

Summary
The iterative formation of nephrons during embryonic development relies on continual replenishment of progenitor cells throughout nephrogenesis. Defining molecular mechanisms that maintain and regulate this progenitor pool is essential to understanding nephrogenesis in developmental and regenerative contexts. Maintenance of nephron progenitors is absolutely dependent on BMP7 signaling, and Bmp7-null mice exhibit rapid loss of progenitors. However, the signal transduction machinery operating downstream of BMP7 as well as the precise target cell remain undefined. Using a novel primary progenitor isolation system, we have investigated signal transduction and biological outcomes elicited by BMP7. We find that BMP7 directly and rapidly activates JNK signaling in nephron progenitors resulting in phosphorylation of Jun and ATF2 transcription factors. This signaling results in the accumulation of cyclin D3 and subsequent proliferation of PAX2+ progenitors, inversely correlating with the loss of nephron progenitors seen in the Bmp7-null kidney. Activation of Jun and ATF2 is severely diminished in Bmp7-null kidneys, providing an important in vivo correlate. BMP7 thus promotes proliferation directly in nephron progenitors by activating the JNK signaling circuitry.
Keywords: Kidney development, Cap mesenchyme, BMP, SMAD, JNK
INTRODUCTION
Development of the mammalian permanent kidney, or metanephros, is initiated at embryonic day (E) 10.5 in the mouse. Reciprocal interactions between the ureteric bud and the metanephric mesenchyme, both derivatives of the intermediate mesoderm, drive and organize the process of kidney development (Dressler, 2006; Saxén, 1987; Vainio and Lin, 2002). Nephron progenitors, known collectively as cap mesenchyme, originate from the metanephric mesenchyme and are induced by the ureteric bud to differentiate, ultimately giving rise to the epithelial components of the nephron, the functional unit of the kidney. Cap mesenchyme in turn induces branching of the ureteric bud, which forms the collecting duct system of the mature kidney. To sustain nephrogenesis throughout kidney development, nephron progenitors must continually be replenished while simultaneously allowing differentiation of new nephrons. In the developing kidney, the formation of new nephrons occurs exclusively in the cortical aspect of the organ where cap mesenchyme is situated adjacent to collecting duct tips. This region, known as the nephrogenic zone (NZ), can be considered the niche for nephron progenitors, delicately balancing their fates until nephrogenesis is complete.
A number of growth factors regulate this balance, including members of the Wnt family, fibroblast growth factors (FGFs) and bone morphogenetic proteins (BMPs) (Carroll et al., 2005; Dudley et al., 1995; Grieshammer et al., 2005; Luo et al., 1995; Perantoni et al., 2005). BMPs belong to the transforming growth factor β (TGFβ) superfamily of cytokines that regulate an array of cellular processes during development, including proliferation and differentiation (Massague, 1998). BMPs signal through cell surface serine/threonine kinase receptors, which activate receptor-associated SMAD transcription factors (R-SMADs) through phosphorylation. TGFβ and activin signal through R-SMAD2 and R-SMAD3, whereas BMPs activate R-SMAD1, R-SMAD5 and R-SMAD8. Upon phosphorylation, R-SMADs form a complex with SMAD4, resulting in nuclear accumulation and modification of target gene transcription. Although the SMAD signaling circuitry is undoubtedly the best characterized to date, components of the mitogen-activated protein kinase (MAPK) pathway, most prominently Jun (also known as c-jun) N-terminal kinases (JNKs) and p38, can be activated by both TGFβ and BMPs (Derynck and Zhang, 2003; Massague and Chen, 2000; Yamaguchi et al., 1995).
In the developing kidney, BMP7 is required for replenishment of the nephron progenitor compartment during nephrogenesis (Dudley et al., 1999). Despite normal initiation of metanephric development and formation of a limited number of nephrons, progenitors are prematurely depleted in the Bmp7-null mouse, resulting in severe renal dysplasia (Dudley et al., 1995). Definition of the signaling events underlying this phenotype is an essential step towards understanding pathways regulating nephron progenitor renewal. Surprisingly, we have found that the NZ is essentially unresponsive to SMAD-mediated transcriptional activation (Blank et al., 2008). This intriguing finding prompted us to investigate whether alternate signaling pathways, activated by BMP7, regulate progenitor cell survival and proliferation. Using a novel system to purify cells from the NZ of developing kidneys, we have investigated both the identities of signaling pathways activated by BMP7 in the NZ, and the biological consequences of this pathway activation. In analogy with findings made using an in vivo BMP reporter (Blank et al., 2008), cells isolated from the nephrogenic zone (NZ), collectively referred to as NZ cells, appear refractory to SMAD-mediated transcriptional activation. However, BMP7 rapidly activates JNK signaling, resulting in phosphorylation of both Jun and ATF2. Furthermore, BMP7 promotes proliferation of PAX2+ nephron progenitor cells through a mechanism involving TAK1-mediated JNK activation. In vivo, phosphorylation of both Jun and ATF2 in the cap mesenchyme of Bmp7-null kidneys is significantly impaired compared with that in the wild type. We therefore propose that BMP7 promotes proliferation of nephron progenitors by directly activating the JNK-Jun-ATF2 signaling axis in these cells. Our findings translate directly to the phenotype observed in the Bmp7-null mouse, providing a novel explanation for the deficiency of nephron progenitors during kidney development.
MATERIALS AND METHODS
Isolation of nephrogenic zone cells
Kidneys from E17.5 embryos were dissected into HBSS (Gibco) and the capsule was removed. Subsequently kidneys were incubated in PBS containing 1% pancreatin (Sigma) and 0.25% collagenase A (Roche) for 15 minutes at 37°C on a rocking platform. Cells in suspension were removed; 5 μl DNase (Invitrogen) was added together with fetal calf serum (FCS, HyClone) to a final concentration of 5%. Remaining cell aggregates were dissociated mechanically by pipetting a few times and then cells were incubated for another 10 minutes at 37°C on a rocking platform. Cells were pelleted and washed once in HBSS containing 5% FCS. Before plating cells were filtered through a 40 μm cell strainer (BD Biosciences).
Cell culture
Nephrogenic zone cells (NZCs) were cultured under serum-free conditions in keratinocyte SFM (Gibco) supplemented with 2 mM l-glutamine (Invitrogen), 100 U/ml penicillin (Sigma), 100 μg/ml streptomycin (Sigma). Cells were seeded at a concentration of 0.4-0.6×106 cells/ml on plates pre-coated with fibronectin (BD Biosciences). Recombinant human BMP7 (R&D Systems) was used at a concentration of 50 ng/ml. JNK inhibitor was purchased from Calbiochem and used at a concentration of 10 μM. TAK1 inhibitor was used at a concentration of 0.5 μM (Analyticon Discovery). Dorsomorphin (Sigma) was used at 20 μM.
Flow cytometry
Dead cells were excluded by staining with 7-aminoactinomycin D (7-AAD, Sigma). Samples were collected on a FACSCalibur (BD) and data were subsequently analyzed using FlowJo software.
