

Summary
Nicotinamide adenine dinucleotide (NAD+) is a central molecule in cellular metabolism and an obligate co-substrate for NAD+-consuming enzymes, which regulate key biological processes such as longevity and stress responses. Although NAD+ biosynthesis has been intensely studied, little analysis has been done in developmental models. We have uncovered novel developmental roles for a nicotinamidase (PNC), the first enzyme in the NAD+ salvage pathway of invertebrates. Mutations in the Caenorhabditis elegans nicotinamidase PNC-1 cause developmental and functional defects in the reproductive system; the development of the gonad is delayed, four uterine cells die by necrosis and the mutant animals are egg-laying defective. The temporal delay in gonad development results from depletion of the salvage pathway product NAD+, whereas the uv1 cell necrosis and egg-laying defects result from accumulation of the substrate nicotinamide. Thus, regulation of both substrate and product level is key to the biological activity of PNC-1. We also find that diet probably affects the levels of these metabolites, as it affects phenotypes. Finally, we identified a secreted isoform of PNC-1 and confirmed its extracellular localization and functional activity in vivo. We demonstrate that nicotinamide phosphoribosyltransferase (Nampt), the equivalent enzyme in nicotinamide recycling to NAD+ in vertebrates, can functionally substitute for PNC-1. As Nampt is also secreted, we postulate an evolutionarily conserved extracellular role for NAD+ biosynthetic enzymes during development and physiology.
Keywords: Pnc1, Npt1, Nicotinic acid, PBEF, Visfatin
INTRODUCTION
Nicotinamide adenine dinucleotide (NAD+) is a crucial reductant in cellular biochemistry. Shuttling of protons between NAD+ and its reduced form NADH is vital during oxidative phosphorylation. NAD+ is also a co-substrate for enzymes called NAD+ consumers, which have an impact on key biological processes, including stress responses and lifespan (Sauve, 2008). The role of NAD+ in oxidative phosphorylation suggests that NAD+ biosynthesis is a ubiquitous cellular process that is crucial to the metabolism of all cells. However, the regulatory activity of NAD+ and its metabolite nicotinamide on NAD+ consumers suggests that NAD+ biosynthetic pathways may elicit specific biological responses. We are interested in probing the functions of the NAD+ salvage pathway and distinguishing between these proposed biological functions in the context of development of a multicellular organism.
NAD+ biosynthesis is achieved via multiple pathways (Fig. 1), including de novo synthesis from amino acid substrates and `salvage' pathways from preformed substrates, such as nicotinamide (NAM), nicotinic acid (NA) and nicotinamide riboside (Bieganowski and Brenner, 2004). The Preiss-Handler pathway converts NA to NAD+ and is conserved among all eukaryotes (Preiss and Handler, 1958a; Preiss and Handler, 1958b). Two alternative enzymatic pathways exist for specifically recycling NAM to NAD+: a pathway through a single intermediate and a longer pathway that converges with the Preiss-Handler pathway (Fig. 1) (Magni et al., 1999). Vertebrate genomes encode nicotinamide phosphoribosyltransferase (Nampt), which converts NAM to nicotinamide mononucleotide (NMN) to initiate the shorter pathway, dictating the nature of the salvage pathway in these species. By contrast, fungi and invertebrates, including Caenorhabditis elegans, encode nicotinamidases, which convert NAM to nicotinic acid, dictating use of the longer salvage pathway (Fig. 1) (Magni et al., 1999; Revollo et al., 2007b; Yang et al., 2006).
Fig. 1.
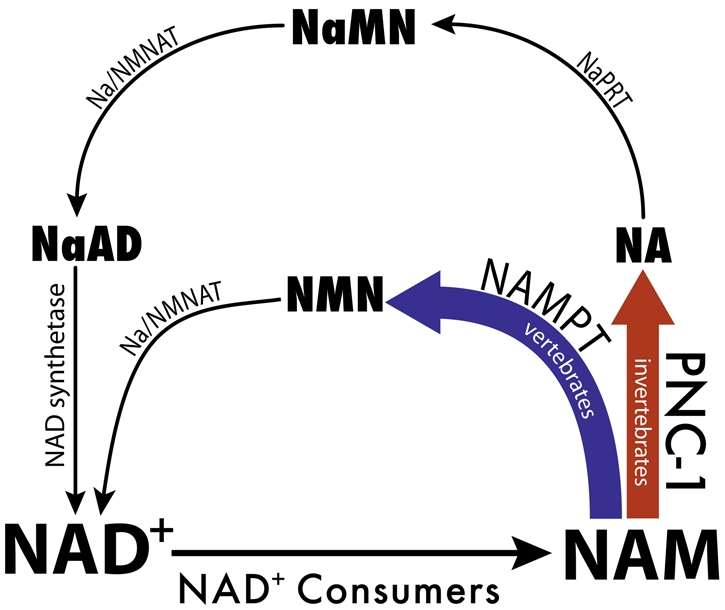
NAD+ salvage biosynthesis pathways. NAD+ consumer enzymes hydrolyze NAD+ and generate NAM. NAM is recycled to reform NAD+ via pathways that differ between invertebrates and vertebrates. PNC (nicotinamidase, in bold red) is found only in invertebrates and fungi. Nampt (purple) is found only in higher eukaryotes. The Preiss-Handler pathway (thin black arrows) is used by all eukaryotes to synthesize NAD+ from NA. NaMN, nicotinic acid mononucleotide; NaAD, nicotinic acid adenine dinucleotide; Na/NMNAT, dual function nicotinic acid/nicotinamide adenyl transferase; NaAD, nicotinic acid adenine dinucleotide; NMN, nicotinamide mononucleotide; NaPRT, nicotinic acid phosphoribosyl transferase.
Although Nampt and nicotinamidases are distinct enzymes, they have equivalent functions in producing NAD+ from NAM. Each has an impact on the concentration of NAD+ as well as their substrate NAM, which has independent biological activity. NAM is both a product of (Landry et al., 2000; Tanny and Moazed, 2001) and a non-competitive feedback inhibitor of (Avalos et al., 2005; Landry et al., 2000; Sauve et al., 2005; Sethi et al., 1996) NAD+ consumers. Because of the predicted impact of nicotinamidases and Nampt on the levels of NAD+ and NAM, these enzymes are hypothesized to have biological regulatory activity and, in particular, to regulate the NAD+-dependent NAD+ consumers, such as sirtuins and PARPs (Yang et al., 2006).
The regulatory activity of NAD+ salvage pathways on sirtuins is well established. High levels of NAM inhibit yeast Sir2 deacetylase activity and its function in transcriptional silencing and longevity (Anderson et al., 2003; Gallo et al., 2004). In addition, perturbation of the salvage pathway at points that do not perturb NAM levels and alterations in the NAD+/NADH ratio also have an impact on sirtuin activity, suggesting that in vivo regulation of NAD+ levels or flux through the salvage pathway affects sirtuin activity (Anderson et al., 2002; Belenky et al., 2007; Lin et al., 2000; Lin et al., 2004; Sandmeier et al., 2002; Smith et al., 2000). In mammals, Nampt-mediated production of NAD+ contributes to increased activity of the SIRT1 deacetylase (Revollo et al., 2004). Nampt also regulates NAD+ levels in mitochondria, affecting cell survival under stress conditions in a manner dependent on mitochondrial sirtuins SIRT3 and SIRT4 (Yang et al., 2007). Nampt mediated reduction in NAM levels is also implicated in mediating Nampt effects (Fulco et al., 2008). However, the relative importance of increases in NAD+ biosynthesis versus decreases in NAM levels to the biological activity of nicotinamidase and Nampt are still not well understood. Furthermore, our understanding of the role of this salvage pathway in the development of animals is limited.
