

Abstract
Recent studies suggest that the function of the blood brain barrier (BBB) is not static under normal physiological conditions and is likely altered in neurodegenerative disease. Prevailing thinking about CNS function, and neurodegenerative disease in particular, is neurocentric excluding the impact of factors outside of the CNS. This review challenges this perspective and discusses recent reports suggesting the involvement of peripheral factors including toxins and elements of adaptive immunity that may not only play a role in pathogenesis, but also progression of neurodegenerative diseases. Central to this view is neuroinflammation. Several studies indicate that the neuroinflammatory changes that accompany neurodegeneration affect the BBB or its function by altering transport systems, enhancing immune cell entry, or influencing the BBB’s role as a signaling interface. Such changes impair the BBB’s normal homeostatic function and affect neural activity. Moreover, recent studies reveal that alterations in BBB and its transporters affect the entry of drugs used to treat neurodegenerative diseases. Incorporating BBB compromise and dysfunction into our view of neurodegenerative disease leads to inclusion of peripheral mediators in its pathogenesis and progression. In addition, this changing view of the BBB raises interesting new therapeutic possibilities for drug delivery as well as treatment strategies designed to reinstate normal barrier function.
Keywords: Parkinson’s disease, Alzheimer’s disease, Amyotrophic Lateral Sclerosis, tight junctions, neuroinflammation, neurotoxins
Introduction and perspective
The blood brain barrier (BBB) is an enigma to many and misunderstood by most. It is probably fair to say that descriptions of the BBB receive only casual mention in most classes or texts on neuroscience. As a result, the prevailing perception is that the BBB is a static element of the neurovasculature that merely serves as an impediment to drug delivery. Moreover, the neurocentric view of CNS disorders normally precludes consideration of a role for the vasculature in CNS disease pathogenesis except in a few notable cases (e.g. stroke). However, given the disproportionate degree of cardiac output (20%) delivered to the brain via an extremely large and delicate vascular network, it would be surprising if that vasculature and its barrier function that separates the peripheral compartment from the central compartment was unaffected by neuronal disease. Indeed, emerging evidence from virtually all CNS degenerative disorders document BBB dysfunction in the disease process. Moreover, the perception of the BBB as a static element is undergoing a paradigm shift as well, such that it should now be viewed as a dynamic interface that is not only responsive to vascular signals, but signals from the parenchyma as well. In addition, the cells of the BBB produce prostaglandins, nitric oxide, and cytokines that affect CNS function. A more thorough understanding of the BBB and an appreciation of its dynamic nature is therefore needed to better understand the pathophysiology of CNS disease.
This review will provide an overview of the normal structure and function of the BBB and point out examples of how barrier dysfunction or barrier compromise might contribute to CNS disease and its treatment. Being newcomers to this field, we have been struck by the theoretical implications barrier dysfunction would have on disease progression. Thus, if the BBB becomes leaky as a result of neuroinflammation caused by the dopamine (DA) neurodegeneration in Parkinson’s disease (PD; our primary field of interest), elements of the peripheral compartment (e.g., immune mediators) may very well participate in the disease process. Moreover, we and others showed that this barrier compromise alters antiparkinsonian drug delivery (Carvey et al. 2005;Westin et al. 2006;Monahan AJ and Carvey PM 2008). Alternatively, does a leaky BBB allow central signals (e.g., chemokines) to more effectively recruit peripheral elements to a site of neuroinflammation? Based on this experience, we feel that a greater appreciation for barrier function/dysfunction would likely influence the way the reader thinks about his or her primary CNS disorder. We will therefore also raise rhetorical questions about how BBB dysfunction could affect CNS diseases in general. Given the numerous reviews about the various elements that comprise the BBB, our intent is not to create a comprehensive review of the literature, but rather, to provoke an appreciation of how BBB dysfunction may contribute to an understanding of CNS disease.
I: Characteristics of the BBB as an interface between the CNS and periphery
The BBB was first identified by experiments such as the one by Paul Ehrlich who noted that basic dyes injected into the circulatory system failed to stain the brain, while staining most other organs (Ehrlich 1904). Subsequent studies by one of his students showed that injection of the dye into the CSF stained all cells in the brain, but not organs within the periphery (Goldmann 1913). These experiments led to the belief that the BBB segregated the central and peripheral compartments effectively preventing molecules in blood from entering brain and molecules in brain from entering blood unless they were lipid soluble. Subsequent studies demonstrated that this was not necessarily the case since numerous transport systems are present on the luminal and abluminal surfaces of the brain endothelial cells (BECs) that make up part of the BBB. In addition, it was soon recognized that barrier integrity was not simply a consequence of a physical barrier, but also involved transport mechanisms (e.g., p-glycoprotein [P-gp]) that exclude certain lipid soluble molecules from brain. Enzymes located in BECs (e.g., Cyptochrome P450-3A4 (CYP-3A4) or monoamine oxidase) metabolize lipid soluble molecules that do penetrate the BECs, preventing entry into brain parenchyma creating another aspect of the barrier. Moreover, it is now clear that there is communication across the BBB including blood-to-brain and brain-to-blood signaling suggesting that BECs act as a signaling interface between the brain and blood. Thus, BECs release prostaglandins and nitric oxide in response to systemic infections to initiate anorexia (Langhans 2007) and neuroinflammation activates signals on the capillary walls to induce immune cell entry. In addition, cytokines are released by the BECs in response to inflammation on either side of the barrier (Verma et al. 2006). The BBB function is therefore best described as a physical and metabolic barrier with selective transport systems acting as a signaling interface between brain and blood.
Ia: Components of the BBB
The structure of the BBB is comprised of highly specialized BECs which make contact with pericytes by peg and socket junctions (Ho 1985;Diaz-Flores et al. 1991). The pericyte is also embedded in a vascular basement membrane which connects with the astrocytic end-feet processes of the glia limitans (Krueger and Bechmann 2009;Balabanov and Dore-Duffy 1998). The glial limitans is composed of astrocytic end feet processes that cover the surface of the brain and spinal cord and is referred to as the glia limitans superficialis when present near the subarachnoid space. A similar structure is present in normal brain parenchyma and is referred to as the glial limitans perivascularis (Owens et al. 2008a). This structure courses near neurons (Figure 1).
Figure 1.
