensitivity and toxicity profiles of DS patients compared to the general population; and epidemiologic studies of protective and adverse environmental risk factors for development of leukemia.
Keywords: Down Syndrome; Lymphoblastic Leukemia, Acute; Megakaryoblastic Leukemia, Acute; Myeloproliferative Disorders; GATA1 Transcription Factor; Janus Kinase 2
Introduction
The spectrum of malignancies in patients with Down syndrome (DS) offers both insights and enigmas. Patients with DS exhibit a unique pattern of malignancies, yielding intriguing insights into cancer biology. These patients also pose distinctive challenges to the oncologist due to their particular profile of treatment-related toxicities. Patients with DS have an excess risk of leukemia, experiencing three distinct disease entities (transient myeloproliferative disorder, acute megakaryoblastic leukemia, and acute lymphoblastic leukemia), and have a reduced risk of solid tumors. This review highlights the epidemiology of cancer in DS; clinical features and biology of the leukemias that are most common in DS; and pharmacogenetic considerations specific to this population.
Epidemiology of Cancer in DS
DS results from trisomy 21 and occurs in approximately 1 in 1,000 births, a number which has decreased over the past several decades due to increased screening and termination of DS pregnancies.[1] While the standardized incidence ratio of cancer in DS does not differ significantly from that of the general population, the distribution of malignancies is strikingly different.[2] In a review of Danish registry data on 2814 individuals with DS, leukemias constituted 60% of malignancies overall, and 97% in patients under 15 years of age. Solid tumors, in contrast, are markedly less frequent in DS individuals across all age groups, with the possible exception of retinoblastoma and germ cell tumors.[2, 3]
While significant advances have occurred in management of the diseases associated with DS, the median age at death remains considerably lower than that of the general population, reported as 49 years in a 1997 study.[4] The most frequent causes of death are non-malignant. Nevertheless, leukemia remains a significant cause of death, particularly in DS children under age 10, who are more than three times more likely to have leukemia listed as a cause of death than are children without DS.[4] Death from malignancies other than leukemia is strikingly less common in DS at all ages.[4]
The first report of leukemia in a DS patient occurred in 1930,[5] and the first systematic study in 1957.[6] Contemporary studies indicate that patients with DS have a 10-20 fold increased relative risk of leukemia, with a cumulative risk of 2% by age 5 and 2.7% by age 30.[2] They constitute approximately 2% of all pediatric ALL, and approximately 10% of pediatric AML. In addition to increased frequency, leukemias in DS also differ in clinical features, timing of occurrence, and response to therapy. Approximately 10% of DS infants exhibit an unusual myelodysplastic disorder known as transient myeloproliferative disorder (TMD). TMD spontaneously regresses, but approximately 20% of these patients later develop AML. AML occurs at a considerably earlier age in DS (median 1.8 versus 7.5 years, and the majority under 5 years),[7] whereas ALL shows a similar age distribution to that in children without DS.[8] Due to the disproportionate frequency of DS AML in under 5-year-olds, the overall ratio of AML to ALL is roughly equal in children with DS, whereas it is approximately 1:4 in children without DS. In DS children over age five, when the incidence of AML declines, the number of ALL cases and ratio of AML:ALL more closely resembles that of the general population. About 70-85% of DS AML cases are acute megakaryocytic leukemia (AMKL), a rare subtype in patients without DS.[7, 9] The increased risk of AMKL in DS children is 500-fold.[10] Additional clinical and biological aspects of leukemias in DS are discussed below.
Epidemiologic Risk Factors for Leukemia in DS
While much investigation in patients with DS has focused on the role of genetic factors related to trisomy 21 in the development of leukemia, another approach has been to examine the role of environmental exposures in triggering leukemogenesis in DS. There is some literature investigating environmental exposures associated with adverse or protective effects. Parental exposures investigated include alcohol intake;[11] household chemical exposures;[12] electromagnetic fields;[13] reproductive history;[14] periconceptional vitamin supplementation.[15] Patient exposures include vitamin supplementation;[16] ionizing radiation;[17] and frequent early childhood infections.[18] The studies cited are retrospective case-control studies based on parental self-report, which will require confirmation using additional methodologies. Moreover, the magnitude of the reported effect sizes are generally small, suggesting that as in leukemia in patients without DS, genetic or as yet unidentified environmental factors must also play a significant role.
Pathogenesis of Malignancies in DS
Why are leukemias more common in DS?
There are an estimated 329 genes localized to the long arm of chromosome 21,[1] and several have been suggested as possible mediators of leukemogenesis through increased dosage effects. These include FPDMM (the cause of autosomal dominant familial platelet disorder), AML1, the IFN-α/β receptor (IFNAR), CRF2-4 (cytokine family 2-4), and phosphoribosylglycinamide formyltransferase (reviewed in [19]). Alternatively, altered folate metabolism in patients with DS may contribute to leukemogenesis by changing methylation and rates of DNA mutation. Two lines of evidence suggest altered folate metabolism in DS.[20] First, mothers of DS children have a higher incidence of polymorphisms associated with reduced activity of the methyltetrahydrofolate reductase (MTHFR) enzyme,[21] and the resultant in utero folate deficiency occurring during these women’s pregnancies may be a risk factor for both DS and development of ALL.[22] Second, increased dosage of the cystathionine β-synthase (CBS) gene on chromosome 21 results in increased CBS expression, which also causes alterations in the folate pathway. Other possible explanations include a general increase in genetic instability caused by trisomy 21 facilitating occurrence of leukemogenic mutations; and disomic homozygosity of a mutated tumor suppressor on chromosome 21.[20]
Why are solid tumors less common in DS?
The basis for the decreased incidence of solid tumors in DS remains uncertain. In a recent elegant work by Sussan and colleagues, mouse models of DS were used to dissect out the protective effect of the DS genetic background against development of intestinal tumors.[23] Ts65Dn mice, which are trisomic for mouse orthologues of about half the human chromosome 21 genes, were crossed to ApcMin mice, which are predisposed to develop intestinal tumors similar to those in familial adenomatous polyposis. The Ts65Dn- ApcMin mice developed significantly fewer intestinal tumors. Further transgenic crosses demonstrated that trisomy of just 33 orthologous genes was protective, and monosomy of those 33 genes led to increased tumors. Additional studies pinpointed the ETS2 gene as largely responsible for both dosage effects, with a decreased number of intestinal tumors occurring with increased ETS2 dosage and an increased number with decreased dosage. Copper-zinc superoxide dismutase is also a candidate tumor suppressor, since its location on chromosome 21 leads to increased dosage.
Another protein implicated as protective against development of solid tumors in DS is endostatin, a cleavage product of collagen XVIII encoded by the COL18A1 gene on chromosome 21, which is present in the serum at higher levels in DS.[24] Since endostatin has been shown to be a potent inhibitor of angiogenesis in many solid tumors, Zorick and colleagues hypothesize that higher levels of endostatin in DS may be responsible for the lower incidence of solid tumors. Finally, environmental factors may play a role, since DS individuals have less likelihood of tobacco and alcohol use and occupational carcinogen exposures.