Immunofluorescence
Cells were fixed in 4% paraformaldehyde directly in the cell culture well for 15 minutes at room temperature and subsequently permeabilized for 10 minutes in PBS containing 0.3% Triton X-100 (VWR International). Cells were blocked in PBS containing 5% chicken and/or goat or donkey serum (Jackson ImmunoResearch). Primary antibodies were diluted in blocking solution as above at the following dilutions: phospho-SMAD1/SMAD5/SMAD8, 1:50 (Cell Signaling Technology); phospho-Jun, 1:50 (Cell Signaling Technology); phospho-histone-H3, 1:100 (Cell Signaling Technology); Pax8, 1:100 (Proteintech Group); Pax2, 1:100 (Zymed); E-cadherin, 1:50 (BD Transduction Laboratories); Six2, 1:50 (Santa Cruz). Alexa Fluor 488- and/or 568-conjugated secondary antibodies were used for detection of labeled cells (Molecular Probes). Nuclei were stained using DAPI (Molecular Probes).
Immunofluorescence (paraffin-embedded sections)
Paraffin-embedded sections were processed as previously described (Blank et al., 2008). Sections were blocked in 1× Tris-buffered saline (TBS) containing 0.1% Tween (Sigma), 1% bovine serum albumin (BSA; Jackson ImmunoResearch), 5% goat serum (Jackson ImmunoResearch) for 30 minutes. Incubation with streptavidin-biotin blocking solutions was carried out according to the manufacturer's instructions (Vector Laboratories). Subsequently, sections were incubated for 2 hours at room temperature in the presence of primary antibodies diluted in blocking solution as follows: phospho-Jun (1:100), phospho-ATF2 (1:100), phospho-SMAD1/SMAD5/SMAD8 (1:50), phospho-histone-H3 (1:100), Pax2 (1:200). Biotinylated dolichos biflorus agglutinin (DBA; Vector Laboratories) was diluted 1:200. Secondary antibodies were used as above and Alexa Fluor 488-conjugated streptavidin (Invitrogen) was used at 1:200. Nuclei were stained using DAPI as above. Sections were mounted using Vectashield (Vector Laboratories).
Quantitative RT-PCR
RNA was extracted using Trizol (Invitrogen) according to manufacturer's protocol. Samples were treated with DNase using a DNA-free kit (Ambion). cDNA synthesis was carried out in the presence of Oligo(dT) using Superscript II reverse transcriptase (Invitrogen) according to the manufacturer's protocol. Quantitative PCRs were performed on a iQ iCycler using iQ SYBR Green Supermix (Bio-Rad). Each assay was performed in triplicate and normalized to β-actin levels. β-actin forward primer, GGCTGTATTCCCCTCCATCG; reverse primer CCAGTTGGTAACA - ATGCCATGT. Id1 forward primer, CCTAGCTGTTCGCTGAAGGC; reverse primer, CTCCGACAGACCAAGTACCAC.
Western blots
NZCs were stimulated with indicated growth factors. Total protein was extracted as previously described (Blank et al., 2008). Antibodies against phosphorylated SMAD1/SMAD5/SMAD8, phosphorylated JNK, phosphorylated p38, phosphorylated Jun, phosphorylated ATF2, cleaved caspase-3, cyclin D3 and XIAP were all from Cell Signaling Technology, anti-β-tubulin was purchased from Santa Cruz, anti-TAK1 was from Upstate, anti-TAB1 from ProSci and anti-HA from Covance.
X-gal staining
Cells were pre-fixed for 15 minutes in 1% formaldehyde, 0.2% glutaraldehyde. After brief washing cells were stained in 0.5 mg/ml X-gal at 37°C overnight.
Adenoviral vectors and transduction
The previously described dominant-negative kinase-defective form of TAK1 (Yamaguchi et al., 1995) (kindly provided by Jun Ninomya-Tsuji) was cloned into pENTR D/TOPO (Invitrogen). Subsequently, the Gateway Technology System (Invitrogen) was used to produce pAd/CMV/V5-dnTAK1 for production of adenovirus, according to the manufacturer's instructions. Transduction of NZCs was carried out for 36 hours at an MOI of 50 in fibronectin-coated wells using serum-free medium as described.
Mouse strains
Animal care in accordance with the National Research Council Guide for the Care and Use of Laboratory Animals was approved by the Institutional Animal Care and Use Committee of Maine Medical Center.
Statistical analyses
Data were analyzed using Student's t-test. P<0.05 was considered significant.
RESULTS
Isolation and characterization of cells derived from the nephrogenic zone niche
To study signaling pathways that regulate progenitor cell fate decisions in the developing kidney, an in vitro system for culture of NZ cells, in isolation from the collecting duct, or inducer, is required. A protocol for limited enzymatic digestion was devised to allow dissociation of E17.5 NZ cells with minimal contamination of cells from collecting ducts and differentiating nephrons. The identities of cells within the population of single cells isolated from the outermost layer of the kidney was determined using reporter mouse strains marking distinct cellular compartments within the NZ (Fig. 1A). The contribution of stromal cells was assessed by isolating NZ cells from the stroma-specific Foxd1+/lacZ reporter strain (Hatini et al., 1996). Foxd1+ cells represented 36.4±2.8% of total NZ cells. NZ cells isolated from Bmp7+/lacZ mice, which specifically express β-galactosidase in cap mesenchyme and collecting duct cells revealed that Bmp7 was actively expressed in 52.7±8.7% of NZ cells. To determine the contribution of cells either derived from Bmp7-expressing cells, or actively expressing Bmp7, we analyzed NZ cells isolated from the Bmp7+/Cre;R26-YFP reporter mouse by flow cytometry (Fig. 1B). On average, the contribution of YFP was 64.8±5.34% (Fig. 1C). The slightly higher contribution of lineage-marked YFP+ cells was not significantly different from cells actively expressing Bmp7, but might suggest that NZCs contain a small proportion of differentiated YFP+/Bmp7- cells. To control for possible contamination of the NZ cell population with collecting duct cells that also express Bmp7, we harvested NZ cells from HoxB7-Cre;R26-YFP mice. HoxB7 is exclusively expressed in collecting duct cells and consequently YFP is restricted to cells of the collecting duct lineage. FACS analysis showed less than 0.04% contamination of collecting duct cells in NZ cell preparations (Fig. 1D). Similarly, X-gal staining of residual kidneys of Bmp7+/lacZ genotype following enzyme digestion, showed exposed but intact collecting duct tips that were free of the overlying NZ (Fig. 1E). The viability of NZ cells at harvest, evaluated by FACS analysis of 7AAD exclusion, was 95% on average (Fig. 1F).
Fig. 1.

Characterization of the NZ cell population from E17.5 embryonic kidneys. (A) Schematic representation of the E17.5 mouse kidney and the major cell populations of the NZ. Cell compartments of mouse reporter strains expressing β-galactosidase are shown in blue and YFP is shown in yellow. CM, cap mesenchyme; CD, collecting duct; NZ, nephrogenic zone. (B) Flow cytometry of NZ cells. (C) Representative flow cytometry histogram showing 70.7% YFP+ cells among NZ cells from Bmp7+/cre;R26-YFP. (D) Representative flow cytometry revealing <0.04% YFP+ collecting duct cells among NZ cells from HoxB7-Cre;R26-YFP. (E) Intact collecting duct tips of residual kidneys from Bmp7+/lacZ embryos following NZ cell isolation. The residual kidneys were stained in X-gal solution. LacZ+ collecting ducts are blue. (F) Representative flow cytometry of 7AAD-stained NZ cells showing 94.8% viable cells. (G) NZ cell purity at harvest. Numbers represent average percentage values ± s.d. YFP indicates positive cells from Bmp7+/cre;R26-YFP (n=3), Bmp7+ cells were calculated from Bmp7+/lacZ (n=3), Foxd1+ cells were calculated from Foxd1+/lacZ (n=3). 7AAD+ indicates percentage of dead cells (n=5). (H) Immunofluorescence (IF) staining of NZ cells following 16 hours of culture. SIX2-labeled cells are shown in red, nuclei are stained blue with DAPI. Scale bars: 50 μm. (I) IF staining of NZ cells after 16 hours of culture showing overlap between SIX2 (red) and PAX2 (green) expression. Insets show individual color channels. DAPI counterstain (blue) shows nuclei. Scale bar: 50 μm.