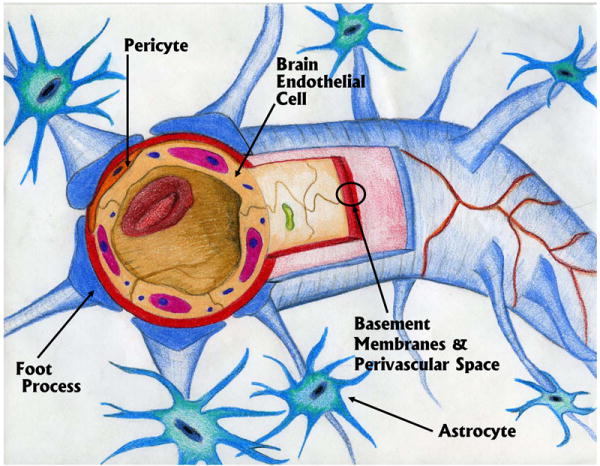
Depiction of a capillary with the main components of the blood brain barrier.
Together with neurons and microglia, the BBB comprises a construct called the neurovascular unit (Hawkins and Davis 2005). Thus, there are interactions among these cells that regulate vascular blood flow to the neuron(s). The pericyte is widely thought to regulate vascular blood flow. In areas of the vessel where pericytes exists, Peppiatt et al. demonstrated via live-imaging in rat retinal and cerebellar slices that ATP and noradrenalin induces constriction of the capillaries. And while GABA blockers also led to constriction of the corresponding capillary, glutamate suppressed it. These observations supports the role of the pericyte in driving contractility of the vessel and therefore its function as a modulator of vascular blood flow (Peppiatt et al. 2006). Biogenic amine containing neurons (i.e. norepinephrine [NE] and serotonin [5HT]) in particular, course in close proximity to blood vessels in many locations (Rennels and Nelson 1975;Kapadia and de Lanerolle 1984;DiCarlo, Jr. et al. 1984) suggesting their potential involvement in BEC regulation, although this remains a question. Indeed, it is thought that NE regulates barrier integrity. Thus, loss of ascending fibers from the locus ceruleus led to significant down regulation of tight junction proteins and barrier compromise (Kalinin et al. 2006). The gliovascular unit is a similar construct (Wolburg et al. 2009), involving only astrocytes and vessels and is thought to be more involved in capillary regulation. The concepts embodied in the neurovascular and gliavascular units suggest active involvement of the brain parenchyma in BEC function. However, these concepts embody more than just vascular flow regulation and imply interplay among the cells to control a number of additional functions including transport of water-soluble substrates, tight junction (TJ) integrity between the BECs, angiogenesis, and metabolic regulation. Takano et al., for example, have demonstrated abnormalities in astrocytes and microvascular control in mouse models of AD using two-photon in vivo imaging of astrocytic Ca2+ signaling (Takano et al. 2007). It is therefore not a conceptual leap to assume that neuronal dysfunction could lead to vascular changes, which, in turn, participate in the changes caused by that neuronal dysfunction. Moreover, since neuronal function is heterogeneous throughout the brain, it follows that BEC function and BBB integrity and function is heterogeneous in the brain. Thus, are regional degenerative changes in PD or Alzheimer’s disease (AD) a consequence of a unique neurovascular environment? If biogenic amines regulate BEC function and BBB integrity, do changes in these neurotransmitters that occur as part of normal brain function, in psychiatric disorders, or in neurodegenerative diseases, alter barrier integrity heterogeneously? Since all neurodegenerative diseases release proinflammatory cytokines, nitric oxide, and neurosteroids, which have been shown to affect barrier function, do they affect the integrity and function of the BBB in areas of active inflammation as well? These concepts have not been adequately studied.
Ia1: Brain Endothelial Cells (BECs) and the microvascular network of the brain
The primary cell responsible for barrier integrity discussed in this review is the brain capillary endothelial cell and from here on will be referred to as brain endothelial cell (BEC) (although it is important to note that epithelial cells form the blood-CSF barrier, similarly play a significant role in blood-brain communication (Redzic et al. 2005). As is true of endothelial cells (EC(s)) throughout the body, the ECs’ phenotypic characteristics are markedly influenced by the tissue in which it resides (Garlanda and Dejana 1997). However, unlike most other ECs in the periphery, those forming the BBB have increased numbers of mitochondria (Oldendorf W.H. et al. 1977), lack fenestrations (Fenstermacher and Kaye 1988), have markedly reduced pinocytotic activity (Sedlakova et al. 1999), and form characteristic tight junctions (TJs) with one another that markedly reduce diffusion of molecules across the vessel (Kniesel and Wolburg 2000).
The TJs that are largely responsible for barrier function are formed and regulated by a set of complex interacting proteins (Figure 2). The existence of the TJ forces water soluble macromolecules, which might otherwise enter the brain via passive diffusion, through paracellular space (Stevenson and Keon 1998), to pass through the BEC itself. Thus, unless a specific transport protein exists for a molecule, or it can be trafficked using pinocytosis, it must diffuse across both the luminal and abluminal surfaces of the BEC (transcellular diffusion). It is important to appreciate however, that even though we think of the BBB as absolute, it is not, and if vascular concentrations are high enough, even relatively large molecules will cross, albeit in low concentrations.
Figure 2.
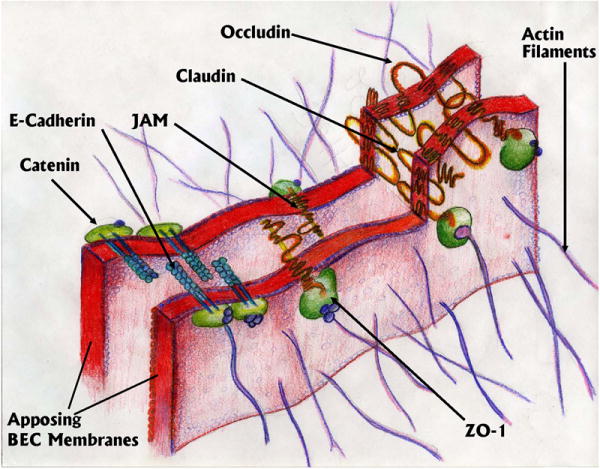
Depiction of a tight junction (TJ) between two apposing segments of adjacent brain endothelial cells showing the major components of the junction.
Occludin, claudins and junctional adhesion molecule (JAM) are the transmembrane proteins of TJs that seal the paracellular space between adjacent plasma membranes of BECs (Figure 2; the proteins that comprise the TJs have been reviewed extensively (Ueno 2007;Wolburg et al. 2009;Wolburg and Lippoldt 2002). Intracellularly, scaffolding proteins zona occludin 1 (ZO-1), ZO-2, and ZO-3 interact with these molecules while linking to actin filaments within the BEC cytoskeleton that appear critical for barrier integrity (Nico et al. 2003). The extracellular domains of claudins and occludins form the major component of the diffusion barrier in the paracellular space although all the proteins contribute to barrier integrity at some level. Deregulation of claudins and JAM may lead to enhanced permeability in microvessels (Liebner et al. 2000;Wittchen et al. 1999;Martin-Padura et al. 1998). There are adherens junctions below the tight junctions in the basal region of lateral plasma membranes that appear to also contribute to barrier function. Cadherins stabilize adhesion between BECs in the adherens junctions. Intracellularly, catenins also link cadherins to the cytoskeleton (Vorbrodt and Dobrogowska 2003). Alterations in the function of any of these proteins destabilize the junctions, facilitating paracellular diffusion although compensatory change following down-regulation or knock–out of one of these proteins has created numerous paradoxes suggesting poor understanding of the TJ as a whole.