Myeloid Disorders in DS
Transient Myeloproliferative Disorder (TMD)
Clinical presentation, treatment and outcomes
TMD is an intriguing disorder unique to DS, which occurs in approximately 10% of DS infants. Symptoms range from asymptomatic leukocytosis to massive organomegaly and fatal liver and/or respiratory failure. TMD is most often incidentally diagnosed in a well-appearing DS infant, although it can present with a clinical picture resembling leukemia, with expansion of a clonal population of blasts causing leukocytosis, hepatosplenomegaly, effusions, and liver fibrosis. It is also an occasional cause of stillbirth. TMD exhibits megakaryoblastic morphology and immunophenotype indistinguishable from AMKL (Figure 1), but the natural history of TMD is one of spontaneous regression over several months. Mild cases do not require treatment, whereas symptomatic cases may require supportive care measures and occasionally chemotherapy. Although spontaneous regression occurs, about 20% of patients with TMD later develop true AMKL, generally by age five years.
Figure 1. Bone marrow aspirate and biopsy findings in acute megakaryoblastic leukemia (AMKL).
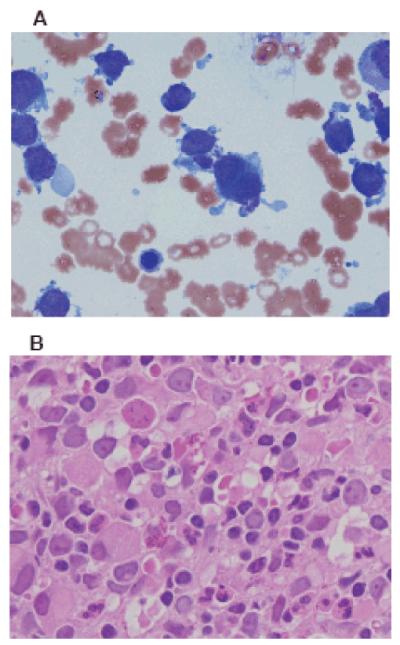
A) Bone marrow aspirate demonstrating megakaryoblast clumping and abundant cytoplasm with budding projections. Wright-Giemsa stain, 100x magnification. Courtesy of Dr. Donald Mahoney. B) Bone marrow biopsy demonstrating clusters of megakaryoblasts, atypical megakaryocytes, megakaryocytic differentiation, and fibrosis. H&E stain, 400x magnification. Courtesy of Dr. Andrea Sheehan.
In the largest prospective evaluation of TMD to date, Klusmann and colleagues reported on 146 children registered in AML-BFM study group trials from 1993-2006.[25] They reported a 5-year EFS of 63 ±4% and overall survival of 85 ±3%. Early death occurred in 15% and development of AMKL in 23%, similar to an earlier Pediatric Oncology Group (POG) study of 47 DS neonates with TMD.[19] Ascites and bleeding diathesis were associated with early death in both studies; additional risk factors in the AML-BFM study were leukocytosis and preterm delivery. Importantly, the AML-BFM study suggested that low-dose cytarabine (0.5-1.5 mg/kg for 3-12 days) improved outcome in patients with clinically significant leukocytosis, thrombocytopenia, cholestasis, or liver dysfunction. Among children at risk for early death, 5-year EFS was superior in those receiving treatment versus not (52 ±12% versus 28 ±11%). An AML-BFM chemoprevention trial is currently underway in TMD with low-dose cytarabine and minimal residual disease monitoring using GATA1s (see “GATA1s and the Pathogenesis of DS TMD and AMKL,” below), based on the hypothesis that eradication of the GATA1s clone may prevent development of AMKL. Successful detection of GATA1 mutations using real-time quantitative PCR has been demonstrated, although it is complex since mutations are case-specific.[26]
Acute Myeloid Leukemia (AML)
Clinical presentation, treatment and outcomes
The clinical hallmarks of DS AML are the megakaryoblastic phenotype in the majority of cases (Figure 1), and markedly superior outcomes compared to non-DS AML (Table 1). DS patients generally have a low initial white blood cell count, no central nervous system (CNS) involvement, and fewer cytogenetic abnormalities than non-DS AML.
Table 1.
Event-free survival in cooperative group studies in DS AML.
| Study protocol (years) | n (DS) | EFS (DS) | EFS (non-DS) | Comments | Ref. |
|---|---|---|---|---|---|
| AML-BFM 98 (1998-2003) | 66 | 89% ±4% (3-yr) | 53% ±2% (p=0.0001) | [9] | |
| CCG 2891 (1989-1999) | 161 | 77% (6-yr) | 33% (p<0.0001) | [7] | |
| Japan (1987-1997) | 33 | 80% ±7% | DS protocol | All <4yo | [34] |
| Japan (2000-2004) | 72 | 83.3% ±9.1% (4-yr) | DS pr otocol | 70/72 <4yo | [35] |
| MRC AML 10 (1987-1995) | 23 | 70% ±9.6% (5-yr) | 59% ±4.6% (p=0.6) | OS, not EFS | [89] |
| NOPHO 88 (1988-1992) | 15 | 47% (not given) | [32] | ||
| NOPHO 93 (1993-2002) | 37 | 81% ±6% (5-yr) | [90] | ||
| POG 8498 (1984-1989) | 12 | 100% (4-yr) | 33% (p=0.0001) | [27] | |
| POG 8821 (1988-1993) | 27 | 77% ±2.1% (3-yr) | 34% ±2.5% | [91] | |
| POG 9421 (1995-1999) | 57 | 76.9% (5-yr) | 51.9% (p<0.001) | [74] | |
| Toronto (1990-2003) | 18 | 67% ±11% (5-yr) | DS protocol | [36] |
AMKL in DS patients is distinguished by an unusually favorable clinical prognosis, first recognized by POG over a decade ago, and confirmed by the Nordic Society for Pediatric Hematology and Oncology and the Children’s Cancer Group (CCG).[7, 27, 28] Current event-free survival (EFS) for DS AMKL is about 80%, whereas EFS in the rarer subgroup of non-DS AMKL is considerably worse than other AML subtypes at 15-20%.[29] Young age is an important positive prognostic factor in DS AML. The prognostic significance of age may be due to biology, since nearly all DS AMKL occurs in children under 5 years of age and it is this AML subtype that has a favorable outcome. Virtually none of the few AML cases occurring in older DS children are AMKL, and they show no significant survival advantage.[7]
Although DS AML patients generally fare better than other AML subgroups, they suffered increased treatment-related mortality on several intensive AML treatment regimens (reviewed in [30]). Intermediate-intensity therapy maximizes their chance of cure without undue toxicity.[9, 31, 32] The CCG AML trial 2891 included randomization between standard and intensively timed chemotherapy. Children with DS had a higher toxic mortality rate with intensive timing (32%, versus 11% in the non-DS population) [33], and lower post-remission disease-free survival (DFS) when randomized to allogeneic bone marrow transplant (BMT) or autologous BMT compared to chemotherapy, with DFS of 33%, 67%, and 89% respectively.[33] The subsequent COG trial A2971 therefore pursued dose reductions (reviewed in [30]). Recent Japanese protocols have eliminated CNS prophylaxis due to the rarity of CNS involvement in DS AML; the most recent protocol yielded a favorable 4-year EFS of 83 ±9% and had no isolated CNS relapses.[34, 35] Finally, Al-Ahmari and colleagues in Toronto have employed an “ultra-low” therapy in DS AMKL consisting of very low dose cytarabine (10mg/m2/dose), vincristine, and retinyl palmitate, and found no significant difference in outcome compared to standard chemotherapy.[36] While this study was small, retrospective, and non-randomized, DS AMKL sensitivity to ultra-low dose therapy merits further study.