This represents a novel and efficient method to purify cells of the E17.5 NZ, essentially in isolation from cells of the collecting duct (Fig. 1G). Using this purified cell population, we sought to establish an in vitro model of the progenitor cell niche of the developing kidney.
Establishing the nephrogenic zone niche in vitro
To evaluate culture conditions that best maintain nephron progenitor cells in vitro, we evaluated the potential of a number of growth conditions to maintain actively Bmp7-expressing cells after 24-48 hours of culture. A number of different surface coatings [gelatin (Sigma), collagen I, collagen IV, matrigel and fibronectin (all from BD BioSciences)] were tested in combination with different media [knockout-DMEM 10% serum replacement (Gibco); DMEM 10% FCS (Hyclone); keratinocyte serum-free (Gibco)]. All combinations were tested and the combination of fibronectin with keratinocyte serum-free medium resulted in the most robust maintenance of Bmp7-expressing cells after 24 and 48 hours of culture. Therefore, serum-free culture conditions in combination with fibronectin surface coating were chosen for all subsequent experiments. To characterize NZ cells following culture, we analyzed a variety of markers expressed by either undifferentiated cap mesenchyme or differentiating cells in vivo. The homeobox gene Six2 is expressed in cap mesenchyme throughout kidney development and is required for renewal of nephron progenitors (Kobayashi et al., 2008; Self et al., 2006). SIX2 thus serves as a convenient marker for undifferentiated nephron progenitors. Immunofluorescence staining of NZ cells 16 hours after plating revealed a significant portion of SIX2+ cells similar to the frequency of Bmp7-expressing cells (Fig. 1H), confirming that undifferentiated nephron progenitor cells are maintained under these culture conditions. The SIX2+ population was comprised of small rounded cells morphologically distinct from SIX2- cells, which were flattened and larger. Furthermore, PAX2, a transcription factor of the paired-box family that is expressed in condensing mesenchyme in vivo (Dressler and Douglass, 1992), was robustly expressed among NZ cells (see Fig. S1A in the supplementary material). Importantly, SIX2+ cells showed coexpression with PAX2 (Fig. 1I).
Pax8, another member of the paired-box transcription factor family, is upregulated upon differentiation and tubule formation, and is downstream of Wnt4 expression (Stark et al., 1994). Using PAX8 as a marker of nephron differentiation, we found that NZ cells contained a population of differentiated cells with epithelial morphology that formed scattered clusters after 16 hours of culture (see Fig. S1B in the supplementary material). In addition, E-cadherin, which is expressed in developing tubules (Vestweber et al., 1985) was expressed intensely by cells within the epithelial clusters, strongly suggesting that these cells indeed represent differentiating cells undergoing epithelialization (see Fig. S1C in the supplementary material). Interestingly, by filtering the NZ cells at harvest through a 40 μm cell strainer, the majority of PAX8+ and E-cadherin+ cells could be eliminated (see Fig. S1B,C in the supplementary material). To further enrich for undifferentiated nephron progenitors we thus incorporated a filtering step before plating NZ cells.
Taken together, we show that a large population of undifferentiated nephron progenitors faithfully expressing SIX2 and PAX2 are contained within the NZ niche culture. Differentiating cells expressing PAX8 and E-cadherin are confined to cell aggregates, most of which can be eliminated by filtration at the time of harvest.
NZ cells are refractory to SMAD-mediated BMP signaling
The NZ is essentially unresponsive at the transcriptional level to SMAD-mediated BMP signaling (Blank et al., 2008), yet paradoxically Bmp7 is absolutely required to sustain nephrogenesis and continued growth of the kidney during development. In organ explant studies using BMP reporter mice, the cap mesenchyme is refractory to exogenous administration of BMP4, whereas stromal cells respond actively (Blank et al., 2008). To ascertain whether ex vivo NZ cells respond in the same way to BMP stimulation, we isolated NZ cells and stimulated them in vitro with BMP7. Western blot analysis revealed robust phosphorylation of SMAD1/SMAD5/SMAD8, indicating that the SMAD signal transduction machinery can indeed be activated in NZ cells in vitro (Fig. 2A). This is further corroborated by immunofluorescence staining of NZ cells, showing that both SIX2+ and SIX2- populations exhibited nuclear accumulation of phospho-SMAD1/SMAD5/SMAD8 (Fig. 2B). Furthermore, phospho-SMAD1/SMAD5/SMAD8 could be weakly detected in the NZ by immunofluorescence staining of wild-type kidney sections at E17.5 (Fig. 2C). To investigate whether the refractory state of the NZ persisted in vitro at the transcriptional level, we performed quantitative RT-PCR analysis of Id1 in NZ cells. Upon BMP7 stimulation, Id1 was only marginally upregulated (less than twofold) in NZ cells in comparison with mouse embryonic fibroblast (MEF) cells, which exhibited a robust sixfold induction of Id1 under the same conditions (Fig. 2D). Similarly, NZ cells isolated from the BMP reporter remained unresponsive to exogenous BMP7, with the exception of a few scattered cells with stromal or epithelial morphology, which responded robustly to BMP7 treatment, as determined by X-gal staining (Fig. 2E). The less than twofold induction of Id1 observed in NZ cells is therefore probably due to the response of a minority of differentiating cells and stromal cells. Thus, our data indicate that SMAD1/SMAD5/SMAD8 can be activated in cap mesenchyme in vivo and in NZ cells in vitro, but this does not lead to transcriptional activation of Id1 (or Id2 and Id3, data not shown). We conclude that NZ cells in vitro recapitulate the characteristics of BMP signaling in the NZ in vivo. Having verified this, we next explored alternative signaling pathways activated downstream of BMP7 using this primary cell culture system.
Fig. 2.