It is well known that inflammatory mediators in the blood destabilize TJs leading to barrier compromise as part of sepsis (Li et al. 2009). However, our recent studies also indicate that products of microglia, including tumor necrosis factor alpha (TNFα), disrupt barrier integrity from the abluminal side of the barrier suggesting receptor-mediated changes occur on the abluminal surface as well (Zhao et al. 2007). This further reinforces the notion that barrier integrity is influenced by changes in brain parenchyma given that microglia and factors released from microglia such as TNF α are elevated in all neurodegenerative diseases studied to date. Although inflammatory mediated compromise, whether from the luminal or abluminal surface, is conceptually easy to understand, albeit not generally considered, barrier compromise is also associated with peripheral pain. McCaffrey et al., (2008) demonstrated increased BBB permeability due to the disruption in the assemblage of the disulfide–bonded occludins using the lambda-carrageenan-induced peripheral inflammatory pain model (McCaffrey et al. 2008). Using the same model, (Huber et al. 2006) showed an early increase in Inter-cellular Adhesion Molecule (ICAM-1) RNA and protein expression that remained for 48 hours. Immunohistochemistry showed that the induction of ICAM-1 was region specific with increased expression noted in the thalamus and frontal and parietal cortices, which directly correlated with increased expression of activated microglia. This suggests that not only can peripheral pain affect barrier integrity and expression of cell adhesion molecules responsible for infiltration of peripheral cells into brain, but that the effect is not global, implicating regional selectivity.
Normal physiological responses to stress similarly alter the dynamics of the BBB. Stress-induced alterations in the BBB have been studied in several animal models including immobilization (Dvorska et al. 1992), and heat stress in rats (Sharma et al. 1992). Both stressors increase BBB permeability to drugs and neurotransmitters. Emotional stress experienced by soldiers has been shown to increase the paracellular diffusion of the acetylcholine esterase inhibitor pyridostigmine (which normally penetrates brain poorly) causing a three-fold increase in frequency of CNS symptoms compared with controls (Sharabi et al. 1991). Friedman et al (1996) followed up on this phenomenon by using a simulated stress situation (i.e., forced swim protocol) in mice and found that stressed mice required less pyridostigmine (50% reduction in dose) to inhibit acetyl-cholinesterase in brain due to increased BBB permeability (Friedman et al. 1996). Although these studies suggest that stress leads to barrier compromise and increased entry of this quaternary amine, recent studies suggest that stress may also reduce P-glycoprotein (P-gp) activity which may also contribute to the increased entry of pyridostigmine (El Masry and Abou-Donia 2006). Taken together, these studies suggest that non-inflammatory mediators participate in regulation of TJ proteins and their interactions as well as proteins expressed on BECs (i.e. P-gp), raising the possibility that other non-inflammatory mediators may have similar effects.
We normally assume that barrier integrity is static within the CNS and barrier compromise is only evident in pathological conditions. The studies alluded to above challenge that view suggesting that dynamic alterations in the barrier play a role in normal brain function as well. Moreover, we tend to view the barrier dysfunction as only leaky or not-leaky. However, as alluded to above and discussed in greater detail below, a binary view as either leaky or intact is simplistic since its signaling, transport, and secretory functions may change independent of barrier compromise. If barrier integrity or functions are compromised by such subtle activities as pain or stress, what is the potential role of circulating NE and 5HT that would enter brain from the periphery in the brain’s response to these stressors? If the BBB becomes leaky to peripheral proteins including cytokines present in the vasculature at low levels during pain or stress, how do these cytokines influence the normal physiological responses of the brain? If these normal physiological activities alter barrier transport functions, do changes in the entry of insulin or amino acids, for instance, as a result of functional alterations in the BBB, also contribute to normal responses? The implications of a “dynamic barrier” in normal CNS function are potentially critical to our understanding of the overall response to these general stimuli and are, to say the least, understudied and rarely considered.
Ia2: The pericyte
In addition to the BEC, the pericyte is thought to be a critical component of a functional BBB, although we know little about it because it is difficult to isolate. Pericytes are sprinkled (~one pericyte for every 5-6 BECs) along the vascular endothelium in microvascular networks and form gap junctions with BECs suggesting an intimate functional relationship (Fisher 2009), 2009). Pericytes also communicate with other elements of the neurovascular unit through autocrine and paracrine signaling pathways (Nakaoke 2007). These actin-expressing cells wrap numerous processes around the vessel and have a number of vasoactive signaling receptors suggesting their involvement in microvascular capillary flow. If this is true, are these cells responsive to CO2, the well-known major regulator of microvascular flow in the brain? Dore-Duffy (2008) implicated a role for these cells in angiogenesis based on her studies demonstrating characteristic de-differentiation when pericytes were cultured (Dore-Duffy 2008). This suggests a stem cell morphology. Do pericytes de-differentiate and then differentiate into BECs as part of the angiogenic process? Given their intimate contact with BECs, the role of the pericyte in BBB integrity may be critical under normal and pathological conditions. Indeed, a recent study suggests that loss of pericytes may be a critical first step in the disruption of the blood-retinal barrier that leads to blindness in patients with diabetes (Diffley et al. 2009). In addition, Nakagawa et al., (2007) showed that pericytes are critical in maintaining the BEC’s ability to transport insulin in the face of high glucose levels (Nakagawa et al. 2007). These studies suggest a role for pericytes in regulating normal function of the BBB as well.
Ia3: Astrocytes
The astrocytic end feet that engulf the capillary networks of the brain have long been assumed to significantly influence neurovascular structure and integrity. It is well established that astrocytes participate in nutritive and metabolic support of neurons. This is underscored by the close apposition of the astrocytic end-feet on the capillary walls and their close relationship with neurons. Thus, astrocytes are in a position to disperse vascular nutrients away from the vessels in support of neural function. In addition, the astrocytic end feet appear to regulate water transport as evidenced by the polarized expression of aquaporin-4 on the astrocytic terminals (Satoh et al. 2007). It is also thought that the astrocytes secrete soluble factors that induce the formation of TJs in the BECs although these substances are yet to be identified (Abbott 2002b). Janzer and Raff (1987) first demonstrated that purified astrocytes, when transplanted into the anterior eye chamber of the rat, induced BBB-like vascular changes in the invading BECs (Janzer and Raff 1987). In vitro studies suggest that BECs grown in the presence of astrocytes or astrocyte conditioned media increases endothelial cell resistance and increases expression of TJ proteins (Abbott 2002a). If this is true, then why do the vessels in the so-called circumventricular organ (e.g., area postrema, regions of the hypothalamus etc.) that line the ventricles, have astrocytes, but no BBB?