Current DS AML protocols underway in Europe and the U.S. employ a similar theme of therapy reductions to decrease toxicities. The European BFM trial utilizes a standard BFM regimen with cytarabine and anthracycline dose reductions.[37] The U.S. trial, open only to DS children under four years, features high-dose cytarabine, reduced anthracycline, and reduced intrathecal therapy. Both trials eliminate maintenance therapy, cranial irradiation, and stem cell transplantation.
Gata1s and the Pathogenesis of DS TMD and AMKL
A breakthrough in understanding TMD and AMKL occurred with the discovery that the hematopoietic transcription factor GATA1, on the X chromosome, is mutated in these disorders.[38, 39] GATA1 mutations are specific to DS TMD and AMKL.[38] The only instances of GATA1 mutations in patients without DS are in patients with acquired trisomy 21 in their hematopoietic progenitors, or in patients with constitutional mosaicism for trisomy 21.
GATA1 mutations are acquired and case-specific, including insertions, deletions, missense, nonsense, and splice site mutations. Nearly all occur in exon 2 and lead to production of a truncated protein of 40 rather than 50 kDa, called GATA1short or GATA1s. Whereas complete deletion of GATA1 is embryonically lethal, GATA1s causes dysregulation of megakaryopoiesis. The GATA1 mutation present in TMD is generally the same as that associated with a given patient’s subsequent AMKL, although AMKL often exhibits additional cytogenetic abnormalities, suggesting that TMD is a preleukemic condition which originates in utero and subsequently evolves into AMKL via additional genetic events (see “Cooperating genetic events,” below). Because TMD and AMKL appear to arise from the same clone, it has been proposed that they be classified as a single discrete entity, “myeloid leukemia of Down syndrome,” in the World Health Organization classification.[40] Additional evidence suggesting that GATA1 mutations arise in utero includes detection of mutations in blood cells from neonatal screening cards[41] and in blasts from identical twins with acquired trisomy 21, presumably due to transfer of cells bearing the mutation from one twin to the other via shared circulation in utero.[42]
Effects of GATA1 and GATA1s in hematopoiesis
GATA1 regulates maturation of megakaryocytes, erythroid cells, mast cells, and eosinophils. GATA1s promotes abnormal proliferation of megakaryocyte progenitor cells in the yolk sac and fetal liver.[43] A family carrying a germline mutation generating GATA1s provided a fortuitous demonstration that GATA1s is not leukemogenic in the absence of trisomy 21: mutation carriers had anemia, thrombocytopenia, and neutropenia but not leukemia.[44] GATA1s knock-in experiments in mouse models also indicate that the mutation alone is not sufficient to cause leukemia.[43] Some in vitro data suggest that GATA1 deficiency also contributes to leukemogenesis.[45] Thus, both loss of wild-type GATA1 and expression of mutant GATA1s appear necessary for development of DS AMKL. Importantly, restoration of wild-type GATA1 in a DS AMKL cell line led to erythroid differentiation.[46]
Cooperating genetic events
Since GATA1 appears necessary but not sufficient for development of AMKL, other as yet unidentified cooperating genetic events must contribute to leukemogenesis. In the largest study to date of gene expression differences between DS and non-DS AMKL, Bourquin et al found that DS AMKL and non-DS AMKL formed distinct gene expression clusters.[47] Two smaller gene expression studies identified genes with differential expression in TMD compared to DS AMKL,[48, 49] but these findings were not confirmed by Bourquin et al. Unexpectedly, Bourquin et al. found that RUNX1 showed decreased expression in DS AMKL despite the increased dosage due to chromosome 21 location. Decreased RUNX1 expression occurs in other leukemias and may contribute to leukemogenesis in DS AMKL as well. They did identify several chromosome 21 genes with increased expression in DS AMKL, including BACH1 (a repressor of megakaryopoiesis and possible GATA1 target) and SON (a MYC homologue). Walters et al identified activation mutations of JAK3 in a small subset of DS AMKL cases and provided mechanistic evidence that these mutations have potential to effect malignant transformation.[50] However, JAK3 mutations have been detected in only a handful of DS AMKL cases.[50-53] Other genes with important roles in non-DS AML such as FLT3, KIT, and c-MPL have been found to be mutated in some DS cases in one recent study[54] but not in others.[51, 53] Thus, the crucial genetic events controlling the evolution of TMD into AMKL remain uncertain.
Acute Lymphoblastic Leukemia (ALL)
Epidemiology and molecular genetics of DS ALL
While DS ALL is not a unique disease entity as are DS AMKL and TMD, it differs importantly from non-DS ALL. The incidence of DS ALL follows roughly the same age peak and range as in the general pediatric population, with the exception that almost no cases occur in infants under one year. The immunophenotype and cytogenetics of DS ALL are also distinctive.[55-61] T-cell and mature B-cell (Burkitt’s) ALL are virtually absent in DS, and recurrent chromosomal abnormalities are much less common, including hyperdiploidy, TEL-AML1, E2A-PBX1, Philadelphia chromosome, and MLL rearrangements. The frequency of TEL-AML1 and hyperdiploidy, the most common lesions in non-DS ALL, is a subject of current debate. One recent study of 215 DS ALL cases reported frequencies of approximately 10% for each.[62] However, a recent POG review of 80 cases found comparable incidence of hyperdiploidy (7.7% with trisomy 4 and 10), but only 2.5% incidence of TEL-AML1,[63] and a review of cases from the Italian Association of Pediatric Hematology and Oncology (AIEOP) likewise identified only one case in 44 (2.2%) with TEL-AML1.[61] Cytogenetic features observed with increased frequency in DS ALL include +X, t(8;14)(q11;q32), and del(9p).[62] That DS ALL has distinctive biologic features is supported by the recent discovery of activating somatic JAK2 mutations in approximately 20% of DS ALL, apparently specific to this subgroup.[64] This notable discovery is the first evidence of an event occurring uniquely in DS which may play a role in development of ALL. Nearly all the JAK2 point mutations in DS ALL occur at a common site, arginine 683, distinct from the V617F site commonly mutated in polycythemia vera and myeloproliferative disorders.[65]
Outcomes in DS ALL
Overall survival has been 10-20% lower in DS compared to non-DS ALL patients on most protocols worldwide. However, several of the most recent reports suggest that DS and non-DS outcomes are comparable when favorable risk patients are excluded from analysis, since favorable risk patients are underrepresented in DS ALL.[59, 60, 66] Table 2 summarizes outcomes for DS ALL in recent major series.
Table 2.