NZ cells recapitulate characteristics of BMP-signaling in the nephrogenic zone. (A) Western blot analysis of phosphorylated SMAD1/SMAD5/SMAD8 in NZ cells stimulated with or without 50 ng/ml BMP7 for 30 minutes. NZ cells were pre-incubated overnight with or without 20 μM dorsomorphin, a BMP-specific inhibitor, as indicated. β-tubulin was used as a loading control. (B) IF staining of NZ cells using anti-phospho-SMAD1/SMAD5/SMAD8 (red) in conjunction with anti-SIX2 (green). DAPI was used as nuclear stain. NZ cells were incubated without (i) or with 50 ng/ml BMP7 (ii) for 30 minutes. Note extensive signaling in both SIX2+ and SIX2- populations. Scale bars: 50 μm. (C) IF staining of WT kidney section at E17.5. White lines indicate the NZ. Phospho-SMAD1/SMAD5/SMAD8 (red); phospho-HH3 (pHH3; green); DAPI (blue) stains nuclei. Note weak nuclear phospho-SMAD1/SMAD5/SMAD8 throughout the NZ. Scale bar: 50 μm. (D) Quantitative RT-PCR of Id1 expression in NZ cells and MEFs stimulated with 50 ng/ml BMP7 for 6 hours. Values are normalized to β-actin and represented as fold increase relative to unstimulated cells. Average values ± s.d. (NZ cell, n=5; MEF, n=1). (E) NZ cells harvested from BMP-reporter embryos were stimulated without (i) or with 50 ng/ml BMP7 (ii) for 16 hours. β-galactosidase+ cells (blue) indicate cells actively responding to BMP by SMAD-mediated transcriptional activation. Insets show magnifications of representative fields for each condition. Black arrows indicate responding β-galactosidase+ cells with epithelial- or stromal-like morphology. White arrows denote non-responding progenitor cells.
BMP7 activates the JNK signaling pathway
Members of the TGFβ superfamily of ligands have previously been reported to activate MAPK signaling pathways, including both p38 and JNK signaling branches, through a TAK1-mediated mechanism (Yamaguchi et al., 1995). Interestingly, TAK1 is expressed in the developing kidney (Jadrich et al., 2003) and western blot analyses indicate that TAK1, as well as other components involved in mediating TAK1-signals, such as XIAP and TAB1 (Yamaguchi et al., 1999), are expressed in NZ cells (Fig. 3A). We therefore investigated the activation of p38 and JNK pathways in response to BMP7 in NZ cells. Whereas phosphorylation of p38 was not significantly changed by BMP7 treatment at any of the time-points tested (5-30 minutes), JNK isoforms of both 46 kDa and 56 kDa in size were rapidly activated by BMP7, with signaling peaking at 5-10 minutes after BMP7 exposure (Fig. 3B). Importantly, activation of JNK signaling occurred before phosphorylation of SMAD1/SMAD5/SMAD8, which culminated 20-30 minutes after BMP7 exposure, suggesting that JNK activation occurs independently of SMAD signaling (Fig. 3C). Downstream transcriptional targets, including both Jun and ATF2 were phosphorylated within 15-20 minutes after addition of BMP7 (Fig. 3D). BMP7 can thus rapidly and specifically activate the JNK signaling pathway in NZ cells, resulting in downstream activation of both Jun and ATF2. To ascertain whether BMP7-mediated phosphorylation of JNK in NZ cells was dependent on TAK1, we overexpressed a dominant-negative kinase-defective form of TAK1 (Yamaguchi et al., 1995), by adenoviral transduction. Transduction efficiency was monitored by flow cytometry using the corresponding GFP adenoviral vector and reached 55% at 36 hours (see Fig. S2 in the supplementary material). Following transduction, cells were stimulated with or without BMP7 for 5-30 minutes, and the phosphorylation status of JNK was subsequently analyzed by western blotting. NZ cells transduced with the control GFP vector exhibited rapid phosphorylation of JNK (Fig. 3E). However, overexpression of dominant-negative TAK1 resulted in impaired activation of all JNK isoforms (Fig. 3E), verifying that TAK1 is indeed required for BMP7-mediated JNK activation in NZ cells.
Fig. 3.

BMP7 activates JNK signaling in SIX2+ NZ cells. (A) Western blot analysis showing expression of TAB1, TAK1 and XIAP in NZ cells. HEK293 cell lysate was used as a positive control (Blonska et al., 2005; Dan et al., 2004; Ge et al., 2003). β-tubulin shows protein loading. (B) Western blot analyses of activated JNK and p38 in NZ cells stimulated with 50 ng/ml BMP7 for 5-30 minutes. β-tubulin was used as loading control. (C) Western blot analysis of phosphorylated SMAD1/5/8 in NZ cells stimulated with 50 ng/ml BMP7 for 5-45 minutes. β-tubulin shows protein loading. (D) Western blot analyses of phosphorylated forms of Jun and ATF2 in NZ cells stimulated with 50 ng/ml BMP7 for 5-30 minutes. β-tubulin was used as loading control. (E) Western blot analyses of NZ cells transduced with control GFP- or dominant-negative (dn) TAK1 adenoviral (Ad) vectors. Cells were stimulated with 50 ng/ml BMP7 for 5-30 minutes and subsequently analyzed for phosphorylated JNK. Lane C, unstimulated control. HA-tagged dnTAK1 was detected using anti-HA antibody. β-tubulin was used as loading control. (F) IF staining of unstimulated NZ cells (i) or stimulated with 50 ng/ml BMP7 for 10 minutes (ii). Phosphorylated JNK (red), SIX2 (green), DAPI (blue). Arrows indicate SIX2+ cells, which robustly activate the JNK pathway upon BMP7 stimulation. Scale bars: 50 μm.
SIX2+ cells are the primary targets of BMP7-induced JNK activation
To further analyze which population of cells within NZ cells responds to BMP7 by JNK activation, we performed immunofluorescence co-staining for phosphorylated JNK and SIX2. In unstimulated NZ cells, limited activation of JNK signaling was found primarily in the SIX2+ population (Fig. 3F). Upon stimulation with BMP7, SIX2+ cells exhibited a clear nuclear localization of phosphorylated JNK. Weak signaling was also found in the cytoplasm of SIX2- cells. This confirms that the SIX2+ population of nephrogenic progenitors is the primary target of BMP7-activated JNK signaling.
BMP7 mediates proliferation of PAX2+ cells through a TAK1-JNK-dependent mechanism
BMP7 is a maintenance factor for cap mesenchyme (Dudley et al., 1995). However, in the Bmp7-null kidney, both increased cell death and decreased renewal of nephron progenitors can be seen (Dudley et al., 1995; Luo et al., 1995), and a potential role for BMP7 as a proliferative factor has never been dissociated from its role as a possible survival factor. JNK signaling has previously been implicated in proliferation, because MEFs deficient in both Jnk1 and Jnk2 exhibit reduced proliferation (Tournier et al., 2000). Furthermore, the JNK pathway has previously been reported to regulate the size of colonies derived from SALL1+ renal progenitors in vitro (Osafune et al., 2006). To investigate the relationship between BMP7 and proliferation and to ascertain the role of TAK1-mediated JNK activation in mediating this process, we examined the proliferative response of NZ cells to BMP7. Wild-type NZ cells were cultured with or without BMP7 in the presence or absence of a JNK inhibitor or a TAK1 inhibitor for 16-18 hours. Subsequently, phosphorylated histone H3 (phospho-HH3)-positive mitotic cells were counted, and numbers were normalized to the total number of cells in each treatment group. NZ cells cultured in the presence of BMP7 exhibited a significant increase in the number of proliferating cells compared with untreated controls (P=0.002, Fig. 4A). This effect was completely abrogated upon addition of either JNK inhibitor or TAK1 inhibitor, with numbers returning to baseline (Fig. 4A). To determine whether the proliferative effect of BMP7 was taking place in the progenitor population, PAX2+ phospho-HH3+ cells were quantified (Fig. 4B). Among PAX2+ cells, addition of BMP7 resulted in an even larger increase of phospho-HH3+ cells, compared with the total NZ cell population. Addition of inhibitors of either JNK or TAK1 completely eliminated this effect (Fig. 4B). Furthermore, immunofluorescence staining of phospho-HH3 in conjunction with SIX2 revealed that the majority of phospho-HH3+ cells resided within the SIX2+ compartment of NZ cells (Fig. 4C), although a smaller fraction of SIX2- cells was also proliferative (data not shown). In addition, cyclin D3 (CCND3) was increased at the protein level after 2 and 6 hours of BMP7 stimulation (Fig. 4D). This effect was abrogated upon addition of JNK inhibitor (Fig. 4D), further substantiating a role for BMP7 in promoting proliferation through the JNK signaling pathway. Addition of BMP7 or JNK inhibitors did not alter the overall survival of NZ cells, because no difference in 7AAD uptake was observed upon FACS analyses of NZ cells (Fig. 4E). Similarly, the abundance of cleaved caspase-3 was not significantly different between treatment groups, although untreated cells exhibited slightly less cleaved caspase-3 (Fig. 4F). In summary, these data demonstrate that BMP7, via a JNK-dependent mechanism, mediates a proliferative signal rather than a survival signal in SIX2+ NZ cells. Additionally, our data suggest that the kidney phenotype observed in the Bmp7-null mouse derives from a JNK-dependent mechanism ultimately leading to a failure of SIX2+ nephron progenitors to proliferate.