Recent studies suggest that products from pericytes may also be involved in initiating TJ formation (Kim et al. 2009). It is also likely that elements of the basement membrane contributed by the astrocyte may participate in TJ formation by stabilizing the BEC and inducing protein expression changes in the BEC. It is therefore more likely that interactions among the BECs, astrocytes, pericytes and the basement membrane are all required to initiate complete TJ formation explaining the difficulties in identifying the factor(s) involved in TJ formation when one component alone is studied. Regardless, this does not explain why some regions of brain do not have a complete barrier, however. Given the likely involvement of several factors in TJ formation, can disease states that affect astrocyte or pericyte function alter the production of the factor(s) responsible for BBB induction in areas that normally have a BBB? What are the consequences of astrocytosis that commonly occurs in brain injury on proteins responsible for inducing or maintaining barrier integrity (Willis and Davis 2008). Since it appears that barrier integrity as well as function is best characterized as dynamic, what are the relative roles of astrocytes, pericytes, and the basement membrane interactions in these changes?
Ia4: The Basement Membrane
The basement membrane is composed of collagen type 4, elastin, fibrillin, laminin and fibronectin. The basement membrane also includes a large number of extracellular matrix proteins, cell adhesion molecules (CAMs), as well as signaling proteins that form an extensive and complex matrix. This membrane engulfs the BECs and pericytes, but does not act as a significant barrier to diffusion of small molecules, although its anchoring function plays an important role (Persidsky et al. 2006). BECs, astrocytes, and pericytes all probably contribute to formation of the basement membrane that not only serves to anchor the cells in place, but also regulates cellular functions of the BECs through signaling molecules on the abluminal BEC surface. Disruption of the basement membrane can therefore lead to alterations in the cytoskeleton of the BEC that, in turn, affects TJ proteins and barrier integrity. Matrix metalloproteinases (MMPs), and in particular MMP9, significantly affect barrier integrity and are known to digest basement membranes (Rosenberg 2002). Disruption of the basement membrane leads to reduced anchoring of BECs and matrix-BEC signaling that affects TJ integrity leading to barrier compromise. Thus, acute bouts of multiple sclerosis (MS) are associated with increased MMP9 and administering a MMP9 inhibitor prevents flare ups in the experimental allergic encephalomyelitis (EAE) model of MS (Rosenberg 2009). MMPs not only induce barrier compromise by disrupting the basement membrane, but also participate in both the formation and clearance of beta-amyloid deposits in AD (Nalivaeva et al. 2008). Administering inhibitors to the MMP to prevent the now established barrier compromise in AD could have both beneficial and detrimental effects. Interestingly, minocycline (a tetracycline family antibiotic) is known to inhibit the activation of microglia and prevent DA neuron loss in PD animal models, is also an inhibitor of MMPs suggesting that at least part of its neuroprotective effects may involve blockade of MMPs and maintenance of barrier integrity (Yong et al. 2007).
In addition to the basement membrane that delimits the BECs, a second barrier called the glia limitans forms an additional barrier to brain parenchyma (Figure 4). The glia limitans is comprised of astrocytic foot processes, and perhaps microglial foot processes, together with a second basement membrane that create a perivascular space between the basement membrane of the glia limitans and the basement membrane of the BECs. Although the basement membranes by themselves do not likely prevent small molecules from entering brain, they do serve to restrict passage of cells including immune cells from freely entering brain. Moreover, the presence of the two basement membranes sandwiching a perivascular space creates a microenvironment that we know little about except that it creates an area where immune cells cluster during inflammation; a phenomenon called perivascular cuffing (see below). Is there a unique microenvironment in this perivascular space that regulates the signaling between astrocytes and BECs (in paracrine fashion) that is important to barrier integrity? Is this microenvironment altered in neurodegenerative disease? Like the pericyte, we know little about this BBB element.
Figure 4.
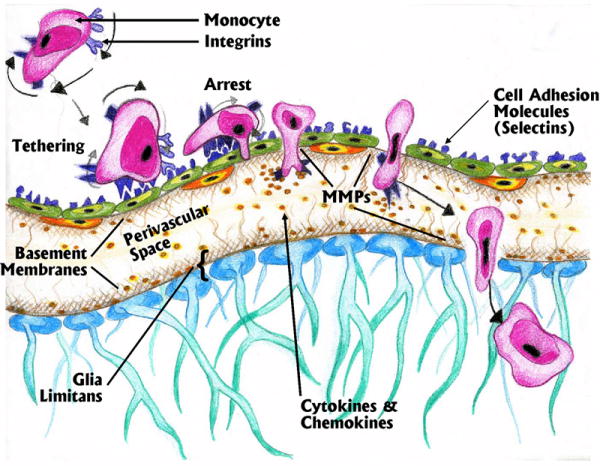
Various phases of transmigration of peripheral monocytes across the BBB based on interactions between integrins and cell adhesion molecules. This receptor interaction process also requires involvement of matrix metalloproteinases (MMPs) to digest the basement membranes on the abluminal surface of the post-capillary venule as well as the basement membrane of the glia limitans.
II: The BBB in development and aging
As is true of many functions of the BBB, very little is known about its early development, and what we do know has been extrapolated from the rodent and chicken. The development of the BBB occurs when angioblasts derived from the mesoderm enter the head region and begin to form the perineural vascular plexus, which occurs in the rodent at embryonic day 9. The perineural vascular plexus covers the neuroectoderm and begins to invade the highly proliferative neuroectoderm via vascular sprouts at embryonic day 11 in rodents and embryonic day 2 in the chicken (Engelhardt 2003). Onset of angiogenesis in the brain occurs at embryonic Day 11 in the rodent and embryonic day 2 in the chick, and continues at maximal rate into early postnatal periods followed by down-regulation in the adult brain (Robertson et al. 1985) (Engelhardt 2003). The sprouting process appears to occur at specific locations, possibly meeting local demands as structural development occurs within the brain (Bar 1980). As development continues, BECs become connected by small TJs forming small, thin walled vessels that evolve into capillaries. The tightness of the BBB appears to occur regionally when angiogenesis is occurring and increases gradually (Robertson et al. 1985).