We report here on distinct biological roles for a nicotinamidase in C. elegans development. Caenorhabditis elegans has two pnc (pyrazinamidase and nicotinamidase) genes, pnc-1 and pnc-2. Overexpression of PNC-1 has a positive effect on survival under conditions of oxidative stress, and pnc-1(RNAi) is reported to decrease lifespan (van der Horst et al., 2007). However, no alleles of pnc-1 or pnc-2 have been previously analyzed, and the RNAi studies failed to reveal developmental defects. We have identified mutations in PNC-1 that surprisingly indicate multiple yet specific roles for the protein in the development and function of the reproductive system. We present results that highlight the dual biological functions of the pathway in promoting NAD+ biosynthesis and in modulating NAM levels.
We also demonstrate functional similarity between PNC-1 and the vertebrate salvage protein Nampt using rescue assays. Nampt is expressed in limited tissues in mice and is secreted by a non-classical mechanism to function as an extracellular enzyme (Revollo et al., 2007b). We find that C. elegans PNC-1 is probably expressed in a limited number of cells and that it has a secreted isoform that can function extracellularly. We postulate an evolutionarily conserved role for secreted NAD+ biosynthetic enzymes and discuss the implications of our results to models for NAD+ salvage function on an organism-wide level.
MATERIALS AND METHODS
Caenorhabditis elegans culture
N2 is the wild-type strain. Strains were grown under standard conditions with OP50 Escherichia coli as a food source at 20°C (Brenner, 1974), except where supplemented with nicotinamide (Alfa Aesar, Ward Hill, MA, USA) and nicotinic acid (Calbiochem, San Diego, CA, USA). We added filter sterilized 1 M stock solutions of NAM and NA (NA solution adjusted to pH 5.4 with 10 N NaOH for solubility) to cooled NGM media at the appropriate dilution immediately before pouring culture plates. We transferred L4 hermaphrodites to supplemented plates and observed their progeny for defects. β-Nicotinamide mononucleotide (Sigma, St Louis, MO, USA), 50 μM, was prepared in water immediately before injection into the body cavity of late L2 animals. To prepare dead food, we spotted standard culture plates with 250 μl of the same culture of OP50. Lawns were grown for either 2 or 3 days and then half were irradiated for 10 minutes using a BioRad GS Gene Linker UV cross-linker. We streaked random plates to check culture viability. We bleached gravid hermaphrodites on dead or live control plates, transferred progeny to a fresh plate of the same condition and scored during L4 or adulthood, as appropriate for the phenotype.
Phenotype assessment
Egg-laying defective (Egl): L4 hermaphrodites were placed on an individual plate and observed for 3 to 4 days. We report the incidence of normal egg laying, scored as percentage of hermaphrodites in the population that lay eggs and escape death due to internal hatching of progeny.
uv1 necrosis: within the uterus of adults, inIs179[ida-1::GFP] is specifically expressed in the uv1 cells. Reduction in the number of GFP-positive cells in the uterus of inIs179; pnc-1(ku212) animals is due to necrosis of the uv1 cells (Huang and Hanna-Rose, 2006). The percentage of normal uv1 cells is the ratio of observed number of inis179 GFP-positive uv1 cells to expected number (four per animal). Sample sizes indicate number of animals scored.
Gonad delay: uterine and vulval morphogenesis normally proceed simultaneously, and mid-L4 animals have an open lumen in both organs. However, most pnc-1 mutants with a typical mid-L4 vulva have not yet developed a uterine lumen (Huang and Hanna-Rose, 2006). We report the percentage of animals with an open lumen in both organs to indicate percentage of normal temporal development.
Transgene production
We generated constructs by PCR amplification of the desired fragment and insertion into a Fire Lab Kit expression vector (A. Fire, S. Xu, J. Ahnn and G. Seydoux, personal communication) followed by sequence confirmation of insert. See Table 1 for plasmid construction details. We generated transgenes by microinjection of plasmid, YAC or cosmid into the germline of unc-119(ed3) hermaphrodites with 60-90 ng/μl unc-119(+) (Maduro and Pilgrim, 1995) as a co-transformation marker.
Table 1.
Constructs and primers used in this study

cDNAs for candidate genes during transformation rescue were obtained from Dr Yuji Kohara (National Institute of Genetics, Japan) or by amplification from a mixed stage C. elegans cDNA library (Stratagene, La Jolla, CA, USA). cDNA for gene Y38C1AA.3a (PNC-1a) was amplified from the library. We used RT-PCR to obtain PNC-1b and PNC-2 cDNAs. To extract total RNA, we washed animals off plates using DEPC-H2O followed by TRIzol (Invitrogen, Carlsbad, CA, USA) extraction, chloroform treatment and isopropanol precipitation and resuspended the RNA in 25 μl of DEPC-H2O. We used 5 μl of total RNA as template for SuperScript One-Step RT-PCR System with Platinum Taq DNA Polymerase (Invitrogen) using a 60-minute cDNA synthesis and 35 cycles of PCR. Primers are detailed in Table 1.
Nicotinamidase activity assay
Expression of His-tagged PNC proteins (Table 1) was induced in BL21 (DE3) cells with 200 μM IPTG at 18°C for 20 hours. Protein was harvested using sonication and purified using Ni-NTA agarose (Qiagen, Valencia, CA, USA). Nicotinamidase activity was assayed as described (Anderson et al., 2003). Briefly, recombinant wild-type or mutant PNC protein (between 5 and 20 μg, determined using a Bradford assay) was combined with either 0 or 8 mM NAM in a final volume of 400 μl buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl and 1 mM MgCl2) and incubated at 30°C. Ammonia production was assessed using an Ammonia Assay Kit (Sigma) and corrected by subtracting the 0 mM NAM control according to the manufacturer's instructions. We performed triplicate experiments at 30 minutes and calculated specific activity (nmol of ammonia produced/minute/mg of PNC).
RESULTS
pnc-1 mutants have defects in gonad development and egg-laying ability
We isolated the ku212 allele in a screen for egg-laying defective mutants and originally named the gene cog-3, connection of gonad defective (Huang and Hanna-Rose, 2006). ku212 mutants have a delay in global gonad development relative to the soma and an egg-laying defect. Additionally, four uterine cells, called uterine vulval 1 (uv1) cells, undergo necrotic death before full differentiation (Huang and Hanna-Rose, 2006).
We mapped ku212 near the left telomere of chromosome IV. One of eight transgenes bearing sequences from YAC Y38C1 rescued the ku212 developmental defects (Table 2) and a second transgene conferred weak rescue (not shown), confirming that the gene resides within the first 400 kb of chromosome IV. However, none of the available cosmids from this region rescued ku212. Furthermore, ten transgenes bearing sequences from Y66H1, which overlaps Y38C1, failed to rescue (not shown). Two regions in Y38C1 are not cloned in cosmids or included in Y66H1, leaving these regions untested. Thus, we used an unconventional method to identify the gene by testing the ability of candidate, uncloned genes to rescue ku212 when expressed from the broadly active sur-5 promoter. Broad expression of Y38C1AA.3a rescues the uv1 cell death, the egg-laying function and the temporal delay in development of the gonad of ku212 mutants (Table 2).