Event-free survival in cooperative group studies in DS ALL.
| Study protocol (years) | n (DS) | EFS (DS) | EFS (non-DS) | Comments | Ref. |
|---|---|---|---|---|---|
| BFM (1981-1995) | 61 | 58% ±8% (6-yr) | 70 ±1% (p=0.14) | 3.3% induction deaths; 6.6% “treatment deaths” |
Dordelmann et al. [56] |
| CCG (1989-1995) | 179 | 68.1% ±3.9% (10-yr) | 77.2 ±0.5% (p<0.001) |
3% induction deaths | Whitlock et al. [60] |
| CCG 1961 (1996-2002) | 51 | 69.1% ±8.4% (5-yr) | 70.4 ± 1.5% | High-risk patients only; 8.7% induction deaths |
Hastings et al. [66] |
| CCG 1952 (1996-2000) | 59 | 79.6% | 84.3% (p=0.04) | Stand.-risk patients only; increased hospitalizations |
Bassal et al. [59] |
| MRC UKALL X, XI (1985-97) |
55 | 53% | 63% (p=0.1) | 11% remission deaths | Chessells et al. [57] |
| NOPHO (1984-2001) | 64 | 51% ±7% (10-yr) | 70 ± 1% | 3.1% remission deaths | Zeller et al. [58] |
| POG & St Jude (1979-92) | 37 | 65% ±14.5% (4-yr) | 74 ± 1.6% (p=0.21) | Pui et al. [55] |
Although DS survival may be comparable on modern protocols, treatment-related toxicities occur with greater frequency and severity (reviewed in [67]). The most common toxicities in DS patients are infection, mucositis, and hyperglycemia. The current front-line COG standard- and high-risk ALL protocols were temporarily suspended to DS patients in 2005 due to excess deaths related to infection. Protocol amendments included replacement of dexamethasone with prednisone in the high-risk induction, substitution of discontinuous for continuous dexamethasone during delayed intensification, leucovorin rescue at 48 hours following intrathecal methotrexate, and increased supportive care measures including consideration of hospitalization in times of neutropenia. These modifications resulted in no further deaths among DS children on the standard-risk protocol,[68] but the high-risk protocol was subsequently closed to enrollment of DS children, due to continued excess deaths (personal communication, Eric Larsen). These grim events highlight the difficulty of effecting improvements in survival of children with DS ALL since improvements in disease control through intensification of therapy may come at the cost of increased treatment-related morbidity and mortality. New approaches are wanted for improving treatment of ALL in this group of patients, which take into account differences in disease biology and host response to therapy.
Pharmacogenetics in DS
DS AML
The favorable prognosis of DS AMKL may in part be attributable to the increased (2- to23-fold) sensitivity of DS AMKL blasts to antileukemic agents including ara-C, anthracyclines, and etoposide.[68, 69] Taub and colleagues have provided evidence for the mechanisms underlying this increased sensitivity.[69, 70] The increased sensitivity to ara-C has been hypothesized to be due to increased dosage of at least two genes localized to chromosome 21: cystathionine β-synthetase (CBS) and copper/zinc superoxide dismutase (SOD). CBS has effects on the reduced folate pathway leading to increased activation of ara-C to the active intracellular metabolite ara-CTP, and decreased competition with ara-C for incorporation into DNA (reviewed in [71]). DS AMKL cells also exhibit lower levels of cytidine deaminase (CDA), the enzyme responsible for ara-C degradation, apparently due to inhibitory effects of GATA1s on the CDA promoter, thus providing a mechanistic link between GATA1 mutations and enhanced ara-C sensitivity in DS AMKL.[72] Another factor leading to enhanced chemosensitivity in DS AMKL may be the increased dosage of the SOD gene on chromosome 21. SOD increases the generation of hydroxyl free radicals, causing increased susceptibility of DS cells to apoptosis, which may enhance chemosensitivity, particularly for anthracyclines.[69]
Cardiotoxicity is an adverse effect of serious concern in DS, particularly in AML where anthracyclines are an integral element of most treatment protocols. Krischer and colleagues reported a 3.4-fold relative risk of anthracycline-related cardiotoxicity in patients with DS.[73] O’Brien and colleagues recently reported that an alarmingly high 17.5% of children with DS AML treated on POG protocol 9421 developed symptomatic cardiomyopathy, and three died of congestive heart failure.[74] CHD was not found to be a risk factor for cardiomyopathy. The high cumulative anthracycline dosage on this protocol (535 mg/m2) likely contributed to the high incidence of cardiomyopathy, as well as increased host sensitivity to oxidative stress.[75] A recent BFM review of cardiotoxicity reported a much lower incidence of cardiomyopathy in approximately 4% of DS AML patients, which was comparable to that observed in non-DS patients with de novo AML.[76] Potential factors contributing to the low rate of cardiotoxicity of the BFM protocols include a one-third dose reduction of anthracycline therapy in DS patients (yielding a cumulative dose of 200-300 mg/m2); use of anthracyclines associated with less risk of cardiotoxicity (idarubicin and liposomal daunorubicin); and use of continuous infusion dosing schedules yielding lower peak levels.
DS ALL
Unlike DS AMKL, DS ALL blasts do not demonstrate enhanced chemosensitivity with conventional ALL chemotherapeutic agents.[68, 77] In DS ALL, the principal areas of pharmacogenetic research to date have been ara-C (discussed above) and methotrexate, which has long been noted to cause increased toxicity in DS patients.[78-81] This is likely due to an extra copy of the reduced folate carrier gene on chromosome 21, which is responsible for intracellular transport of methotrexate, leading to higher intracellular methotrexate levels at a given dose level compared to non-DS patients, and hence both increased leukemic sensitivity and increased somatic toxicity to the host.[71, 79, 82] A recent study of vincristine pharmacokinetics indicated no significant differences in DS children.[83] Pharmacokinetic and pharmacodynamic studies of other ALL chemotherapeutic agents in DS patients are lacking. An in vitro study of effects of several ALL agents on DS cells did not demonstrate excessive cytotoxicity, although in vivo effects could differ.[84]
The toxicity of greatest concern in DS ALL is infection, but it remains unclear which chemotherapeutic agent or agents are most responsible. Concern regarding dexamethasone, which is known for its potent immunosuppression, prompted recent COG study amendments for DS patients mandating prednisone in HR induction steroid randomization and discontinuous timing of dexamethasone in delayed intensification. Underlying deficits in the DS host immune system are another element that likely make an important contribution to increased infectious complications.[85-88]
Conclusion
DS exhibits a unique profile of malignancies, with differences in disease incidence, biology, and response to treatment. The study of cancer predisposition syndromes has yielded many important insights in cancer biology, from Li-Fraumeni syndrome and p53 to familial retinoblastoma and Rb. The recent discoveries of the roles of GATA1 in AMKL and JAK2 in ALL are important breakthroughs, but unsolved puzzles remain. In TMD and AMKL, it remains to determine the events subsequent to GATA1 mutation that cause malignant transformation in a subset of TMD cases; and to translate our current understanding of GATA1 into further clinical advances. In ALL, the challenge remains to determine the alternative events associated with leukemogenesis in the four-fifths of cases without JAK2 mutations; and to devise treatment strategies with less frequent and severe toxicities than those in current practice. As oncologists increasingly tailor their treatment in this era of personalized medicine, patients with DS require recognition and further study due to the unique aspects of malignancies in this genetic context.