Whether or not the developing brain vasculature is leaky has been a question of debate. Studies similar to those of Ehrlich, showed that plasma proteins enter fetal brain in higher amounts than adult brain (Wakai and Hirokawa 1978); (Stewart and Hayakawa 1987;Risau 1991). However, others argue that very little plasma protein enters fetal and newborn brain, arguing that even in utero, the BBB is impermeable to proteins from blood (Mollgard et al. 1988). Additionally, permeability to ions as measured by electrical resistance of small pial blood vessels is higher in the fetal brain than the adult brain suggesting barrier integrity (Butt et al. 1990) (Keep et al. 1995). Saunders et al., (2008) recently discussed the issues surrounding barrier integrity during development and suggested that both TJs and integrity develop early. However, they also suggested that the maturity of the various transporters on BEC were not well characterized raising the possibility that transporter immaturity could contribute to toxic exposure during development (Saunders et al. 2008). This does not preclude the possibility that the same types of peripheral factors that affect the BBB during adulthood, are not at work during development. Given that fetal insults can produce long-term effects on brain function (Carvey et al. 2003) (Barlow et al. 2007), alterations in barrier permeability or function, especially if they are regional, could contribute to neurodegenerative diseases that have focal pathology (e.g., AD and PD) in later life. Indeed, we have shown that prenatal exposure to lipopolysaccharide (LPS), administered systemically to the adult female rat or mouse, leads to selective loss of dopamine and serotonergic neurons, but not gabaergic neurons (Ling et al. 2009) in the young adult. Moreover, the changes in the brain in response to this insult appear to be regional. Does this occur because of some unique vulnerability of these types or neurons during development, or does LPS and the proinflammatory cytokines it produces affect selected regions of the brain because systemic LPS produced a localized effect on BECs because their functional development was different from other regions of the developing brain?
Surprisingly, little is known about the effects of aging on the BBB, although a general consensus is that barrier integrity is normal in the aged brain, albeit more readily disrupted by insult. Thus, recent studies in aged mice reveal that traumatic brain injury is greater in aged animals and associated with significantly slower repair of the barrier (Onyszchuk et al. 2008). Similarly, DiNapoli et al., (2008) demonstrated increased damage and slower recovery of barrier integrity in the middle cerebral artery occlusion model of stroke (DiNapoli et al. 2008). The SAMP8 mouse model of accelerated aging and AD, revealed increased extravasation of IgG into the hippocampus in aged vs. young animals indicating altered function of BBB in aged senescence-accelerated mice (Pelegri et al. 2007). However, these changes may more readily reflect the fact that many strains of SAM mice are contaminated with virus (Banks et al., 2000).
Aged rats (18 mo) exhibit evidence of reduced TJ proteins and alterations in barrier integrity compared with young adults (Campbell et al. 2007). Chan-Ling and colleagues (2007) demonstrated an age-related dysfunction in the blood-retinal barrier in aged rats that was associated with increased entry of phagocytic macrophages (Chan-Ling et al. 2007). Bartels and colleagues (2008) demonstrated a significant down-regulation of P-gp in aged humans suggesting a possible mechanism for age-related neurodegeneration. They argued that reductions in P-gp would increase brain exposure to neurotoxins normally excluded by these xenobiotic transporters. Thus, there is some evidence of barrier dysfunction in the aged brain, but as can be seen, the area needs greater study (Bartels et al. 2008). Given that age is the most important risk factor for several neurodegenerative diseases, should altered barrier function and perhaps even barrier compromise, be added to the list of age-related phenomena that are considered risk factors for these diseases? Do age-related changes in transporter function lead to increased entry of xenobiotics? Does reduced function lead to increased entry of peripheral free radicals or immune mediators that promote further neuroinflammation? Does an age-related barrier dysfunction represent a tipping point where the ability of brain parenchyma becomes overwhelmed by peripheral factors leading to degenerative change as proposed by Zlokovic (2008)? These issues represent potentially important questions that are normally not considered in the pathogenesis of CNS disease by the major stake-holders in the field.
III: The Transport Systems of the BBB
The movement of water soluble molecules or large molecular weight substances from the lumen of a blood vessel to the parenchyma or vice versa is largely regulated by polarized transport systems within BECs. The blood-to-brain influx transport systems supply nutrients such as glucose, amino acids and nucleotides from the blood to the brain, while the brain-to-blood efflux transport systems remove metabolites and neurotoxic compounds from the brain parenchyma to the blood (Ohtsuki 2004). In addition, drug efflux pumps, located on the luminal wall of the BECs, transport lipid soluble xenobiotics that enter BECs back into blood. These transport systems thus not only facilitate supply of water-soluble substances needed for normal brain function, but prevent potentially harmful substances from entering as well. The various transport systems thus play a critical role in brain homeostasis. Although the list of transporters is ever expanding, it appears that they can be conveniently classified into four categories (ion transporters, Na+ dependent, Na+-independent, and active transporters (see Figure 3)).
Figure 3.
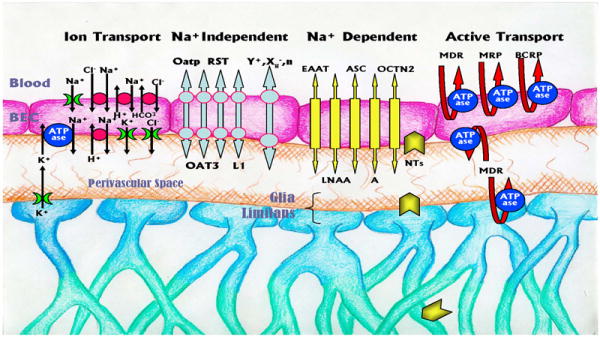
Transport systems of the BBB are categorized into four distinct groups based on their substrates (ion transporters), Na+ dependence (dependent and independent), and the ABC class of transporters (Active Transport). Although the Na+ dependent and independent transporters are depicted as one transport system, this likely does not reflect reality, and two separate coupled transporters that may or may not have similar structures are more likely involved. Certain types of transporters have also been detected in astrocytes including Na+ dependent neurotransmitter transporters (NTs) and P-gp. K+ flux from the astrocytes depicts potassium-spatial buffering. There are numerous additional transport systems not depicted. (See text for details).