Table 2.
Loss of pnc-1 activity causes specific reproductive defects
|
uv1 cell
specified†
|
Normal gonad
timing†
|
Normal
egg-laying†
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Strain | % | n | P¶ | % | n | P¶ | % | n | P¶ |
| ku212; psEx199 (control)‡,§ | 9 | 48 | - | 42 | 19 | - | 29 | 42 | - |
| ku212; psEx44 (Yac Y38C1) | 98 | 99 | *** | 97 | 31 | *** | 100 | 30 | *** |
| ku212; psEx124 (sur-5::PNC-1a) | 95 | 38 | *** | 100 | 21 | *** | 100 | 26 | *** |
| ku212; psEx125 (sur-5::PNC-1a) | 98 | 47 | *** | 100 | 16 | ** | 100 | 26 | *** |
| ku212; psEx152 (sur-5::PNC-1b) | 99 | 23 | *** | 100 | 21 | *** | 72 | 89 | *** |
| ku212; psEx155 (sur-5::PNC-1b) | 80 | 22 | *** | 100 | 17 | ** | 67 | 22 | ** |
| pk9605; psEx200 (control) | 4 | 75 | - | 54 | 26 | - | 25 | 81 | - |
| pk9605; psEx166 (sur-5::PNC-1a) | 96 | 77 | *** | 100 | 54 | *** | 79 | 47 | *** |
| pk9605; psEx167 (sur-5::PNC-1b) | 93 | 47 | *** | 100 | 31 | *** | 72 | 36 | *** |
Phenotypes are scored as described in Materials and methods. n, number of animals scored.
Transgenes are named psEx##, as detailed in Table 1.
Control transgenes, as detailed in Table 1.
P-value calculated using Fischer's exact test, comparing the sample with the respective transgenic control. **P<0.005, ***P<0.0001.
Y38C1AA.3 encodes PNC-1 (Pyrazinamidase and NiCotinamidase) (van der Horst et al., 2007), a 335 amino acid protein with a catalytic isochorismatase hydrolase domain most homologous to nicotinamidases, as predicted by SMART (Letunic et al., 2004) (Fig. 2). Interestingly, cDNA evidence indicates two pnc-1 isoforms, which are identical except for alternate first exons (Fig. 2). Exon 1a of pnc-1a encodes 20 amino acids that comprise a signal peptide (predicted by SignalP 3.0) (Bendtsen et al., 2004) that is cleaved after the fourth amino acid encoded by exon 2, resulting in probable extracellular localization for PNC-1a. By contrast, the first exon of the pnc-1b isoform is untranslated with translation predicted to start at the first codon of exon 2. Thus, PNC-1b and mature cleaved PNC-1a are identical with the exception of the first four amino acids, and we predict that PNC-1b is cytoplasmic or cytonuclear using WoLFPSORT (Horton et al., 2007). EST clones (http://www.wormbase.org) verify the expression of both isoforms.
Fig. 2.

The pnc-1 gene locus. The use of alternative first exons, exon 1a and exon 1b, leads to production of two isoforms, pnc-1a and pnc-1b. exon 1a encodes the bulk of a predicted cleaved signal peptide (red). Exon 1b is non-coding (light blue) and translation of PNC-1b would start at the first codon (ATG) of exon 2. Green hatched boxes indicate the extent of the conserved catalytic domain. Vertical dashed lines indicate the position and nature of the lesions associated with ku212 and pk9605. The horizontal dashed lines left of the genes illustrate the extent of genomic sequence used in transcriptional GFP fusion reporters.
We used RT-PCR to recover pnc-1a cDNA from ku212 mutants for allele identification, further confirming expression of this isoform. Sequence analysis shows a G to A transition in codon 328. This lesion introduces a stop codon in exon four, truncating the protein by only eight amino acids (Fig. 2). We verified the lesion by sequencing an amplified genomic fragment. Based on transgenic rescue and sequencing data, we conclude that ku212 is an allele of pnc-1 and that ku212 would affect both pnc-1 isoforms (Fig. 2).
Cuppen et al. generated alleles of pnc-1 using target-selected mutagenesis (Cuppen et al., 2007). We isolated four pnc-1 alleles from their library: pk9598(L109F), pk9603(E188K), pk9609(S228F) and pk9605. pk9598, pk9603 and pk9609 introduce missense mutations and display no phenotypes (not shown). pnc-1(pk9605) introduces a premature stop codon (W201X), resulting in truncation of 135 amino acids (Fig. 2). This lesion removes half of the conserved catalytic domain from both PNC-1 isoforms (Fig. 2), including two of three active site residues, and is a presumed null allele. Like pnc-1(ku212), pnc-1(pk9605) hermaphrodites exhibit uv1, egg-laying and gonad development defects that can be rescued by expressing PNC-1a or PNC-1b (Table 2), further confirming that the observed defects in gonad development and egg laying result from loss of PNC-1 activity.
Whereas pk9605 is a putative loss-of-function allele, the nature of the ku212 allele is less clear. The uv1, gonad and egg-laying defects are recessive, and overexpression of the protein encoded by pnc-1a(ku212), PNC-1a(W328X), in wild type does not produce any detectable phenotypes (not shown), indicating that PNC-1a(W328X) does not have a dominant effect. Furthermore, overexpression of PNC-1a(W328X) does not rescue pk9605 (Table 3), suggesting that the mutant protein retains little function.
Table 3.
Nicotinamidase activity is crucial for PNC-1 functions in reproductive development
|
uv1 cell
specified†
|
Normal gonad
timing†
|
Normal
egg-laying†
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Strain | % | n | P | % | n | P | % | n | P |
| pk9605; psEx200 (control)‡,§ | 4 | 75 | - | 54 | 26 | - | 25 | 81 | - |
| pk9605; psEx183 [sur-5::PNC-1 (W328X)] | 7 | 77 | NS | 34 | 32 | NS | 10 | 42 | NS |
| pk9605; psEx188 [sur-5::PNC-1 (W328X)] | 4 | 53 | NS | 44 | 27 | NS | 17 | 47 | NS |
| pk9605; psEx168 [sur-5::PNC-1 (C265A)] | 11 | 54 | ** | 30 | 20 | NS | 38 | 29 | NS |
| pk9605; psEx169 [sur-5::PNC-1 (C265A)] | 1 | 32 | NS | 50 | 20 | NS | 31 | 35 | NS |
| pk9605; psEx187 (sur-5::PNC-2) | 59 | 40 | *** | 100 | 16 | ** | 79 | 48 | *** |
| pk9605; psEx197 (sur-5::PNC-2) | 63 | 25 | *** | 97 | 33 | *** | 85 | 27 | *** |
| pk9605; psEx180 (sur-5::yeast Pnc1p) | 77 | 70 | *** | 89 | 28 | * | 55 | 42 | ** |
| pk9605; psEx184 (sur-5::yeast Pnc1p) | 72 | 20 | *** | 91 | 12 | * | 98 | 41 | *** |
| pk9605; psEx170 (sur-5::humanNAMPT) | 15 | 28 | ** | 95 | 21 | ** | 7 | 31 | NS |
¶P-value calculated using Fischer's exact test, comparing the sample with the respective transgenic control. *P<0.05, **P<0.005, ***P<0.0001; NS, not significant.
Phenotypes are scored as described in Materials and methods. n, number of animals scored.
Transgenes are named psEx##, as detailed in Table 1.