Acknowledgement
This work was supported by a National Institutes of Health Pediatric Oncology Clinical Research Training Grant (Grant CA90433-06, PI David Poplack) and in part by 1 U10 CA098543-01 (PI Gregory Reaman).
Footnotes
Disclaimers: None
- Identify malignancies for which children with Down syndrome are at increased and decreased risk.
- Discuss the clinical and biologic features of transient myeloproliferative disease and acute megakaryoblastic leukemia in children with DS.
- Discuss the clinical and biologic features of acute lymphoblastic leukemia in children with DS.
References
- [1].Roizen NJ, Patterson D. Down’s syndrome. Lancet. 2003 Apr 12;361(9365):1281–1289. doi: 10.1016/S0140-6736(03)12987-X. [DOI] [PubMed] [Google Scholar]
- [2].Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet. 2000 Jan 15;355(9199):165–169. doi: 10.1016/S0140-6736(99)05264-2. [DOI] [PubMed] [Google Scholar]
- [3].Patja K, Pukkala E, Sund R, et al. Cancer incidence of persons with Down syndrome in Finland: a population-based study. Int J Cancer. 2006 Apr 1;118(7):1769–1772. doi: 10.1002/ijc.21518. [DOI] [PubMed] [Google Scholar]
- [4].Yang Q, Rasmussen SA, Friedman JM. Mortality associated with Down’s syndrome in the USA from 1983 to 1997: a population-based study. Lancet. 2002 Mar 23;359(9311):1019–1025. doi: 10.1016/s0140-6736(02)08092-3. [DOI] [PubMed] [Google Scholar]
- [5].Brewster HF, Cannon HE. Acute lymphatic leukemia: Report of a case in eleventh month mongolina idiot. New Orleans Med Surg J. 1930;82:872–873. [Google Scholar]
- [6].KRIVIT W, GOOD RA. Simultaneous occurrence of mongolism and leukemia; report of a nationwide survey. AMA J Dis Child. 1957 Sep;94(3):289–293. doi: 10.1001/archpedi.1957.04030040075012. [DOI] [PubMed] [Google Scholar]
- [7].Gamis AS, Woods WG, Alonzo TA, et al. Increased age at diagnosis has a significantly negative effect on outcome in children with Down syndrome and acute myeloid leukemia: a report from the Children’s Cancer Group Study 2891. J Clin Oncol. 2003 Sep 15;21(18):3415–3422. doi: 10.1200/JCO.2003.08.060. [DOI] [PubMed] [Google Scholar]
- [8].Hitzler JK. Acute megakaryoblastic leukemia in Down syndrome. Pediatr Blood Cancer. 2007 Dec;49(7 Suppl):1066–1069. doi: 10.1002/pbc.21353. [DOI] [PubMed] [Google Scholar]
- [9].Creutzig U, Reinhardt D, Diekamp S, et al. AML patients with Down syndrome have a high cure rate with AML-BFM therapy with reduced dose intensity. Leukemia. 2005 Aug;19(8):1355–1360. doi: 10.1038/sj.leu.2403814. [DOI] [PubMed] [Google Scholar]
- [10].Zipursky A, Thorner P, De HE, et al. Myelodysplasia and acute megakaryoblastic leukemia in Down’s syndrome. Leuk Res. 1994 Mar;18(3):163–171. doi: 10.1016/0145-2126(94)90111-2. [DOI] [PubMed] [Google Scholar]
- [11].Mejia-Arangure JM, Fajardo-Gutierrez A, Flores-Aguilar H, et al. Environmental factors contributing to the development of childhood leukemia in children with Down’s syndrome. Leukemia. 2003 Sep;17(9):1905–1907. doi: 10.1038/sj.leu.2403047. [DOI] [PubMed] [Google Scholar]
- [12].Alderton LE, Spector LG, Blair CK, et al. Child and maternal household chemical exposure and the risk of acute leukemia in children with Down’s syndrome: a report from the Children’s Oncology Group. Am J Epidemiol. 2006 Aug 1;164(3):212–221. doi: 10.1093/aje/kwj203. [DOI] [PubMed] [Google Scholar]
- [13].Mejia-Arangure JM, Fajardo-Gutierrez A, Perez-Saldivar ML, et al. Magnetic fields and acute leukemia in children with Down syndrome. Epidemiology. 2007 Jan;18(1):158–161. doi: 10.1097/01.ede.0000248186.31452.be. [DOI] [PubMed] [Google Scholar]
- [14].Puumala SE, Ross JA, Olshan AF, et al. Reproductive history, infertility treatment, and the risk of acute leukemia in children with down syndrome: a report from the Children’s Oncology Group. Cancer. 2007 Nov 1;110(9):2067–2074. doi: 10.1002/cncr.23025. [DOI] [PubMed] [Google Scholar]
- [15].Ross JA, Blair CK, Olshan AF, et al. Periconceptional vitamin useand leukemia risk in children with Down syndrome: a Children’s Oncology Group study. Cancer. 2005 Jul 15;104(2):405–410. doi: 10.1002/cncr.21171. [DOI] [PubMed] [Google Scholar]
- [16].Blair CK, Roesler M, Xie Y, et al. Vitamin supplement use among children with Down’s syndrome and risk of leukaemia: a Children’s Oncology Group (COG) study. Paediatr Perinat Epidemiol. 2008 May;22(3):288–295. doi: 10.1111/j.1365-3016.2008.00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Linabery AM, Olshan AF, Gamis AS, et al. Exposure to medical test irradiation and acute leukemia among children with Down syndrome: a report from the Children’s Oncology Group. Pediatrics. 2006 Nov;118(5):e1499–e1508. doi: 10.1542/peds.2006-0644. [DOI] [PubMed] [Google Scholar]
- [18].Canfield KN, Spector LG, Robison LL, et al. Childhood and maternal infections and risk of acute leukaemia in children with Down syndrome: a report from the Children’s Oncology Group. Br J Cancer. 2004 Nov 29;91(11):1866–1872. doi: 10.1038/sj.bjc.6602223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Massey GV, Zipursky A, Chang MN, et al. A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children’s Oncology Group (COG) study POG-9481. Blood. 2006 Jun 15;107(12):4606–4613. doi: 10.1182/blood-2005-06-2448. [DOI] [PubMed] [Google Scholar]
- [20].Gurbuxani S, Vyas P, Crispino JD. Recent insights into the mechanisms of myeloid leukemogenesis in Down syndrome. Blood. 2004 Jan 15;103(2):399–406. doi: 10.1182/blood-2003-05-1556. [DOI] [PubMed] [Google Scholar]
- [21].James SJ, Pogribna M, Pogribny IP, et al. Abnormal folate metabolism and mutation in the methylenetetrahydrofolate reductase gene may be maternal risk factors for Down syndrome. Am J Clin Nutr. 1999 Oct;70(4):495–501. doi: 10.1093/ajcn/70.4.495. [DOI] [PubMed] [Google Scholar]