Understanding the function of these transporters requires an appreciation for BEC polarity. Because of the BBB, substances in blood must cross both the luminal and abluminal surfaces of BECs. The induction of TJs leads to this organization such that different transport proteins are present on either side of the cell, although how this occurs is currently unknown. Transport of a water-soluble substance into the BEC from the blood requires a second transport system on the abluminal surface to carry it from the BEC cytoplasm into the perivascular space and vice versa (Figure 3). For example, facilitative transporters for glutamate and glutamine are present on the luminal wall of the BEC while three Na+-dependent excitatory amino acid transporters (EAATs) and a Na+-dependent glutamine transporter are found on the abluminal wall (Bernacki et al. 2008). Thus, transport systems appear to be coupled as is true of many transport systems within the body. Indeed, some transporters (e.g. Glut1) are more prevalent on the abluminal wall to ensure a blood-to-brain flux of needed glucose. Because of this polarity, it is possible that alterations on the vascular side can disrupt transport function on one side of the cell without compensating alterations on the abluminal side with potential negative consequences. Thus, Salkeni et al., (2008) demonstrated that LPS exposure affected P-gp resulting in significant increases in the entry of P-gp substrates (i.e. verapamil; (Salkeni et al. 2008). Given this result, it is possible to consider that neuroinflammation may preferentially affect transporters on the abluminal surface independent of the luminal surface? If this were to occur, would it disrupt the polarity of coupled transport? Uncoupling these functions might have profound effects on transport functions potentially leading to disruption of BEC function and altered brain homeostasis that could complicate an already pathogenic process. Such dysfunction may contribute to the abnormal transport of β-amyloid protein in AD (see below).
IIIa: Ion transport systems
Given that many of the transport systems derive their energy from Na+ counter-transport and neuronal function is extremely dependent upon Na+ currents, ion transport systems at the BBB are critical to normal brain function. As is typical of many cells, the transport of ions are coupled to maintain electrical neutrality within the BEC while also regulating [H+] (Betz 1986); (Somjen 2002),; Figure 3). Na+-K+ ATPase pumps located on the abluminal surface maintain high sodium concentrations in the brain interstitial fluid to help drive Na+ counter-transporters and neuronal depolarization. A Na+-H+ counter-transporter also participates. In order to maintain electrical neutrality as well as pH, these abluminal transporters work in concert with a Na+-Cl- symporter as well as Na+-H+ and Cl--HCO3- counter-transporters on the abluminal surface. What happens to the homeostatic integrity of this system in neuroinflammation is unknown. Moreover, the impact on neuronal function associated with reductions in extracellular Na+ following barrier compromise has not been studied.
In addition, to ionic flux across the BEC layer, no discussion of ionic regulation within the CNS would be complete without a discussion of K+ spatial buffering in astrocytes. High levels of extracellular K+ produced by neuronal depolarization leads to further depolarization of neurons as well as glia, and thus regulation of extracellular K+ is critical. If extracellular K+ is not regulated, it can lead to further depolarization of neurons and thereby disrupt the normal flow of excitation regulated through synaptic activity. Astrocytes play an important role in regulating extracellular K+ by taking up K+ and distributing it to other areas of the brain where neuronal activity is not as high. The mechanisms responsible for potassium spatial buffering are not completely understood. However, K+ channel density is very high at the end feet of astrocytes where excess K+ is released into the perivascular space and where it can be subsequently transported into BECs using the Na+-K+ ATPase pump and eventually out of the brain. As discussed below, epileptic seizures are known to affect expression of P-gp. Does the associated increase in electrical activity produce increased levels of K+ within interstitial fluid that overwhelms these ionic transporters, resulting in changes that, in turn, affect other transport systems?
IIIb: Na+ independent transporters
The solute carrier family (SoLute Carrier family) of transporters consist of 300 genes and 43 families of membrane transporters that are either Na+ dependent or independent and can transport an extensive variety of solutes that otherwise do not cross the BBB. Of importance to BBB function (as well as blood-CSF barrier function) are those that are heavily expressed on luminal or abluminal surfaces of the BBB or both.
A number of organic anion transporting polypeptides (Oatp/SLCO) and organic anion transporters (Oat/SLC22A) are found in brain (Figure 3). Of the fourteen members of the Oatp family, only Oatp1a4 (Oatp2), Oatp1a5 (Oatp3) and Oatp1c1 (Oatp14) have been identified in brain microvessel (Tamai et al. 2000). Substrates for this family of transporters include bile salts, organic dyes, steroid conjugates, thyroid hormones, neuroactive peptides and various drugs. Within the SLC22 sub-family of transporters, the organic anion transporter OAT3 and renal-specific transporter (RST) have also been identified in the BBB (Enomoto et al. 2002a;Enomoto et al. 2002b). OAT3 in rat and mouse BECs transport homovanillic acid (HVA), the main dopamine metabolite, as well as other neurotransmitter metabolites out of brain (Mori et al. 2003;Ohtsuki et al. 2002). Since these transporters are saturable and subject to competition by other organic anions, build up of other anionic metabolites of neurotransmitters would inhibit HVA transport by OAT3 leading to accumulation of this metabolite. Interpretation of animal studies by examination of the so called neurotransmitter activity ratio (e.g., [HVA]/[DA]) assumes a dependence on neurotransmitter release. Thus, it is assumed that loss of DA neurons leads to increased DA release by remaining neurons with subsequent increased catabolism leading to increases in the [HVA]/[DA] ratio. However, this increase could also reflect reduced clearance of HVA because of competition at OAT3 or dysfunction of OAT3 due to the neuroinflammatory process associated with DA neuron degeneration. As investigators who have routinely published these ratios as an index of DA activity, we have never considered this alternative interpretation until we began to gain a better understanding of the role of the BBB in neurodegenerative disease. Moreover, if the BBB is compromised in neurodegenerative disease, do cationic or anionic metabolites need a transporter for extrusion from the brain, or will they simply diffuse down a concentration gradient into blood paracellularly?
The uricosuric agent probenecid is an OAT competitive inhibitor. At one time probenecid was widely used to assess acidic monoamine metabolites and cyclic nucleotides in the CSF as an index of CNS neurotransmitter activity (Wood 1980). The assumption was that 24 hour production of NE and 5HT metabolites in the presence of inhibition of their efflux from CSF could be diagnostic of depression. However, if the BBB is dynamic, and function of its transport systems are influenced by CNS disease, interpretation of measures such as these would be extremely complicated.
Transport of essential amino acids into brain occurs by facilitative, Na+-independent transporters called the L1system (Figure 3). L1 transporters are found on both sides of the BEC, but like Glut1 described above, are present at much higher concentrations on the luminal side. In addition, Na+ independent transport systems include the y+ system for cationic amino acids, the xG- system for acidic amino acids, and the n (SNAT3 and SNAT5) amino acid transporters that are key mediators of glutamine transport which can function independent of Na+. In addition to identical substrates, these facilitative transporters can transport a number of xenobiotics due to their low substrate specificity
IIIc: Na+ dependent transporters
A large number of Na+ transporters are present in the BBB as well as in the choroid plexus and transfer a large number of water-soluble solutes. These include the Large Neutral Amino Acid transporters (LNAA), the ASC system, the alanine preferring A system for small neutral amino acids, and the Excitatory Amino Acid transporters (EAAT system). In addition, the organic cation/carnitine transporter, OCTN2 is also Na+ dependent (Betz and Goldstein 1978;Sanchez del Pino et al. 1995). These are coupled transporters with Na+ dependence occurring on the abluminal surface utilizing the Na+ concentration created by the Na+-K+ ATPase pump (O’Kane and Hawkins 2003).