Control transgene expresses unc-119 and sur-5::GFP, as detailed in Table 1.
To directly test the enzymatic activity of PNC-1 and the mutant proteins encoded by ku212 and pk9605, we made recombinant proteins and assayed nicotinamidase activity. Recombinant PNC-1 has robust nicotinamidase activity, whereas we observed no activity for PNC-1(W328X) or PNC-1(W201X) (Fig. 3). We conclude that PNC-1 is a nicotinamidase and that pk9605 is a loss-of-function allele. Whereas the molecular nature of the ku212 allele may not suggest loss of function, the lack of rescue activity or nicotinamidase activity of PNC-1(W328X) suggest that ku212 is also a strong reduction-of-function allele.
Fig. 3.
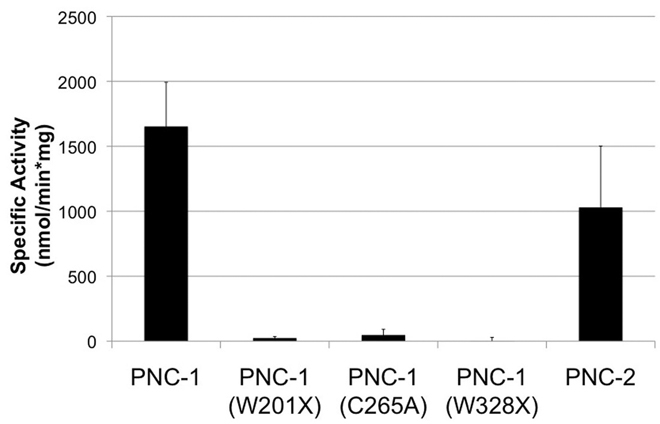
PNC-1 is a nicotinamidase. The average specific activity from three assays is plotted with error bars indicating s.d. PNC-1 and PNC-2 have nicotinamidase activity, but the W201X, C265A and W328X mutants do not.
Nicotinamidase activity is crucial for pnc-1 function in development
Nicotinamidase mutants have not previously been associated with animal developmental defects. Thus, we sought to test the hypothesis that the lack of nicotinamidase activity of PNC-1, and not some other unknown function, is responsible for the novel developmental defects revealed in pnc-1 mutants. We took a transgenic approach and attempted to rescue pnc-1 mutants with catalytically inactive PNC-1 and two alternate nicotinamidases.
Yeast Pnc1p residue C167 is required for catalytic function (Du et al., 2001; Gallo et al., 2004; Hu et al., 2007) and corresponds to C265 of C. elegans PNC-1. We mutated C265 to alanine and confirmed that this mutation eliminates catalytic activity in vitro (Fig. 3). We expressed PNC-1b(C265A) as a GFP fusion from the sur-5 promoter and tested its ability to rescue pnc-1(pk9605) mutant phenotypes. Although we observe robust expression of PNC-1b(C265A)::GFP in duplicate transgenic lines (not shown), neither transgene rescued pnc-1(pk9605) animals (Table 3).
If the nicotinamidase activity of PNC-1 is crucial to its ability to promote reproductive development and function, we would expect that expression of another nicotinamidase would substitute for PNC-1. Indeed, S. cerevisiae Pnc1 (YGL037C) was able to robustly rescue the pnc-1(pk9605) defects when expressed from the widely active sur-5 promoter (Table 3). Additionally, the C. elegans genome encodes a second nicotinamidase, PNC-2 (Y57G11C.47). We isolated a PNC-2 cDNA from wild-type animals using RT-PCR, demonstrating that PNC-2 is an expressed gene, and confirmed its nicotinamidase activity in vitro (Fig. 3). Broad expression of PNC-2 also rescues pnc-1(pk9605) (Table 3). Thus, other nicotinamidases can substitute for PNC-1. We conclude that nicotinamidase activity is crucial to the PNC-1 developmental functions.
NAD+ depletion and substrate buildup separately contribute to pnc-1 mutant phenotypes
NAD+ salvage pathway activity is expected to have an impact on the levels of both NAD+ and NAD+ precursors. In mutants lacking PNC-1 nicotinamidase activity, nicotinamide (NAM) conversion to nicotinic acid (NA) would be compromised. As a result, we would expect NAM concentration to rise and NA concentration to fall in tissues where PNC-1 functions. Additionally, as NAM is not being recycled into NAD+, the level of NAD+ may also decrease. The relevance of each independent effect to the biological activity of nicotinamidase in yeast or to the activity of the equivalent mammalian protein Nampt has been debated. To investigate the relative importance of perturbations on the concentration of both the substrate and product when PNC-1 activity is compromised, as well as to further confirm that the lack of nicotinamidase activity causes the observed pnc-1 mutant phenotypes, we used a pharmacological approach to determine which perturbations underlie which pnc-1 mutant phenotypes.
To partially mimic the effect of pnc-1 mutation, we supplemented wild-type cultures with NAM. Wild-type hermaphrodites raised in the presence of 12.5 mM (not shown) or 25 mM NAM (Fig. 4A), have highly penetrant uv1 necrosis and egg-laying defects but do not have delayed gonad development. To assess the contribution of decreased NA levels to pnc-1 mutant phenotypes, we raised pnc-1 mutant animals on culture plates supplemented with NA. The gonad delay phenotype of NA-supplemented pnc-1(pk9605) and pnc-1(ku212) mutants was robustly rescued (Fig. 4B and not shown). However, these pnc-1(pk9605) animals had no change in the penetrance of uv1 necrosis or egg laying. NA did not induce any pnc-1 mutant defects in wild-type animals (not shown). We conclude that the pnc-1 phenotypes are strikingly separable and that NAM and NA concentrations are each physiologically relevant in different circumstances, perhaps in a tissue-dependent manner.
Fig. 4.

Substrate accumulation and product depletion independently affect pnc-1 phenotypes. (A) NAM (25 mM) causes uv1 necrosis and an egg-laying defect in wild-type animals but does not delay gonad development. Higher levels of NAM (up to 75 mM) have moderate deleterious effects on viability and fecundity (not shown). (B) Supplementation with 25 mM NA rescues the gonad temporal delay but not the egg-laying or uv1 necrosis phenotypes of pnc-1 mutants. NA (up to 25 mM) has no effect on wild-type animals. Levels above 50 mM cause sterility and death in wild type and mutants (not shown). Injection of 50 μM NMN into late L2 to early L3 pnc-1 mutants rescues the gonad delay but not the uv1 cell necrosis. Black bars indicate percentage of normal uv1 cell differentiation. Light gray bars indicate percentage of population with normal temporal development of the gonad, and dark gray bars indicate percentage of population with normal egg-laying ability. Actual percentages and sample sizes are indicated either above or on each bar. *, P<0.05, calculated using Fisher's exact test.