- [22].Krajinovic M, Lamothe S, Labuda D, et al. Role of MTHFR genetic polymorphisms in the susceptibility to childhood acute lymphoblastic leukemia. Blood. 2004 Jan 1;103(1):252–257. doi: 10.1182/blood-2003-06-1794. [DOI] [PubMed] [Google Scholar]
- [23].Sussan TE, Yang A, Li F, et al. Trisomy represses Apc(Min)-mediated tumours in mouse models of Down’s syndrome. Nature. 2008 Jan 3;451(7174):73–75. doi: 10.1038/nature06446. [DOI] [PubMed] [Google Scholar]
- [24].Zorick TS, Mustacchi Z, Bando SY, et al. High serum endostatin levels in Down syndrome: implications for improved treatment and prevention of solid tumours. Eur J Hum Genet. 2001 Nov;9(11):811–814. doi: 10.1038/sj.ejhg.5200721. [DOI] [PubMed] [Google Scholar]
- [25].Klusmann JH, Creutzig U, Zimmermann M, et al. Treatment and prognostic impact of transient leukemia in neonates with Down syndrome. Blood. 2008 Mar 15;111(6):2991–2998. doi: 10.1182/blood-2007-10-118810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pine SR, Guo Q, Yin C, et al. GATA1 as a new target to detect minimal residual disease in both transient leukemia and megakaryoblastic leukemia of Down syndrome. Leuk Res. 2005 Nov;29(11):1353–1356. doi: 10.1016/j.leukres.2005.04.007. [DOI] [PubMed] [Google Scholar]
- [27].Ravindranath Y, Abella E, Krischer JP, et al. Acute myeloid leukemia (AML) in Down’s syndrome is highly responsive to chemotherapy: experience on Pediatric Oncology Group AML Study 8498. Blood. 1992 Nov 1;80(9):2210–2214. [PubMed] [Google Scholar]
- [28].Lie SO, Jonmundsson G, Mellander L, et al. A population-based study of 272 children with acute myeloid leukaemia treated on two consecutive protocols with different intensity: best outcome in girls, infants, and children with Down’s syndrome. Nordic Society of Paediatric Haematology and Oncology (NOPHO) Br J Haematol. 1996 Jul;94(1):82–88. doi: 10.1046/j.1365-2141.1996.d01-1761.x. [DOI] [PubMed] [Google Scholar]
- [29].Athale UH, Razzouk BI, Raimondi SC, et al. Biology and outcome of childhood acute megakaryoblastic leukemia: a single institution’s experience. Blood. 2001 Jun 15;97(12):3727–3732. doi: 10.1182/blood.v97.12.3727. [DOI] [PubMed] [Google Scholar]
- [30].Gamis AS. Acute myeloid leukemia and Down syndrome evolution of modern therapy--state of the art review. Pediatr Blood Cancer. 2005 Jan;44(1):13–20. doi: 10.1002/pbc.20207. [DOI] [PubMed] [Google Scholar]
- [31].Lange BJ, Kobrinsky N, Barnard DR, et al. Distinctive demography, biology, and outcome of acute myeloid leukemia and myelodysplastic syndrome in children with Down syndrome: Children’s Cancer Group Studies 2861 and 2891. Blood. 1998 Jan 15;91(2):608–615. [PubMed] [Google Scholar]
- [32].Abildgaard L, Ellebaek E, Gustafsson G, et al. Optimal treatment intensity in children with Down syndrome and myeloid leukaemia: data from 56 children treated on NOPHO-AML protocols and a review of the literature. Ann Hematol. 2006 May;85(5):275–280. doi: 10.1007/s00277-005-0045-5. [DOI] [PubMed] [Google Scholar]
- [33].Lange B. The management of neoplastic disorders of haematopoiesis in children with Down’s syndrome. Br J Haematol. 2000 Sep;110(3):512–524. doi: 10.1046/j.1365-2141.2000.02027.x. [DOI] [PubMed] [Google Scholar]
- [34].Kojima S, Sako M, Kato K, et al. An effective chemotherapeutic regimen for acute myeloid leukemia and myelodysplastic syndrome in children with Down’s syndrome. Leukemia. 2000 May;14(5):786–791. doi: 10.1038/sj.leu.2401754. [DOI] [PubMed] [Google Scholar]
- [35].Kudo K, Kojima S, Tabuchi K, et al. Prospective study of a pirarubicin, intermediate-dose cytarabine, and etoposide regimen in children with Down syndrome and acute myeloid leukemia: the Japanese Childhood AML Cooperative Study Group. J Clin Oncol. 2007 Dec 1;25(34):5442–5447. doi: 10.1200/JCO.2007.12.3687. [DOI] [PubMed] [Google Scholar]
- [36].Al-Ahmari A, Shah N, Sung L, et al. Long-term results of an ultra low-dose cytarabine-based regimen for the treatment of acute megakaryoblastic leukaemia in children with Down syndrome. Br J Haematol. 2006 Jun;133(6):646–648. doi: 10.1111/j.1365-2141.2006.06097.x. [DOI] [PubMed] [Google Scholar]
- [37].Zwaan MC, Reinhardt D, Hitzler J, et al. Acute leukemias in children with Down syndrome. Pediatr Clin North Am. 2008 Feb;55(1):53–70. x. doi: 10.1016/j.pcl.2007.11.001. [DOI] [PubMed] [Google Scholar]
- [38].Wechsler J, Greene M, McDevitt MA, et al. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet. 2002 Sep;32(1):148–152. doi: 10.1038/ng955. [DOI] [PubMed] [Google Scholar]
- [39].Mundschau G, Gurbuxani S, Gamis AS, et al. Mutagenesis of GATA1 is an initiating event in Down syndrome leukemogenesis. Blood. 2003 Jun 1;101(11):4298–4300. doi: 10.1182/blood-2002-12-3904. [DOI] [PubMed] [Google Scholar]
- [40].Hasle H, Niemeyer CM, Chessells JM, et al. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia. 2003 Feb;17(2):277–282. doi: 10.1038/sj.leu.2402765. [DOI] [PubMed] [Google Scholar]
- [41].Ahmed M, Sternberg A, Hall G, et al. Natural history of GATA1 mutations in Down syndrome. Blood. 2004 Apr 1;103(7):2480–2489. doi: 10.1182/blood-2003-10-3383. [DOI] [PubMed] [Google Scholar]
- [42].Rainis L, Bercovich D, Strehl S, et al. Mutations in exon 2 of GATA1 are early events in megakaryocytic malignancies associated with trisomy 21. Blood. 2003 Aug 1;102(3):981–986. doi: 10.1182/blood-2002-11-3599. [DOI] [PubMed] [Google Scholar]
- [43].Li Z, Godinho FJ, Klusmann JH, et al. Developmental stage-selective effect of somatically mutated leukemogenic transcription factor GATA1. Nat Genet. 2005 Jun;37(6):613–619. doi: 10.1038/ng1566. [DOI] [PubMed] [Google Scholar]
- [44].Hollanda LM, Lima CS, Cunha AF, et al. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat Genet. 2006 Jul;38(7):807–812. doi: 10.1038/ng1825. [DOI] [PubMed] [Google Scholar]