We tend to view these transporters as carriers of nutrients. However, neurotransmitter transporters are also present on BECs. For example, GAT2/BGT-1 is present on the abluminal wall and extrudes γ-amino-butyric acid (GABA) into blood (Tetsuka et al. 2001). The norepinephrine transporter is localized on the abluminal membrane while serotonin transporters are present on both the abluminal and luminal walls membrane (Wakayama et al. 2002). These transporters likely play a role in the clearance of excess levels of neurotransmitters while other transporters carry their metabolites out of brain.
The excitatory acidic amino transporters (EAAT1-3) are located on astrocytes as well as the abluminal surface of the BECs. These transporters remove glutamate from the interstitial fluid of the brain preventing potential excitotoxicity (Beretta et al. 2003). EAAT transporters on BECs thus play an important role in homeostatic regulation of glutamate concentrations. Although it is likely that EAAT transporters on astrocytes play a more important role in sequestering glutamate from its potential local excitotoxic effects, transporters on the BECs likely play a significant role in determining the level in brain over all. Since glutamate toxicity is implicated in so many of the neurodegenerative diseases, the role of abluminal glutamate transport cannot be under-appreciated. Thus, if neuroinflammation induces inflammation of BECs, what are the consequences on glutamate clearance from brain and if glutamate transfer is compromised, does it produce glutamate-mediated, excitotoxic neuron loss.
IIId: Active efflux pumps
In contrast to the facilitative transport systems described above, the ATP-binding cassette (ABC) superfamily of transporters are membrane bound and require ATP to transport solutes across the BBB. They are appropriately classified as active transporters since they can extrude molecules against a concentration gradient and require ATP. Included in this family of proteins are the Multidrug Resistant Proteins (MDRs), the MDR associated proteins (MRPs) and Breast Cancer Resistance protein (BCRP). The first MDR protein identified was P-gp, a 170 kDa transporter that has a variety of substrates (Juliano and Ling 1976). P-gp, also referred to as MDR1, is highly expressed on the BBB (Kamiie et al. 2008); (Bernacki et al. 2008) and found localized to the luminal membrane of the BEC where substrates are secreted from the BEC back into blood (123, Bernacki, J 2008). P-gp has also recently been found on the abluminal surface, although we know little about its function at this location (Bendayan et al. 2006). Normally, P-gp on the luminal surface participates in cholesterol, lipid, glucocorticoid and peptide transport out of brain, but its broad substrate specificity allows binding and subsequent transport of xenobiotics (Terasaki and Hosoya 1999). Although it plays an important role in physiological function, its evolution as a de-toxicant defense mechanism cannot be discounted.
Many lipid soluble drugs have structures capable of binding to P-gp and hence, their entry into brain is inhibited. As is true of drugs intentionally developed to be water-soluble so their systemic administration only has peripheral effects (e.g., pyridostigmine used in the management of myasthenia gravis), substrate binding to P-gp has also been used to advantage. A classic example is loperamide. This drug takes advantage of the constipating effects of the opioid class of drugs, but since it is a substrate for P-gp, it does not enter brain readily. In the management of PD, the DA antagonist domperidone is effective at reducing peripheral DA agonist side effects (e.g. epigastric distress) without exacerbating PD symptoms, because it is a substrate for P-gp and thus does not readily enter brain. However, we demonstrated that the neuroinflammation that accompanies DA neuron loss in the rat model of PD, led to increased entry of domperidone despite up-regulation of P-gp on the BBB (Carvey et al. 2005). We interpreted this to suggest that increased entry occurred because of barrier compromise. In addition, Westerlund and colleagues (2009) recently showed polymorphisms in P-gp were prevalent in PD patients suggesting reduced capacity to exclude possible DA neurotoxins. P-gp also participates in the extrusion of amyloid beta from brain parenchyma, the accumulation of which is thought to contribute to AD (Westerlund et al. 2009;Kuhnke et al. 2007). If there are polymorphisms in P-gp or induction of this protein occurs as part of neuroinflammation, what are its effects on the progression of the degenerative process? Strategies to inhibit P-gp or uncouple it from ATP is an important emerging area of research in CNS drug delivery (see (Hennessy and Spiers 2007). Interestingly, grapefruit juice is a well known inhibitor of P-gp, although not all varieties are equally as potent (Uno and Yasui-Furukori 2006). Since grapefruit juice consumption is more prevalent in some locales, is there an interaction between grapefruit juice and P-gp toxin substrate specificity that creates a regional risk factor for neurodegenerative disease? Since grapefruit juice is also an inhibitor of CYP-3A4, the possibilities of these dietary, regional, and biotransformation interactions become very complex and difficult to interpret within epidemiological data.
In addition to P-gp, there are twelve additional members of the MRP family (MRP1-9). Their primary role is to transport organic anions. MRP 1 and 2 are the most studied of the 12 members (see (Begley 2004), for review). MRP 1 transports leukotriene C4 while MRP2 removes anionic glucuronides and glutathione conjugates and unconjugated organic anions into bile (Rappa et al. 1999;Konig et al. 1999;Bernacki et al. 2008). It is now well established that epilepsy increases MRP2 expression leading to reduced effectiveness of MRP2 substrate antiseizure drugs (Loscher 2005). Using cDNA arrays, Dombrowski, and colleagues (2001) showed a 225% increase in MRP2 gene expression in BECs from patients with drug resistant temporallobe epilepsy (TLE). This suggests that up-regulation of MRP2 expression may play an important role in resistance to certain antiepileptic drugs (e.g., phenytoin and levetiracetam) by decreasing the permeability of these drugs across the BBB. Given these results, it should be questioned how electrical changes within the brain and BECs alter expression of transporters (Dombrowski et al. 2001). Are other transporters up-regulated or down-regulated by changes in electrical current that could subject the individual to toxic levels of drugs or environmental neurotoxins? Given that astrocytes play a significant role in regulating interstitial fluid neurotransmitter and ion concentrations and the intimate relationship between astrocytes and BECs, are changes in the BBB that occur in epilepsy mediated by astrocytes? What impact do alterations in other MRP transporters have on the pathogenesis of neurodegenerative diseases and their therapies?