To further probe the underlying basis of the distinct gonad delay phenotype associated with a deficiency of the NAD+ precursor NA, we sought to determine if NA rescue results from an increase in NA concentration per se or from an indirect effect on NAD+ biosynthesis via restoration of the salvage pathway. We specifically tested the model that maintenance of NAD+ biosynthesis but not NA production is key to prevention of the gonad developmental delay by restoring an alternative salvage pathway in the mutants without restoring pnc-1 activity or affecting NA concentration. In vertebrates, Nampt catalyzes the conversion of NAM to nicotinamide mononucleotide (NMN), which is subsequently converted to NAD+ by a dual function enzyme, nicotinic acid/nicotinamide mononucleotide adenyltransferase Na/NMNAT (Fig. 1). Na/NMNAT is also used to convert NaMN to NaAD in the invertebrate salvage pathway. Thus, although no Nampt is encoded in the C. elegans genome, Na/NMNAT is present. By expressing human Nampt from the sur-5 promoter, we expect to provide the enzyme needed to form NAD+ from NAM via an alterative vertebrate-like pathway (Fig. 1). We found that sur-5p::Nampt rescued the gonad developmental delay of pnc-1 mutants (Table 3). The Nampt transgene also partially rescued uv1 cell necrosis (Table 3), as expected if NAM is being partially depleted. We also raised wild-type and pnc-1(pk9605) animals on media supplemented with NMN, the product of Nampt. NMN rescued gonad development of the mutants (not shown), but it also induced uv1 cell necrosis in wild type, probably because NMN is hydrolytically unstable (Kobayashi and Maudsley, 1969) and degrades into NAM during culture. To reduce the chance of hydrolysis, we directly injected 50 μM NMN into the body cavity of young animals. Injection of NMN into wild type had no effect on gonad development and minimal effect on uv1 cells (not shown), indicating little hydrolysis to NAM. By contrast, injection of NMN into pnc-1(pk9605) mutants rescued the gonad delay but not the uv1 defect (Fig. 4B). These data support the hypothesis that maintenance of NAD+ levels can prevent the developmental delay in the gonad. These experiments also highlight the similarity in biological function of Nampt and nicotinamidase.
Viability of diet impacts penetrance of pnc-1 phenotypes
As NAD+ is such a pivotal biomolecule in an array of processes, it seems surprising that loss of pnc-1 results in specific reproductive defects of varying penetrance but no other obvious deleterious effects. We considered alternative sources of NAD+ that could affect NAD+-dependent phenotypes, including PNC-2 mediated salvage, de novo synthesis from tryptophan and the E. coli food source.
We are presently unable to directly test the contribution of PNC-2 to the development and physiology of C. elegans, because there are no loss-of-function alleles of pnc-2, and RNAi of pnc-2 has no effect on either wild type or pnc-1 mutants. We tested the efficiency of pnc-2 RNAi by examining the effectiveness of pnc-2(RNAi) in preventing PNC-2-mediated rescue of pnc-1(ku212). PNC-2 expression restores uv1 cell specification to 94% (n=33) in the ku212 mutant background. pnc-2(RNAi) only weakly prevents rescue by PNC-2, reducing uv1 specification to 62% (n=96), suggesting that pnc-2(RNAi) results in only a weak knockdown. Although PNC-2 may catalyze NAM to NAD+ salvage in some tissues to promote viability, the endogenous activity of PNC-2 is not sufficient to compensate for the reproductive defects.
We next considered the de novo NAD+ synthesis pathway in C. elegans. Blast analysis using the yeast and mammalian sequences for the six enzymes that mediate de novo NAD+ synthesis in eukaryotes revealed no homolog to quinolinate phosphoribotransferase (QPRTase) (Altschul et al., 1997). QPRTase converts quinolinic acid to NaMN, converging with the salvage pathway, and is essential for de novo synthesis from tryptophan. The apparent loss of QPRTase in C. elegans has also been reported by Rongvaux et al., and suggests that C. elegans use salvage as their primary method of generating NAD+ (Rongvaux et al., 2003). Thus, the lack of more deleterious phenotypes is not due to compensatory activity of a de novo synthesis pathway.
Lastly, we examined the effects of diet on pnc-1 mutants by comparing the phenotypes of animals grown on live versus dead E. coli. Growth on live versus dead E. coli had no effect on the development of wild type and resulted in no new detectable developmental phenotypes in mutants. However, we observed a marked increase in the penetrance of the gonad-timing defect of pnc-1 mutants grown on dead food (Fig. 5). Combined with our demonstration that NAD+ synthesis defects underlie the gonad development phenotype, this suggests that NAD+ synthesis by the bacterial food source partially compensates for pnc-1 reproductive defects.
Fig. 5.

The E. coli food source partially compensates for pnc-1 during gonad development. The penetrance of the gonad-timing defect is dramatically increased in pnc-1 mutants fed dead OP50 E. coli. Black bars indicate percentage of normal uv1 cell differentiation. Light gray bars indicate percentage of population with normal egg-laying ability, and dark gray bars indicate percentage of population with normal temporal development of the gonad. Actual percentages and sample sizes are indicated either above or on each bar. *, P<0.05, calculated using Fisher's exact test.
The pnc-1a and pnc-1b promoters are active in distinct locations and in only a few specific cells
Two broad models could explain the specific defects we observe when perturbing the NAD+ salvage pathway: (1) the pathway could elicit tissue-specific responses because of the intrinsic genetic program of specific cells and tissues (for example tissue-specific expression of an NAD+ consumer); or (2) perturbation of the pathway could elicit specific responses due to tissue-specific sensitivities to metabolism. To begin to investigate these alternatives, we examined the localization of the PNC-1 proteins to see if they were generally expressed as predicted for a ubiquitously functioning metabolic pathway or were more limited in their expression pattern, suggesting potential regulatory activity. We used GFP reporters to examine expression patterns of each PNC-1 isoform.
We isolated a pnc-1a promoter consisting of exon 1a and the intervening region between exon 1a and the adjacent gene (Fig. 2). We fused this promoter region to GFP with or without a nuclear localization signal (NLS) to form pnc-1a::exon1a::GFP and made two transgenic lines with each construct. We observed consistent expression patterns in a limited set of cells in the four extrachromosomal transgenic lines. We detected robust GFP expression in head and pharyngeal muscle (Fig. 6A and not shown), the two ASK neurons (Fig. 6A), the two distal tip cells (Fig. 6B) and the three rectal gland cells (RectD, RectVL and RectVR) (Fig. 6C). Whereas the four uv1 cells did not express pnc-1a::exon1a::GFP, four adjacent uv2 cells had moderate levels of GFP expression (Fig. 6D). Low levels of pnc-1a::exon1a::GFP expression were also detected in the vulval muscle (not shown).
Fig. 6.

PNC-1a and PNC-1b are differentially expressed and localized. (A-E′) Nomarski photomicrographs (A-E) and corresponding fluorescence images (A′-E′). The pnc-1a promoter is active in a limited number of cells. (A,A′) Head muscle and the ASK neurons (arrow, ASKL) in psEx142. (B,B′) The distal tip cells (arrow, anterior DTC) in psEx140. (C,C′) Rectal gland cells, rectVL (arrow), rectVR and rectD (not shown) in psEx140. (D,D′) The uv2 cells (arrows, left side) in psEx140. Note that the subcellular localization in different cell types is similar and is never nuclear even though the transgene in A′ contains a nuclear localization sequence. (E,E′) The pnc-1b promoter (psEx156) is active in a number of head neurons (and head and vulva muscle, not shown). Scale bar: 10 μm.
Using PROSCAN 1.7 (Prestridge, 1995), we predict a separate promoter for pnc-1b between exons 1a and 1b. To examine the activity of this PNC-1b promoter, we fused the intervening region between exon 1a and exon 1b to GFPNLS (Fig. 2) and again examined an extrachromosomal transgenic line for GFP expression. pnc-1b::exon1b::GFPNLS transgenic animals had extensive expression in the neurons of the pharynx and the nerve ring (Fig. 6E), although never in ASK or within the gonad. Within the egg-laying apparatus, we detected only weak expression in the vulval muscle of a minority of animals (not shown). Thus, the pnc-1b reporter overlaps in expression pattern with the pnc-1a reporter only in head muscle and perhaps vulval muscle. These distinct and tissue-limited expression patterns seem more consistent with a specific regulatory function of PNC-1 than a general metabolic function.