- [45].Stachura DL, Chou ST, Weiss MJ. Early block to erythromegakaryocytic development conferred by loss of transcription factor GATA-1. Blood. 2006 Jan 1;107(1):87–97. doi: 10.1182/blood-2005-07-2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Xu G, Nagano M, Kanezaki R, et al. Frequent mutations in the GATA-1 gene in the transient myeloproliferative disorder of Down syndrome. Blood. 2003 Oct 15;102(8):2960–2968. doi: 10.1182/blood-2003-02-0390. [DOI] [PubMed] [Google Scholar]
- [47].Bourquin JP, Subramanian A, Langebrake C, et al. Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene expression profiling. Proc Natl Acad Sci U S A. 2006 Feb 28;103(9):3339–3344. doi: 10.1073/pnas.0511150103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lightfoot J, Hitzler JK, Zipursky A, et al. Distinct gene signatures of transient and acute megakaryoblastic leukemia in Down syndrome. Leukemia. 2004 Oct;18(10):1617–1623. doi: 10.1038/sj.leu.2403466. [DOI] [PubMed] [Google Scholar]
- [49].McElwaine S, Mulligan C, Groet J, et al. Microarray transcript profiling distinguishes the transient from the acute type of megakaryoblastic leukaemia (M7) in Down’s syndrome, revealing PRAME as a specific discriminating marker. Br J Haematol. 2004 Jun;125(6):729–742. doi: 10.1111/j.1365-2141.2004.04982.x. [DOI] [PubMed] [Google Scholar]
- [50].Walters DK, Mercher T, Gu TL, et al. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell. 2006 Jul;10(1):65–75. doi: 10.1016/j.ccr.2006.06.002. [DOI] [PubMed] [Google Scholar]
- [51].Kiyoi H, Yamaji S, Kojima S, et al. JAK3 mutations occur in acute megakaryoblastic leukemia both in Down syndrome children and non-Down syndrome adults. Leukemia. 2007 Mar;21(3):574–576. doi: 10.1038/sj.leu.2404527. [DOI] [PubMed] [Google Scholar]
- [52].Klusmann JH, Reinhardt D, Hasle H, et al. Janus kinase mutations in the development of acute megakaryoblastic leukemia in children with and without Down’s syndrome. Leukemia. 2007 Jul;21(7):1584–1587. doi: 10.1038/sj.leu.2404694. [DOI] [PubMed] [Google Scholar]
- [53].Norton A, Fisher C, Liu H, et al. Analysis of JAK3, JAK2, and C-MPL mutations in transient myeloproliferative disorder and myeloid leukemia of Down syndrome blasts in children with Down syndrome. Blood. 2007 Aug 1;110(3):1077–1079. doi: 10.1182/blood-2007-03-080374. [DOI] [PubMed] [Google Scholar]
- [54].Malinge S, Ragu C, la-Valle V, et al. Activating mutations in human acute megakaryoblastic leukemia. Blood. 2008 Aug 28; doi: 10.1182/blood-2008-01-136366. [DOI] [PubMed] [Google Scholar]
- [55].Pui CH, Raimondi SC, Borowitz MJ, et al. Immunophenotypes and karyotypes of leukemic cells in children with Down syndrome and acute lymphoblastic leukemia. J Clin Oncol. 1993 Jul;11(7):1361–1367. doi: 10.1200/JCO.1993.11.7.1361. [DOI] [PubMed] [Google Scholar]
- [56].Dordelmann M, Schrappe M, Reiter A, et al. Down’s syndrome in childhood acute lymphoblastic leukemia: clinical characteristics and treatment outcome in four consecutive BFM trials. Berlin-Frankfurt-Munster Group. Leukemia. 1998 May;12(5):645–651. doi: 10.1038/sj.leu.2400989. [DOI] [PubMed] [Google Scholar]
- [57].Chessells JM, Harrison G, Richards SM, et al. Down’s syndrome and acute lymphoblastic leukaemia: clinical features and response to treatment. Arch Dis Child. 2001 Oct;85(4):321–325. doi: 10.1136/adc.85.4.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zeller B, Gustafsson G, Forestier E, et al. Acute leukaemia in children with Down syndrome: a population-based Nordic study. Br J Haematol. 2005 Mar;128(6):797–804. doi: 10.1111/j.1365-2141.2005.05398.x. [DOI] [PubMed] [Google Scholar]
- [59].Bassal M, La MK, Whitlock JA, et al. Lymphoblast biology and outcome among children with Down syndrome and ALL treated on CCG-1952. Pediatr Blood Cancer. 2005 Jan;44(1):21–28. doi: 10.1002/pbc.20193. [DOI] [PubMed] [Google Scholar]
- [60].Whitlock JA, Sather HN, Gaynon P, et al. Clinical characteristics and outcome of children with Down syndrome and acute lymphoblastic leukemia: a Children’s Cancer Group study. Blood. 2005 Dec 15;106(13):4043–4049. doi: 10.1182/blood-2003-10-3446. [DOI] [PubMed] [Google Scholar]
- [61].Arico M, Ziino O, Valsecchi MG, et al. Acute lymphoblastic leukemia and Down syndrome: presenting features and treatment outcome in the experience of the Italian Association of Pediatric Hematology and Oncology (AIEOP) Cancer. 2008 Jun 2; doi: 10.1002/cncr.23587. [DOI] [PubMed] [Google Scholar]
- [62].Forestier E, Izraeli S, Beverloo B, et al. Cytogenetic features of acute lymphoblastic and myeloid leukemias in pediatric patients with Down syndrome: an iBFM-SG study. Blood. 2008 Feb 1;111(3):1575–1583. doi: 10.1182/blood-2007-09-114231. [DOI] [PubMed] [Google Scholar]
- [63].Maloney KW, Carroll WL, Carroll A, et al. Comparison of the biology of Down syndrome (DS) acute lymphoblastic leukemia (ALL) and non-DS ALL: Children’s Oncology Group study P9900. J Clin Oncol. 2008 May 20;26(May 20 suppl):10003a. [Google Scholar]
- [64].Bercovich D, Ganmore I, Scott LM, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet. 2008 Sep 19; doi: 10.1016/S0140-6736(08)61341-0. [DOI] [PubMed] [Google Scholar]
- [65].James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005 Apr 28;434(7037):1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- [66].Hastings C, Whitlock JA, La M, Seibel N. Improved Outcome of Children with Down Syndrome and High Risk Acute Lymphocytic Leukemia: A Report of CCG-1961. Blood. 2007;110:586a. [Google Scholar]
- [67].Whitlock JA. Down syndrome and acute lymphoblastic leukaemia. Br J Haematol. 2006 Dec;135(5):595–602. doi: 10.1111/j.1365-2141.2006.06337.x. [DOI] [PubMed] [Google Scholar]
- [68].Zwaan CM, Kaspers GJ, Pieters R, et al. Different drug sensitivity profiles of acute myeloid and lymphoblastic leukemia and normal peripheral blood mononuclear cells in children with and without Down syndrome. Blood. 2002 Jan 1;99(1):245–251. doi: 10.1182/blood.v99.1.245. [DOI] [PubMed] [Google Scholar]