Fig. 4.

BMP7 promotes proliferation of PAX2+ NZ cells. (A) Number of mitotic phospho-histone H3 (phospho-HH3)-positive cells are shown normalized to total number of cells in each treatment group. Values represent averages ± s.e.m. (n=3). Analysis was carried out after 16-18 hours of stimulation. *P=0.002 for control versus BMP7-treated samples. **P=0.005 for BMP7 treated versus BMP7+JNK inhibitor or BMP7+TAK1 inhibitor treated samples. (B) Number of phospho-HH3+ PAX2+ cells normalized to total cell numbers in each treatment group. Values represent averages ± s.e.m. (n=3). Analysis was carried out after 16-18 hours of stimulation. *P=0.03 for control versus BMP7-treated samples. **P=0.006 for BMP7-treated versus BMP7+JNK inhibitor-treated samples. ***P=0.04 for BMP7-treated versus BMP7+TAK1 inhibitor-treated samples. (C) Representative images of stained NZ cells, revealing that SIX2+ (red) cells proliferate (phospho-HH3+, green) after 16-18 hours of BMP7 stimulation. DAPI stained nuclei are blue. Scale bar: 10 μm. (D) Western blot analysis of NZ cells reveals upregulation of cyclin D3 after 2 and 6 hours of BMP7 exposure. Addition of JNK inhibitor eliminates the increase in cyclin D3 in response to BMP7. β-tubulin was used as loading control. (E) Representative FACS analyses of 7AAD-stained NZ cells following 16-18 hours of stimulation, as indicated. No significant difference in the contribution of 7AAD+ dead cells could be detected between treatment groups. (F) Western blot analysis of cleaved caspase-3 in NZ cells following 16-18 hours of stimulation as indicated. β-tubulin was used as loading control.
Activated Jun and ATF2 localize to cap mesenchyme in vivo
To investigate the spatial distribution of phosphorylated Jun and ATF2 within the NZ in vivo, we performed immunofluorescence co-staining with DBA lectin, marking collecting ducts, on E17.5 wild-type kidney sections. Cap mesenchyme was identified by co-staining adjacent sections with PAX2 and DBA lectin (Fig. 5A,A′). Phosphorylated Jun and ATF2 were both localized in nuclei of cap mesenchyme cells (Fig. 5B-C′). Other cell types, including collecting ducts and differentiating nephrons, also displayed nuclear distribution of phosphorylated Jun and ATF2. This is in agreement with the in vitro data showing activation of JNK signaling in SIX2+ cells upon BMP7 exposure.
Fig. 5.

Activated Jun and ATF2 localize to cap mesenchyme in the E17.5 kidney. Representative IF staining of E17.5 WT kidneys. DAPI counterstain shows nuclei (blue). (A,A′) Pax2 (red), DBA lectin (green). PAX2+ DBA- cells indicate cap mesenchyme (CM) adjacent to DBA+ collecting duct (CD) tips. (B,B′) Phosphorylated Jun (red); DBA lectin (green). (C,C′) Phosphorylated ATF2 (red); DBA lectin (green). Nuclear staining of both activated Jun and ATF2 is present in cap mesenchyme adjacent to collecting duct tips. NZ, nephrogenic zone; RV, renal vesicle. Scale bars: 50 μm.
Reduced proliferation and decreased activation of Jun and ATF2 in Bmp7-null kidneys
The phenotype of Bmp7-null mice manifests at or around E14, before which induction and development proceeds relatively normally (Dudley et al., 1995; Luo et al., 1995). Furthermore, there is no detectable difference in apoptosis between wild-type and Bmp7-null kidneys before E13.5 (Dudley and Robertson, 1997). To study a potential role for BMP7 as a proliferative factor in vivo before the onset of the overt phenotype, we compared the number of mitotic cells per kidney section from three separate individuals of each wild-type and Bmp7-null genotype at E12.5. The number of mitotic cells within metanephric mesenchyme was significantly reduced in Bmp7-null kidneys, supporting the in vitro findings that BMP7 functions as a proliferative growth factor (Fig. 6A). Furthermore, immunofluorescence co-staining with anti-phospho-SMAD1/SMAD5/SMAD8 revealed a significant amount of nuclear phospho-SMAD1/SMAD5/SMAD8 in both wild-type and Bmp7-null kidneys (Fig. 6B), suggesting that a failure to activate SMAD-mediated signaling is not the causative factor behind the proliferative defect of cap mesenchyme in the Bmp7-null kidney. By contrast, phosphorylation of Jun and ATF2 was severely impaired specifically in cap mesenchyme adjacent to collecting duct tips of Bmp7-null kidneys (Fig. 7A-D). Activation of Jun and ATF2 was observed in collecting ducts of both wild-type and Bmp7-null kidneys (Fig. 7A-D), suggesting that other growth factors activate JNK signaling in cells of the collecting duct lineage. Collectively, these data confirm that BMP7 is required for proliferation and proper activation of Jun and ATF2 in cap mesenchyme, whereas phosphorylation of SMAD1/SMAD5/SMAD8 remains independent of BMP7.
Fig. 6.

Decreased proliferation but maintained phosphorylation of SMAD1/SMAD5/SMAD8 in Bmp7-null kidneys. (A) Number of mitotic phospho-HH3+ cells per kidney section at E12.5 (n=3). Values are shown as average ± s.d. Phospho-HH3+ cells in collecting ducts were excluded. Bmp7-null kidneys exhibit a significant reduction in phospho-HH3+ cells compared with WT littermates (P=0.005). (B) Representative IF staining of E12.5 WT (i) and Bmp7-null (ii) kidneys. Phospho-HH3+ (green); phospho-SMAD1/SMAD5/SMAD8 (red); DAPI stained nuclei (blue). Kidneys are outlined with a dashed line. Arrowheads indicate mitotic phospho-HH3+/phospho-SMAD1/SMAD5/SMAD8+ cells. Note the absence of phospho-HH3+ cells in the Bmp7-null kidney. Scale bars: 50 μm.
Fig. 7.