Evidence for secretion of the PNC-1a isoform
In light of the proposed extracellular role of Nampt, the presence of a putatively secreted isoform of PNC-1 is intriguing. We thus investigated potential extracellular localization of PNC-1a. First, we investigated the authenticity of the signal peptide by examining the subcellular localization of the GFP fusions to exon 1a. We constructed pnc-1a::exon1a::GFP transgenes that differ with respect to the presence or absence of a nuclear localization signal in the GFP. SignalP 3.0 (Bendtsen et al., 2004) predicts that in the context of the GFP fusion without the four residues from PNC-1 exon 2, the signal peptide would not be cleaved, leading to localization of the GFP protein to membranes of the secretory structures, regardless of the presence of the latter NLS. As predicted, we observed no difference in subcellular localization of the GFP expressed from the two transgenes. Fluorescence is always distinctly non-nuclear, even in the presence of the NLS (Fig. 6A), and is observed in a punctate pattern (e.g. Fig. 6A′,C′), consistent with localization to the membranes of the secretory system.
By expressing a secreted GFP-protein fusion in muscle, others have previously visualized protein secretion into the C. elegans body cavity and subsequent uptake by coelomocytes (Fares and Greenwald, 2001). As a second method to explore the subcellular localization of the protein isoforms, we likewise expressed PNC-1a::GFP and PNC-1b::GFP fusions with the muscle-specific myo-3 promoter. We examined GFP fusion protein localization in two transgenic lines for each isoform. All four transgenes restored normal phenotypes to between 97 and 100% of transgenic ku212 animals (n=13-32 animals per strain), demonstrating expression of each translational GFP fusion. However, they had strikingly distinct expression patterns. The myo-3::PNC-1b transgenes caused intense GFP fluorescence throughout the cytoplasm and nucleus of muscle cells (e.g. Fig. 7A). By contrast, there was no GFP fluorescence in the muscles of animals with the myo-3::pnc-1a transgenes (Fig. 7B), as expected after efficient secretion. Upon expression at a higher level, PNC-1a::GFP visibly accumulated in the body cavity and within the coelomocytes (Fig. 7C). We conclude that PNC-1a is a secreted isoform.
Fig. 7.

PNC-1a is secreted. (A-B′) Nomarski photomicrographs (A,B) and corresponding fluorescent images (A′,B′) of a body wall muscle cell in an animal with a functional (A) myo-3::PNC-1b::GFP reporter or (B) myo-3::PNC-1a::GFP reporter. Fluorescent images in A and B were acquired and processed using identical conditions. Both transgenes efficiently rescue all pnc-1 mutant defects (not shown). (C,C′) Nomarski (C) and fluorescent (C′) images of L4 animal with higher levels of the myo-3::PNC-1a::GFP transgene demonstrating accumulation of the translational fusion in the coelomocytes (arrows) and body cavity (arrowheads). Scale bars: 10 μm in A; 5 μm in C.
DISCUSSION
A novel role for nicotinamidase activity in regulating development
We have revealed a role for the NAM to NAD+ salvage biosynthesis pathway in development: blocking the pathway at the first step caused defects in development and function of the reproductive system in C. elegans. Specifically, mutation of the nicotinamidase PNC-1 caused necrosis of the uv1 cells, delayed development of the gonad and an egg-laying defect. Interestingly, Nampt, the proposed functionally equivalent enzyme of NAD+ metabolism in vertebrates, is required for viability (Revollo et al., 2007a; Yang et al., 2006). Nampt homozygous mutant mice die before embryonic day 10.5 (Revollo et al., 2007b). Thus, a biological role for NAD+ salvage during developmental processes may be conserved.
Does PNC-1 elicit biological responses indirectly via effects on metabolism or directly via control of other cellular processes?
The specific phenotypes in reproductive development and function associated with pnc-1 mutants demonstrate that this NAD+ salvage pathway enzyme elicits tissue-specific biological responses. These responses could arise from tissue-specific sensitivities to optimal metabolism. For example, actively dividing tissues, such as the gonad and its associated germline, or tissues with high metabolic demands, such as the nervous system or muscle, may have higher demands for NAD+. Alternatively, downstream effectors that are expressed in a tissue-specific fashion, e.g. NAD+ consumers, could mediate the biological effects of PNC-1.
Excess NAM caused uv1 cell necrosis and defective egg laying, suggesting that PNC-1 control of NAM levels promotes uv1 cell survival and egg laying via a mechanism independent of NAD+ biosynthesis or general metabolism. A known physiological role for NAM is inhibition of NAD+ consumers such as sirtuins and PARPs (Avalos et al., 2005; Landry et al., 2000; Sethi et al., 1996). Sirtuins have well-established roles in promoting cell survival via a variety of mechanisms (Michan and Sinclair, 2007; Yang et al., 2007). We previously reported that ectopic expression of LIN-3 EGF or constitutive activity of LET-23 EGR receptor rescues uv1 necrosis in pnc-1(ku212) (Huang and Hanna-Rose, 2006). Constitutive activity of LET-23 EGFR also prevents NAM induced uv1 necrosis (not shown). We previously considered the interaction with LET-23 EGFR and LIN-3 EGF, which are required for uv1 cell specification (Chang et al., 1999; Hwang and Sternberg, 2004), as evidence that a defect in inductive EGF signaling between the somatic vulva and the uv1 cells in the gonad could underlie the uv1 necrosis (Huang and Hanna-Rose, 2006). Our current results demonstrating that the gonad delay and the uv1 necrosis phenotypes are separable are not consistent with this model, and we suggest that EGF signaling may provide a survival cue that prevents NAM-induced death. However, it remains to be determined what role NAD+ consumer inhibition may play in promoting uv1 cell death or in mediating any survival signal in this system.
In contrast to the uv1 and egg-laying phenotypes, the temporal delay in gonad development results from compromised salvage NAD+ biosynthesis. As mentioned previously, developing tissues may have a more stringent need for NAD+ and may be more susceptible to suboptimal metabolism. However, we see no phenotypes in the development of other tissues, including the vulva, which develops with a temporal pattern similar to that of the gonad, or in the function of the nervous system, a tissue with high metabolic demands, suggesting that a purely metabolic explanation may be insufficient. We speculate that different NAD+ consumers, which may be differentially sensitive to regulation by either NAM or NAD+ levels, mediate the phenotypic effects of both too much NAM and too little NAD+ biosynthesis.
Our finding that the pnc-1 product and substrate have separable biological roles is particularly interesting in light of longevity studies involving nicotinamidase. Yeast Pnc1 is a positive regulator of the longevity protein Sir2, the founding member of the sirtuin family (Anderson et al., 2003). As sirtuins use NAD+ as a substrate generating NAM, which can act as a non-competitive inhibitor, Pnc1 can potentially promote sirtuin activity by increasing levels of the substrate NAD+ through salvage and also by lowering levels of the inhibitor NAM. It is currently debated whether Pnc1 regulation of NAD+ or NAM levels is most relevant to activation of sirtuins (Bitterman et al., 2002; Gallo et al., 2004; Lin et al., 2000; Lin et al., 2004). We demonstrate that pnc-1 processing of NAM and production of NAD+ are not mutually exclusive roles; NAM processing is key in some processes, whereas NAD+ biosynthesis (or maintaining flow through the salvage pathway) is crucial for others.