- [69].Taub JW, Huang X, Matherly LH, et al. Expression of chromosome 21-localized genes in acute myeloid leukemia: differences between Down syndrome and non-Down syndrome blast cells and relationship to in vitro sensitivity to cytosine arabinoside and daunorubicin. Blood. 1999 Aug 15;94(4):1393–1400. [PubMed] [Google Scholar]
- [70].Ge Y, Dombkowski AA, LaFiura KM, et al. Differential gene expression, GATA1 target genes, and the chemotherapy sensitivity of Down syndrome megakaryocytic leukemia. Blood. 2006 Feb 15;107(4):1570–1581. doi: 10.1182/blood-2005-06-2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Taub JW, Ge Y. Down syndrome, drug metabolism and chromosome 21. Pediatr Blood Cancer. 2005 Jan;44(1):33–39. doi: 10.1002/pbc.20092. [DOI] [PubMed] [Google Scholar]
- [72].Ge Y, Jensen TL, Stout ML, et al. The role of cytidine deaminase and GATA1 mutations in the increased cytosine arabinoside sensitivity of Down syndrome myeloblasts and leukemia cell lines. Cancer Res. 2004 Jan 15;64(2):728–735. doi: 10.1158/0008-5472.can-03-2456. [DOI] [PubMed] [Google Scholar]
- [73].Krischer JP, Epstein S, Cuthbertson DD, et al. Clinical cardiotoxicity following anthracycline treatment for childhood cancer: the Pediatric Oncology Group experience. J Clin Oncol. 1997 Apr;15(4):1544–1552. doi: 10.1200/JCO.1997.15.4.1544. [DOI] [PubMed] [Google Scholar]
- [74].O’Brien MM, Taub JW, Chang MN, et al. Cardiomyopathy in children with Down syndrome treated for acute myeloid leukemia: a report from the Children’s Oncology Group Study POG 9421. J Clin Oncol. 2008 Jan 20;26(3):414–420. doi: 10.1200/JCO.2007.13.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Morawiec Z, Janik K, Kowalski M, et al. DNA damage and repair in children with Down’s syndrome. Mutat Res. 2008 Jan 1;637(12):118–123. doi: 10.1016/j.mrfmmm.2007.07.010. [DOI] [PubMed] [Google Scholar]
- [76].Creutzig U, Diekamp S, Zimmermann M, et al. Longitudinal evaluation of early and late anthracycline cardiotoxicity in children with AML. Pediatr Blood Cancer. 2007 Jun 15;48(7):651–662. doi: 10.1002/pbc.21105. [DOI] [PubMed] [Google Scholar]
- [77].Frost BM, Gustafsson G, Larsson R, et al. Cellular cytotoxic drug sensitivity in children with acute leukemia and Down’s syndrome: an explanation to differences in clinical outcome? Leukemia. 2000 May;14(5):943–944. doi: 10.1038/sj.leu.2401753. [DOI] [PubMed] [Google Scholar]
- [78].Kalwinsky DK, Raimondi SC, Bunin NJ, et al. Clinical and biological characteristics of acute lymphocytic leukemia in children with Down syndrome. Am J Med Genet Suppl. 1990;7:267–271. doi: 10.1002/ajmg.1320370753. [DOI] [PubMed] [Google Scholar]
- [79].Garre ML, Relling MV, Kalwinsky D, et al. Pharmacokinetics and toxicity of methotrexate in children with Down syndrome and acute lymphocytic leukemia. J Pediatr. 1987 Oct;111(4):606–612. doi: 10.1016/s0022-3476(87)80131-2. [DOI] [PubMed] [Google Scholar]
- [80].Peeters M, Poon A. Down syndrome and leukemia: unusual clinical aspects and unexpected methotrexate sensitivity. Eur J Pediatr. 1987 Jul;146(4):416–422. doi: 10.1007/BF00444952. [DOI] [PubMed] [Google Scholar]
- [81].Blatt J, Albo V, Prin W, et al. Excessive chemotherapy-related myelotoxicity in children with Down syndrome and acute lymphoblastic leukaemia. Lancet. 1986 Oct 18;2(8512):914. doi: 10.1016/s0140-6736(86)90429-0. [DOI] [PubMed] [Google Scholar]
- [82].Zhang L, Taub JW, Williamson M, et al. Reduced folate carrier gene expression in childhood acute lymphoblastic leukemia: relationship to immunophenotype and ploidy. Clin Cancer Res. 1998 Sep;4(9):2169–2177. [PubMed] [Google Scholar]
- [83].Lonnerholm G, Frost BM, Soderhall S, et al. Vincristine pharmacokinetics in children with down syndrome. Pediatr Blood Cancer. 2008 Jul 9; doi: 10.1002/pbc.21691. [DOI] [PubMed] [Google Scholar]
- [84].Valle M, Plon SE, Rabin KR. Differential in vitro cytotoxicity does not explain increased host toxicities from chemotherapy in Down syndrome acute lymphoblastic leukemia. Leuk Res. 2008 Aug 19; doi: 10.1016/j.leukres.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Gregory L, Williams R, Thompson E. Leucocyte function in Down’s syndrome and acute leukaemia. Lancet. 1972 Jun 24;1(7765):1359–1361. doi: 10.1016/s0140-6736(72)91093-8. [DOI] [PubMed] [Google Scholar]
- [86].Spina CA, Smith D, Korn E, et al. Altered cellular immune functions in patients with Down’s syndrome. Am J Dis Child. 1981 Mar;135(3):251–255. doi: 10.1001/archpedi.1981.02130270043015. [DOI] [PubMed] [Google Scholar]
- [87].Avanzini MA, Monafo V, De AM, et al. Humoral immunodeficiencies in Down syndrome: serum IgG subclass and antibody response to hepatitis B vaccine. Am J Med Genet Suppl. 1990;7:231–233. doi: 10.1002/ajmg.1320370746. [DOI] [PubMed] [Google Scholar]
- [88].Ugazio AG, Maccario R, Notarangelo LD, et al. Immunology of Down syndrome: a review. Am J Med Genet Suppl. 1990;7:204–212. doi: 10.1002/ajmg.1320370742. [DOI] [PubMed] [Google Scholar]
- [89].Craze JL, Harrison G, Wheatley K, et al. Improved outcome of acute myeloid leukaemia in Down’s syndrome. Arch Dis Child. 1999 Jul;81(1):32–37. doi: 10.1136/adc.81.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Lie SO, Abrahamsson J, Clausen N, et al. Long-term results in children with AML: NOPHO-AML Study Group--report of three consecutive trials. Leukemia. 2005 Dec;19(12):2090–2100. doi: 10.1038/sj.leu.2403962. [DOI] [PubMed] [Google Scholar]
- [91].Ravindranath Y, Yeager AM, Chang MN, et al. Autologous bone marrow transplantation versus intensive consolidation chemotherapy for acute myeloid leukemia in childhood. Pediatric Oncology Group. N Engl J Med. 1996 May 30;334(22):1428–1434. doi: 10.1056/NEJM199605303342203. [DOI] [PubMed] [Google Scholar]