Impaired activation of Jun and ATF2 in Bmp7 null kidneys. Representative IF staining of E12.5 (A,A',C,C') WT and (B,B',D,D') Bmp7-null kidneys. (A-B') Phospho-Jun (red). (C-D') Phospho-ATF2 (red). Arrows indicate regions of intense nuclear staining. Asterisks indicate collecting duct tips. MM, metanephric mesenchyme. DAPI stained nuclei are blue. Scale bars: 50 μm.
DISCUSSION
To date, the signaling mechanisms through which BMP7 acts to maintain nephron progenitors have not been elucidated in detail, and the precise target cell of BMP7 has furthermore remained undefined. Using a BMP-reporter mouse, we recently demonstrated that the NZ is essentially unresponsive to SMAD-mediated transcription in vivo (Blank et al., 2008). Owing to the strong effect of Bmp7 inactivation on nephron progenitor maintenance, this surprising finding prompted us to investigate whether BMP7 acts through a SMAD-independent signaling mechanism in the NZ. The most extensively described alternative pathway downstream of TGFβ and BMP is activation of MAPK signaling through TGFβ-activated kinase 1 (TAK1) (Yamaguchi et al., 1995). Activation of p38 and JNK pathways have both been reported downstream of TAK1 (Hanafusa et al., 1999; Shirakabe et al., 1997; Yamaguchi et al., 1995), and immortalized Tak1 mutant embryonic fibroblasts exhibited impaired phosphorylation of JNK (Shim et al., 2005). Mice deficient in Tak1 die at mid-gestation (Jadrich et al., 2006; Shim et al., 2005), precluding analysis of TAK1-mediated signaling in metanephric kidney development. However, TAK1 is expressed in the NZ of the developing kidney, suggesting that activation of MAPK signaling downstream of BMPs might occur in this region of the kidney (Jadrich et al., 2003) (and this study). Previous work by Hu and colleagues indicate that BMP7 can activate p38 signaling in collecting duct cells in vitro (Hu et al., 2004). Similarly, conditional deletion of Bmp7 using a podocyte-specific Cre, resulted in defective p38 signaling, but no effect on phosphorylation of SMAD1/SMAD5/SMAD8 was observed (Kazama et al., 2008). Using our primary cell purification system, we found no evidence that BMP7 activates p38 in the NZ. Instead, several JNK isoforms were rapidly phosphorylated in response to BMP7, resulting in downstream phosphorylation of both Jun and ATF2. Importantly, we show that TAK1 is required for efficient activation of JNK in response to BMP7. SIX2 immunostaining shows that JNK activation takes place directly in the nephron progenitor compartment, indicating that these cells indeed do respond directly to BMP7. Interestingly, phosphorylated forms of the JNK-activated transcription factors Jun and ATF2 are both localized to cap mesenchyme in the developing E17.5 kidney. Furthermore, E12.5 Bmp7-null kidneys displayed sharply reduced JNK-Jun-ATF2 signaling in metanephric mesenchyme compared with the wild type, demonstrating that Bmp7 is required for activation of this signaling axis in nephron progenitors in vivo. Interestingly, phosphorylation of Jun or ATF2 does not appear to be significantly reduced in collecting duct cells of Bmp7-deficient embryos, suggesting that other growth factors function to activate the pathway in this cellular compartment.
JNK protein kinases are encoded by three genes: Jnk1, Jnk2 and Jnk3. Mice lacking genes encoding individual JNK family members are viable, but compound mutants of Jnk1 and Jnk2 exhibit defects in neural tube closure and brain abnormalities and die at mid-gestation (Kuan et al., 1999; Sabapathy et al., 1999). Interestingly, Jnk1-/- Jnk2+/- mutants develop to term but exhibit a number of developmental defects, including deformed renal epithelial cells and nephrons (Weston et al., 2003). The majority of mutant animals die within 48 hours of birth, presumably because of kidney defects. There is thus a clear precedent for the role of JNK in kidney development, and further analysis of JNK compound mutants conditionally deleted within cap mesenchyme comprises an interesting area for future research.
A role for BMP7 as a proliferative factor and as a survival factor have both been proposed, but never distinguished from each other. Our data indicate that BMP7 promotes proliferation of NZ cells in culture through a signaling mechanism dependent on both TAK1 and JNK. Importantly, the majority of proliferating cells reside within the SIX2+ compartment, confirming that BMP7-induced JNK activation directly promotes proliferation of nephron progenitors. A role for JNK signaling in proliferation is well established in vitro: MEF cells deficient in both Jnk1 and Jnk2 exhibit decreased proliferative capacity (Tournier et al., 2000). Similarly, primary embryonic fibroblasts derived from Jun-null embryos display greatly reduced growth in vitro, indicating that Jun might mediate the proliferative signal downstream of JNK1 and JNK2 (Johnson et al., 1993). In vivo, Jun deficiency results in mid-gestation lethality before formation of the NZ (Hilberg et al., 1993; Johnson et al., 1993), and the role of this transcription factor in nephron progenitor maintenance has not been studied. However, conditional deletion of Jun in the liver results in impaired hepatocyte proliferation and decreased liver regeneration upon partial hepatectomy (Behrens et al., 2002). Interestingly, conditional deletion of Smad4 specifically in Bmp7-expressing cells does not lead to impaired proliferation in the developing kidney (Oxburgh et al., 2004), implying that BMP7-mediated SMAD signaling is unlikely to be responsible for the proliferative defect observed in the Bmp7-null kidney. Furthermore, podocyte-specific deletion of Bmp7 results in a growth defect of nephrons and a reduction in Ki67+ proliferating cells in proximal tubules (Kazama et al., 2008). BMP7 thus appears to promote proliferation in several cell types during kidney development. An interesting avenue for future studies will be conditional deletion of Tak1 and Jun in the SIX2+ compartment.
D-type cyclins function to couple extracellular signals to the cell cycle machinery, and are induced by growth factor stimulation in early G1 (Sherr, 1995). Our data show that cyclin D3 is upregulated at the protein level by BMP7 through a JNK-dependent mechanism in vitro. Interestingly, Jun has previously been linked to regulation of cyclin D1 expression (Bakiri et al., 2000). JNK-dependent accumulation of cyclin D3 upon BMP7 stimulation is therefore probably dependent on Jun, although the exact mechanism for this remains to be determined.
Recently, Brunskill and colleagues published a comprehensive atlas of gene expression within the developing mouse kidney (Brunskill et al., 2008). We compared expression of components of the BMP signaling pathway between cap mesenchyme and renal vesicle, the first morphologically distinct stage of nephron differentiation. Interestingly, Jnk2 was downregulated as cap mesenchyme progressed through the renal vesicle stage. By contrast, expression of Id4 and Bambi, known SMAD-responsive BMP target genes, increased as cells advanced to the renal vesicle stage. This corroborates our findings, and indicates that there might be a qualitative switch in the response to BMP stimulation from JNK-mediated signaling to SMAD-mediated signaling in the transition from nephron progenitor to differentiated renal vesicle (Fig. 8). This is furthermore reflected in in vivo BMP reporter activation: SMAD-dependent transcription is silent in cap mesenchyme, but increases in strength from the renal vesicle stage onwards (Blank et al., 2008).
Fig. 8.