Given the central role of NAD+ in the metabolism of all cells, it is somewhat surprising that compromising NAD+ salvage biosynthesis in an organism that lacks de novo synthesis is not more deleterious. While it remains possible that PNC-2 may compensate for loss of PNC-1 activity, PNC-2 is predicted to be expressed at either very low levels or in very few cells, as no ESTs have been found for this gene (http://www.wormbase.org). Our experiments suggest that the C. elegans diet provides either NAD+ or NAD+ precursors, which could account for some general cell metabolism needs.
Live E. coli appear to be the source of NAD+ or NAD+ precursors that can partially compensate for the absence of PNC-1 activity. In the course of our studies, we occasionally observed some variability in the penetrance of the pnc-1 phenotypes. The effect of food viability revealed a necessity for rigorously controlling food quality between experiments to ensure that animals are obtaining the same nutrients. We did not detect an effect of dead food on reproductive development of wild-type animals, suggesting that our NAD+ salvage mutants are uniquely sensitive to the food source. As NAD+ levels are associated with regulating the activity of NAD+ consumers, it is of particular importance to pay attention to food quality when assessing longevity mutants, especially metabolic mutants, which may have exacerbated defects when nutrients are inadequate.
How does an extracellular NAD+ biosynthetic/NAM-processing enzyme affect cellular functions?
A correlation between the limited detected expression patterns of the PNC-1a and b promoters and the function of PNC-1 in development is not obvious. For example, we detected no promoter activity in the uv1 cells, which necrosed in the absence of PNC-1, and in only two cell types within the gonad, which was globally delayed in the absence of PNC-1. However, the PNC-1a isoform was secreted and could, thus, function non-cell-autonomously. Although the secreted isoform is sufficient to rescue all phenotypes, the concentrations of NAD+ and NAM in the cytoplasm and/or cellular organelles presumably elicit phenotypes. Determining the concentrations of NAM, NA and NAD+ in whole C. elegans animals is possible. However, the more relevant question of how these molecules vary between tissues and within tissues over time is more difficult to address with current technology (Sauve, 2008). This question is particularly difficult because both NAM and NA can rapidly diffuse across cellular membranes (Lan and Henderson, 1968). We postulate that secreted PNC-1 produced by one tissue can process excess NAM from other tissues extracellularly with two important effects: (1) NAM levels are generally lowered, even intracellularly; and (2) NA is produced and supplied to tissues for NAD+ biosynthesis. Thus tissues that express PNC-1 could impact NAM and NAD+ levels in tissues that do not, perhaps even in a regulatory fashion. Movement of NAD+ biosynthetic precursors between tissues could account for the observation that expression of PNC-1b in body wall muscle cells, which surround the circumference of the whole animal, efficiently rescues mutant phenotypes. It is also possible that the muscle is the focus of activity for all observed phenotypes, although we consider this scenario unlikely.
Interestingly, the vertebrate equivalent of PNC-1 is also secreted. Nampt is identical to visfatin, which is secreted from visceral fat (Revollo et al., 2007b), and a secreted cytokine called Pre-B cell enhancing factor (PBEF) (Samal et al., 1994). Thus, extracellular NAD+ salvage activity and/or NAM metabolism may be an evolutionarily conserved activity. In vertebrates, Nampt is not expressed in every tissue (Revollo et al., 2007b), suggesting that some tissues may lack intrinsic salvage pathway function, making them selectively sensitive to salvage pathway perturbations. Compensatory salvage activity may be provided globally to the animal via circulation of Nampt in the serum (Revollo et al., 2007a; Samal et al., 1994). Secreted Nampt is enzymatically active and is predicted to provide its product NMN to the pancreas, where Nampt expression is undetectable, to help satisfy its requirements for NAD+ biosynthesis (Revollo et al., 2007b). Our expression analysis suggests a similar scenario in C. elegans. The expression pattern of PNC-1 may be limited. It remains possible that a third promoter could control expression of a protein encoded only by exons 2 through 4 (this would produce a protein identical to PNC-1b) and result in a broader expression pattern. However, we were unable to test this possibility because the pnc-1 genomic region was never successfully cloned in any vector except a YAC, and our repeated attempts to clone this entire region have not been successful. By comparison to the Nampt studies, the tissues that express and secrete significant amounts of PNC-1a could impact NAD+ and NAM levels at distant sites.
NAD+ and NAD+ precursors and metabolites are implicated in regulation of longevity, metabolism and death via regulatory activity on NAD+ consumers. Our results suggest that PNC-1 can function at a distance to maintain both NAD+ biosynthesis and NAM concentrations in tissues. We speculate that some of the developmental defects we observe could be a result of misregulation of NAD+ consumers due to perturbations in NAM and NAD+ concentrations. It will be important to examine NAD+ consumer mutants for these developmental phenotypes.
We thank Edwin Cuppen for generous assistance in isolating pnc-1 alleles. We thank Katie Estes for her work on PNC-2 and Andrew Spracklen for preparation of the clones for protein expression. Thanks to Melissa Long and Cheryl Swayne for technical assistance. We thank Melissa Rolls and Matt Crook for critical reading of the manuscript and members of the Hanna-Rose laboratory for helpful discussions. This research was supported by grants to W.H.-R. from the National Science Foundation (IOS-0718675) and the NIH (R01-GM086786). Deposited in PMC for release after 12 months.
References
- Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. and Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, R. M., Bitterman, K. J., Wood, J. G., Medvedik, O., Cohen, H., Lin, S. S., Manchester, J. K., Gordon, J. I. and Sinclair, D. A. (2002). Manipulation of a nuclear NAD+ salvage pathway delays aging without altering steady-state NAD+ levels. J. Biol. Chem. 277, 18881-18890. [DOI] [PubMed] [Google Scholar]
- Anderson, R. M., Bitterman, K. J., Wood, J. G., Medvedik, O. and Sinclair, D. A. (2003). Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423, 181-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalos, J. L., Bever, K. M. and Wolberger, C. (2005). Mechanism of sirtuin inhibition by nicotinamide: altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. Mol. Cell 17, 855-868. [DOI] [PubMed] [Google Scholar]
- Belenky, P., Racette, F. G., Bogan, K. L., McClure, J. M., Smith, J. S. and Brenner, C. (2007). Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell 129, 473-484. [DOI] [PubMed] [Google Scholar]
- Bendtsen, J. D., Nielsen, H., von Heijne, G. and Brunak, S. (2004). Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340, 783-795. [DOI] [PubMed] [Google Scholar]
- Bieganowski, P. and Brenner, C. (2004). Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 117, 495-502. [DOI] [PubMed] [Google Scholar]
- Bitterman, K. J., Anderson, R. M., Cohen, H. Y., Latorre-Esteves, M. and Sinclair, D. A. (2002). Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 277, 45099-45107. [DOI] [PubMed] [Google Scholar]
- Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C., Newman, A. P. and Sternberg, P. W. (1999). Reciprocal EGF signaling back to the uterus from the induced C. elegans vulva coordinates morphogenesis of epithelia. Curr. Biol. 9, 237-246. [DOI] [PubMed] [Google Scholar]
- Cuppen, E., Gort, E., Hazendonk, E., Mudde, J., van de Belt, J., Nijman, I. J., Guryev, V. and Plasterk, R. H. (2007). Efficient target-selected mutagenesis in Caenorhabditis elegans: toward a knockout for every gene. Genome Res. 17, 649-658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, X., Wang, W., Kim, R., Yakota, H., Nguyen, H. and Kim, S. H. (2001). Crystal structure and mechanism of catalysis of a pyrazinamidase from Pyrococcus horikoshii. Biochemistry 40, 14166-14172. [DOI] [PubMed] [Google Scholar]
- Fares, H. and Greenwald, I. (2001). Regulation of endocytosis by CUP-5, the Caenorhabditis elegans mucolipin-1 homolog. Nat. Genet. 28, 64-68. [DOI] [PubMed] [Google Scholar]
- Fulco, M., Cen, Y., Zhao, P., Hoffman, E. P., McBurney, M. W., Sauve, A. A. and Sartorelli, V. (2008). Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 14, 661-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo, C. M., Smith, D. L., Jr and Smith, J. S. (2004). Nicotinamide clearance by Pnc1 directly regulates Sir2-mediated silencing and longevity. Mol. Cell. Biol. 24, 1301-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton, P., Park, K. J., Obayashi, T., Fujita, N., Harada, H., Adams-Collier, C. J. and Nakai, K. (2007). WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585-W587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, G., Taylor, A. B., McAlister-Henn, L. and Hart, P. J. (2007). Crystal structure of the yeast nicotinamidase Pnc1p. Arch. Biochem. Biophys. 461, 66-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, L. and Hanna-Rose, W. (2006). EGF signaling overcomes a uterine cell death associated with temporal mis-coordination of organogenesis within the C. elegans egg-laying apparatus. Dev. Biol. 300, 599-611. [DOI] [PubMed] [Google Scholar]
- Hwang, B. J. and Sternberg, P. W. (2004). A cell-specific enhancer that specifies lin-3 expression in the C. elegans anchor cell for vulval development. Development 131, 143-151. [DOI] [PubMed] [Google Scholar]
- Kobayashi, Y. and Maudsley, D. V. (1969). Practical aspects of liquid scintillation counting. Methods Biochem. Anal. 17, 55-133. [DOI] [PubMed] [Google Scholar]
- Lan, S. J. and Henderson, L. M. (1968). Uptake of nicotinic acid and nicotinamide by rat erythrocytes. J. Biol. Chem. 243, 3388-3394. [PubMed] [Google Scholar]
- Landry, J., Slama, J. T. and Sternglanz, R. (2000). Role of NAD(+) in the deacetylase activity of the SIR2-like proteins. Biochem. Biophys. Res. Commun. 278, 685-690. [DOI] [PubMed] [Google Scholar]
- Letunic, I., Copley, R. R., Schmidt, S., Ciccarelli, F. D., Doerks, T., Schultz, J., Ponting, C. P. and Bork, P. (2004). SMART 4.0: towards genomic data integration. Nucleic Acids Res. 32, D142-D144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S. J., Defossez, P. A. and Guarente, L. (2000). Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289, 2126-2128. [DOI] [PubMed] [Google Scholar]
- Lin, S. J., Ford, E., Haigis, M., Liszt, G. and Guarente, L. (2004). Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 18, 12-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduro, M. and Pilgrim, D. (1995). Identification and cloning of unc-119, a gene expressed in the Caenorhabditis elegans nervous system. Genetics 141, 977-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magni, G., Amici, A., Emanuelli, M., Raffaelli, N. and Ruggieri, S. (1999). Enzymology of NAD+ synthesis. Adv. Enzymol. Relat. Areas Mol. Biol. 73, 135-182, xi. [DOI] [PubMed] [Google Scholar]
- Michan, S. and Sinclair, D. (2007). Sirtuins in mammals: insights into their biological function. Biochem. J. 404, 1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiss, J. and Handler, P. (1958a). Biosynthesis of diphosphopyridine nucleotide. I. Identification of intermediates. J. Biol. Chem. 233, 488-492. [PubMed] [Google Scholar]
- Preiss, J. and Handler, P. (1958b). Biosynthesis of diphosphopyridine nucleotide. II. Enzymatic aspects. J. Biol. Chem. 233, 493-500. [PubMed] [Google Scholar]
- Prestridge, D. S. (1995). Predicting Pol II promoter sequences using transcription factor binding sites. J. Mol. Biol. 249, 923-932. [DOI] [PubMed] [Google Scholar]
- Revollo, J. R., Grimm, A. A. and Imai, S. (2004). The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 279, 50754-50763. [DOI] [PubMed] [Google Scholar]
- Revollo, J. R., Grimm, A. A. and Imai, S. (2007a). The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr. Opin. Gastroenterol. 23, 164-170. [DOI] [PubMed] [Google Scholar]
- Revollo, J. R., Korner, A., Mills, K. F., Satoh, A., Wang, T., Garten, A., Dasgupta, B., Sasaki, Y., Wolberger, C., Townsend, R. R. et al. (2007b). Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 6, 363-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux, A., Andris, F., Van Gool, F. and Leo, O. (2003). Reconstructing eukaryotic NAD metabolism. BioEssays 25, 683-690. [DOI] [PubMed] [Google Scholar]
- Samal, B., Sun, Y., Stearns, G., Xie, C., Suggs, S. and McNiece, I. (1994). Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol. Cell. Biol. 14, 1431-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandmeier, J. J., Celic, I., Boeke, J. D. and Smith, J. S. (2002). Telomeric and rDNA silencing in Saccharomyces cerevisiae are dependent on a nuclear NAD(+) salvage pathway. Genetics 160, 877-889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve, A. A. (2008). NAD+ and Vitamin B3: From metabolism to therapies. J. Pharmacol. Exp. Ther. 324, 883-893. [DOI] [PubMed] [Google Scholar]
- Sauve, A. A., Moir, R. D., Schramm, V. L. and Willis, I. M. (2005). Chemical activation of Sir2-dependent silencing by relief of nicotinamide inhibition. Mol. Cell 17, 595-601. [DOI] [PubMed] [Google Scholar]
- Sethi, J. K., Empson, R. M. and Galione, A. (1996). Nicotinamide inhibits cyclic ADP-ribose-mediated calcium signalling in sea urchin eggs. Biochem. J. 319, 613-617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J. S., Brachmann, C. B., Celic, I., Kenna, M. A., Muhammad, S., Starai, V. J., Avalos, J. L., Escalante-Semerena, J. C., Grubmeyer, C., Wolberger, C. and Boeke, J. D. (2000). A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. USA 97, 6658-6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanny, J. C. and Moazed, D. (2001). Coupling of histone deacetylation to NAD breakdown by the yeast silencing protein Sir2: Evidence for acetyl transfer from substrate to an NAD breakdown product. Proc. Natl. Acad. Sci. USA 98, 415-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Horst, A., Schavemaker, J. M., Pellis-van Berkel, W. and Burgering, B. M. (2007). The Caenorhabditis elegans nicotinamidase PNC-1 enhances survival. Mech. Ageing Dev. 128, 346-349. [DOI] [PubMed] [Google Scholar]
- Yang, H., Lavu, S. and Sinclair, D. A. (2006). Nampt/PBEF/Visfatin: a regulator of mammalian health and longevity? Exp. Gerontol. 41, 718-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H., Yang, T., Baur, J. A., Perez, E., Matsui, T., Carmona, J. J., Lamming, D. W., Souza-Pinto, N. C., Bohr, V. A., Rosenzweig, A. et al. (2007). Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130, 1095-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]