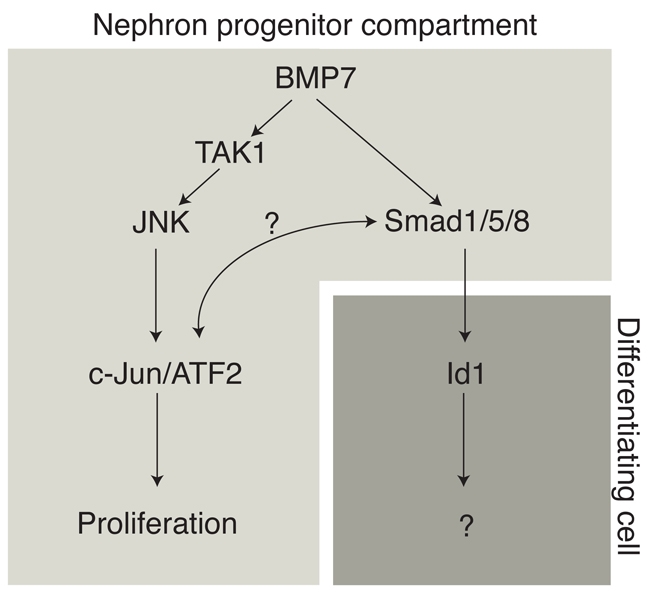
Model of BMP7 signaling in the NZ. BMP7 preferentially functions through the JNK signaling pathway to promote proliferation in the nephron progenitor compartment. The SMAD machinery is silent at the transcriptional level in cap mesenchyme, but becomes increasingly activated as cells progress to the renal vesicle stage and beyond.
Although we could not detect a robust transcriptional response of Id1 upon BMP7 stimulation, our data does not exclude alternative functions of SMAD proteins within the progenitor compartment. We found that SMAD1/SMAD5/SMAD8 could be activated in NZ cells in vitro following BMP7 stimulation, but that this did not lead to transcriptional activation. This data is in agreement with immunostaining of E17.5 kidneys showing weak activation of SMAD1/SMAD5/SMAD8 in the NZ but no downstream transcriptional response as analyzed in the BMP reporter mouse. Whether SMAD and JNK signaling pathways cooperate or negatively regulate each other remains to be investigated, but such a mechanism has been shown for SMAD2 (Pessah et al., 2001) and it is conceivable that a similar mechanism is in place for SMAD1/SMAD5/SMAD8.
In summary, we have analyzed the signaling mechanism, target cells and functional significance of BMP7 during kidney development. We show that BMP7 functions to activate the JNK-Jun-ATF2 signaling axis directly in SIX2+ cap mesenchyme, leading to a proliferative signal through upregulation of cyclin D3.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/cgi/content/full/136/21/3557/DC1
Supplementary Material
This work was supported by R01 DK078161 from NIDDK (L.O.), a Norman S. Coplon extramural grant from Satellite Healthcare (L.O.), and postdoctoral fellowships from the Wenner-Gren Foundations of Sweden and Kungliga Fysiografiska Sällskapet, Lund, Sweden (U.B.). Additional support was provided by Maine Medical Center Research Institute core facilities for Histopathology, Bioinformatics and FACS (supported by 2P20RR18789-06) and the MMCRI Animal Facility. Deposited in PMC for release after 12 months.
References
- Bakiri, L., Lallemand, D., Bossy-Wetzel, E. and Yaniv, M. (2000). Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. EMBO J. 19, 2056-2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens, A., Sibilia, M., David, J. P., Mohle-Steinlein, U., Tronche, F., Schutz, G. and Wagner, E. F. (2002). Impaired postnatal hepatocyte proliferation and liver regeneration in mice lacking c-jun in the liver. EMBO J. 21, 1782-1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank, U., Seto, M. L., Adams, D. C., Wojchowski, D. M., Karolak, M. J. and Oxburgh, L. (2008). An in vivo reporter of BMP signaling in organogenesis reveals targets in the developing kidney. BMC Dev. Biol. 8, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blonska, M., Shambharkar, P. B., Kobayashi, M., Zhang, D., Sakurai, H., Su, B. and Lin, X. (2005). TAK1 is recruited to the tumor necrosis factor-alpha (TNF-alpha) receptor 1 complex in a receptor-interacting protein (RIP)-dependent manner and cooperates with MEKK3 leading to NF-kappaB activation. J. Biol. Chem. 280, 43056-43063. [DOI] [PubMed] [Google Scholar]
- Brunskill, E. W., Aronow, B. J., Georgas, K., Rumballe, B., Valerius, M. T., Aronow, J., Kaimal, V., Jegga, A. G., Grimmond, S., McMahon, A. P. et al. (2008). Atlas of gene expression in the developing kidney at microanatomic resolution. Dev. Cell 15, 781-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll, T. J., Park, J. S., Hayashi, S., Majumdar, A. and McMahon, A. P. (2005). Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev. Cell 9, 283-292. [DOI] [PubMed] [Google Scholar]
- Dan, H. C., Sun, M., Kaneko, S., Feldman, R. I., Nicosia, S. V., Wang, H. G., Tsang, B. K. and Cheng, J. Q. (2004). Akt phosphorylation and stabilization of X-linked inhibitor of apoptosis protein (XIAP). J. Biol. Chem. 279, 5405-5412. [DOI] [PubMed] [Google Scholar]
- Derynck, R. and Zhang, Y. E. (2003). Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577-584. [DOI] [PubMed] [Google Scholar]
- Dressler, G. R. (2006). The cellular basis of kidney development. Annu. Rev. Cell Dev. Biol. 22, 509-529. [DOI] [PubMed] [Google Scholar]
- Dressler, G. R. and Douglass, E. C. (1992). Pax-2 is a DNA-binding protein expressed in embryonic kidney and Wilms tumor. Proc. Natl. Acad. Sci. USA 89, 1179-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley, A. T. and Robertson, E. J. (1997). Overlapping expression domains of bone morphogenetic protein family members potentially account for limited tissue defects in BMP7 deficient embryos. Dev. Dyn. 208, 349-362. [DOI] [PubMed] [Google Scholar]
- Dudley, A. T., Lyons, K. M. and Robertson, E. J. (1995). A requirement for bone morphogenetic protein-7 during development of the mammalian kidney and eye. Genes Dev. 9, 2795-2807. [DOI] [PubMed] [Google Scholar]
- Dudley, A. T., Godin, R. E. and Robertson, E. J. (1999). Interaction between FGF and BMP signaling pathways regulates development of metanephric mesenchyme. Genes Dev. 13, 1601-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge, B., Xiong, X., Jing, Q., Mosley, J. L., Filose, A., Bian, D., Huang, S. and Han, J. (2003). TAB1beta (transforming growth factor-beta-activated protein kinase 1-binding protein 1beta), a novel splicing variant of TAB1 that interacts with p38alpha but not TAK1. J. Biol. Chem. 278, 2286-2293. [DOI] [PubMed] [Google Scholar]
- Grieshammer, U., Cebrian, C., Ilagan, R., Meyers, E., Herzlinger, D. and Martin, G. R. (2005). FGF8 is required for cell survival at distinct stages of nephrogenesis and for regulation of gene expression in nascent nephrons. Development 132, 3847-3857. [DOI] [PubMed] [Google Scholar]
- Hanafusa, H., Ninomiya-Tsuji, J., Masuyama, N., Nishita, M., Fujisawa, J., Shibuya, H., Matsumoto, K. and Nishida, E. (1999). Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-beta-induced gene expression. J. Biol. Chem. 274, 27161-27167. [DOI] [PubMed] [Google Scholar]
- Hatini, V., Huh, S. O., Herzlinger, D., Soares, V. C. and Lai, E. (1996). Essential role of stromal mesenchyme in kidney morphogenesis revealed by targeted disruption of Winged Helix transcription factor BF-2. Genes Dev. 10, 1467-1478. [DOI] [PubMed] [Google Scholar]
- Hilberg, F., Aguzzi, A., Howells, N. and Wagner, E. F. (1993). c-jun is essential for normal mouse development and hepatogenesis. Nature 365, 179-181. [DOI] [PubMed] [Google Scholar]
- Hu, M. C., Wasserman, D., Hartwig, S. and Rosenblum, N. D. (2004). p38MAPK acts in the BMP7-dependent stimulatory pathway during epithelial cell morphogenesis and is regulated by Smad1. J. Biol. Chem. 279, 12051-12059. [DOI] [PubMed] [Google Scholar]
- Jadrich, J. L., O'Connor, M. B. and Coucouvanis, E. (2003). Expression of TAK1, a mediator of TGF-beta and BMP signaling, during mouse embryonic development. Gene Expr. Patterns 3, 131-134. [DOI] [PubMed] [Google Scholar]
- Jadrich, J. L., O'Connor, M. B. and Coucouvanis, E. (2006). The TGF beta activated kinase TAK1 regulates vascular development in vivo. Development 133, 1529-1541. [DOI] [PubMed] [Google Scholar]
- Johnson, R. S., van Lingen, B., Papaioannou, V. E. and Spiegelman, B. M. (1993). A null mutation at the c-jun locus causes embryonic lethality and retarded cell growth in culture. Genes Dev. 7, 1309-1317. [DOI] [PubMed] [Google Scholar]
- Kazama, I., Mahoney, Z., Miner, J. H., Graf, D., Economides, A. N. and Kreidberg, J. A. (2008). Podocyte-derived BMP7 is critical for nephron development. J. Am. Soc. Nephrol. 19, 2181-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, A., Valerius, M. T., Mugford, J. W., Carroll, T. J., Self, M., Oliver, G. and McMahon, A. P. (2008). Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3, 169-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan, C. Y., Yang, D. D., Samanta Roy, D. R., Davis, R. J., Rakic, P. and Flavell, R. A. (1999). The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 22, 667-676. [DOI] [PubMed] [Google Scholar]
- Luo, G., Hofmann, C., Bronckers, A. L., Sohocki, M., Bradley, A. and Karsenty, G. (1995). BMP-7 is an inducer of nephrogenesis, and is also required for eye development and skeletal patterning. Genes Dev. 9, 2808-2820. [DOI] [PubMed] [Google Scholar]
- Massague, J. (1998). TGF-beta signal transduction. Annu. Rev. Biochem. 67, 753-791. [DOI] [PubMed] [Google Scholar]
- Massague, J. and Chen, Y. G. (2000). Controlling TGF-beta signaling. Genes Dev. 14, 627-644. [PubMed] [Google Scholar]
- Osafune, K., Takasato, M., Kispert, A., Asashima, M. and Nishinakamura, R. (2006). Identification of multipotent progenitors in the embryonic mouse kidney by a novel colony-forming assay. Development 133, 151-161. [DOI] [PubMed] [Google Scholar]
- Oxburgh, L., Chu, G. C., Michael, S. K. and Robertson, E. J. (2004). TGFbeta superfamily signals are required for morphogenesis of the kidney mesenchyme progenitor population. Development 131, 4593-4605. [DOI] [PubMed] [Google Scholar]
- Perantoni, A. O., Timofeeva, O., Naillat, F., Richman, C., Pajni-Underwood, S., Wilson, C., Vainio, S., Dove, L. F. and Lewandoski, M. (2005). Inactivation of FGF8 in early mesoderm reveals an essential role in kidney development. Development 132, 3859-3871. [DOI] [PubMed] [Google Scholar]
- Pessah, M., Prunier, C., Marais, J., Ferrand, N., Mazars, A., Lallemand, F., Gauthier, J. M. and Atfi, A. (2001). c-Jun interacts with the corepressor TG-interacting factor (TGIF) to suppress Smad2 transcriptional activity. Proc. Natl. Acad. Sci. USA 98, 6198-6203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabapathy, K., Jochum, W., Hochedlinger, K., Chang, L., Karin, M. and Wagner, E. F. (1999). Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mech. Dev. 89, 115-124. [DOI] [PubMed] [Google Scholar]
- Saxén, L. (1987). Organogenesis of the Kidney. Cambridge: Cambridge University Press.
- Self, M., Lagutin, O. V., Bowling, B., Hendrix, J., Cai, Y., Dressler, G. R. and Oliver, G. (2006). Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 25, 5214-5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr, C. J. (1995). D-type cyclins. Trends Biochem. Sci. 20, 187-190. [DOI] [PubMed] [Google Scholar]
- Shim, J. H., Xiao, C., Paschal, A. E., Bailey, S. T., Rao, P., Hayden, M. S., Lee, K. Y., Bussey, C., Steckel, M., Tanaka, N. et al. (2005). TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 19, 2668-2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakabe, K., Yamaguchi, K., Shibuya, H., Irie, K., Matsuda, S., Moriguchi, T., Gotoh, Y., Matsumoto, K. and Nishida, E. (1997). TAK1 mediates the ceramide signaling to stress-activated protein kinase/c-Jun N-terminal kinase. J. Biol. Chem. 272, 8141-8144. [DOI] [PubMed] [Google Scholar]
- Stark, K., Vainio, S., Vassileva, G. and McMahon, A. P. (1994). Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature 372, 679-683. [DOI] [PubMed] [Google Scholar]
- Tournier, C., Hess, P., Yang, D. D., Xu, J., Turner, T. K., Nimnual, A., Bar-Sagi, D., Jones, S. N., Flavell, R. A. and Davis, R. J. (2000). Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288, 870-874. [DOI] [PubMed] [Google Scholar]
- Vainio, S. and Lin, Y. (2002). Coordinating early kidney development: lessons from gene targeting. Nat. Rev. Genet. 3, 533-543. [DOI] [PubMed] [Google Scholar]
- Vestweber, D., Kemler, R. and Ekblom, P. (1985). Cell-adhesion molecule uvomorulin during kidney development. Dev. Biol. 112, 213-221. [DOI] [PubMed] [Google Scholar]
- Weston, C. R., Wong, A., Hall, J. P., Goad, M. E., Flavell, R. A. and Davis, R. J. (2003). JNK initiates a cytokine cascade that causes Pax2 expression and closure of the optic fissure. Genes Dev. 17, 1271-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi, K., Shirakabe, K., Shibuya, H., Irie, K., Oishi, I., Ueno, N., Taniguchi, T., Nishida, E. and Matsumoto, K. (1995). Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 270, 2008-2011. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, K., Nagai, S., Ninomiya-Tsuji, J., Nishita, M., Tamai, K., Irie, K., Ueno, N., Nishida, E., Shibuya, H. and Matsumoto, K. (1999). XIAP, a cellular member of the inhibitor of apoptosis protein family, links the receptors to TAB1-TAK1 in the BMP signaling pathway. EMBO J. 18, 179-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}