As can be seen, alterations in transport proteins such as P-gp in neurodegenerative disease are attracting more attention given their potential role in pathogenesis and drug therapy. In addition to the traditional view that P-gp as well as other members of the ABC class of transporters are present on the BEC, recent studies (summarized by (Ronaldson et al. 2008) suggest that they are also present on astrocytes. Localization of P-gp on astrocytic end foot processes suggests a role for P-gp at the glia limitans as well. In addition, levels of P-gp in astrocytes as well as BECs are induced by chronic treatment with protease inhibitors such as atazanavir (Perloff et al. 2005) having significant implications in the neurodegenerative changes that occur in AIDS.
In addition to these transporters, there are numerous others that regulate vitamin entry and other trace nutrients. The genomics of transporters has been categorized and a nomenclature has been established. This system has at least 298 members in 43 families and has been recently reviewed by (Hediger et al. 2004). It is important to recognize that as one reads the transporter literature, it is assumed that they are identically expressed across all species. This is clearly not the case however, and there are innumerable examples of greater prevalence of a particular transporter expressed in rats and mice that are not expressed in humans and vice versa. This makes generalization of data from animal studies complex and such generalization should always be made with caution.
IIIe: Receptor-mediated transcytosis
In addition to the transport proteins mentioned above, receptor-mediated transcytosis (RMT) is an important process for moving essential nutrients across the BBB into parenchyma. RMT utilizes the vesicular trafficking machinery of the endothelium to transport nutrients such as iron via the transferrin receptor (Moos 2002) and insulin via the insulin receptor into the parenchmya. RMT requires ligand binding to a specific receptor at the luminal side of the BEC which initiates the process of endocytosis including the formation of intracellular transport vesicles that deliver the dissociated ligand to the parenchyma (Jones and Shusta 2007) Does altered transport of such nutrients worsen or participate in neurological disease? On the other hand, does neurological disease alter the process of transcytosis?
IV: The Metabolic Function of the Blood-Brain-Barrier
Many of the cytochrome P450 (CYP) subfamilies are present in the BBB as well as in circumventricular organs, including the choroid plexus and posterior pituitary. Indeed, it is generally thought that enzymatic levels are higher in the circumventricular regions of the vascular-brain interface where TJs are not present (Ghersi-Egea et al. 1994). This increase in biotransforming enzymes is consistent with the notion that metabolism would need to be higher due to increased paracellular diffusion.
In rodents, CYP 1A1 is expressed in the ECs of the BBB (Dey et al. 1999); (Huang et al. 2000) (Morse et al. 1998). Activities of enzymes involved in drug metabolism, NADPH-cytochrome CYP reductase, cytochrome 1A and 2B and uridine diphosphate glucruronosyl transferase (UGT) have been measured in the immortalized rat brain endothelial cell line RBE4 (Chat et al. 1998). In 2002, Granberg studied the CYP 1A1 and CYP 1B1 in rodent blood brain interfaces and found that CYP 1A1 is constitutively expressed at low concentration in some blood brain interfaces and is highly inducible and functional, however, evidence for CYP 1B1 was inconclusive (Granberg et al. 2003). In human brain, evidence suggests the presence of CYP 1B1 and CYP 2B at the BBB (McFadyen et al. 1997). These studies suggest that BEC from different organisms do not express the same enzymes and further highlight the caution needed when extrapolating data from animals to humans as discussed above. A newly discovered cytochrome CYP, CYP 4X1, has been detected in BECs within the brain, however, its function is unknown (Al Anizy et al. 2006). It is important to recognize, however, that the levels of CYPs in brain (~0.5-2% of that in liver), are unlikely to significantly affect the overall pharmacokinetics of drugs in the body. However, these CYP enzymes may play an important role in regulating the levels of endogenous GABA, which is thought to participate in brain cholesterol homeostasis as well as elimination of retinoids (Hedlund et al. 2001) Astrocytes also express CYP enzymes in high concentrations (Meyer et al. 2007). Since astrocytes and the basement membrane they secrete form the glia limitans, CYP enzymes in these cells form a second metabolic barrier that affects physiological substrates as well as xenobiotics.
Kalaria and Harik (1987) demonstrated that monoamine oxidase was present in brain capillaries although levels in rats were much higher than those isolated from humans (Kalaria and Harik 1987). The existence of aromatic amino acid decarboxylase in the BECs was established soon after the use of levodopa in the management of PD became prevalent (Hardebo et al. 1980). The existence of these enzymes in BECs together with observations that serotonin and noradrenergic transporters are present on the surface of BECs in the brain (Wakayama et al. 2002) may be more related to the possible role of serotonin and norepinephrine in regulating vascular flow then a xenobiotic role. Regardless, the relative roles of these enzymes as well as the CYP enzymes in relation to changes that may occur as part of the neurodegenerative process and subsequent effects on the BECs is virtually unknown. It appears that the CYP enzymes in the BBB may also be inducible as they are in the liver. Although the low levels of CYP enzymes normally present in the BBB represents only a small fraction present in the body, does induction of these enzymes in the BBB, the last point of entry into brain, have significant therapeutic implications in the management of psychiatric or neurodegenerative disorders? Very little work has been done in this area.
V: The Blood Brain Barrier as a signaling interface
Data is beginning to accumulate suggesting that the BECs comprising the BBB may serve a signaling function. Although the polarized transport of molecules described above can be viewed as representing a signaling mechanism, recent evidence suggests that receptor activation can influence barrier integrity as well as generate signals that are transferred into brain parenchyma and vice versa.
Polakis (2008) recently reviewed signaling pathways that appear to regulate the integrity of TJ proteins. Calcium, RhoGTPases, G-protein signaling, and various kinases have been shown to regulate TJ proteins, including the claudins (Hawkins and Davis 2005); (Persidsky et al. 2006) and recent studies by Liebner et al. (2008) strongly implicate the Wnt signaling pathway (Liebner et al. 2008). These studies provide a means through which luminal or abluminal signals can affect TJs and therefore barrier integrity under physiological as well as pathological circumstances. However, other studies suggest that the BECs can transduce signals from the lumen causing release of mediators on the abluminal surface as well.
Gosselin and Rivest (2008), demonstrated that Interleukin-6 (IL-6) receptors on BECs mediated activation of microglia through prostaglandin E2 signaling suggesting a role for the BEC in transmitting systemic inflammatory events into the brain (Gosselin and Rivest 2008). Banks (2006) summarized several of these studies. Thus, LPS treatment of BEC monolayers in transwell chambers led to the release of IL-6, IL-10, TNFα, and granulocyte-macrophage colony stimulating factor from the abluminal surface of the cells suggesting that a vascular signal caused the release of cytokines into brain parenchyma. Adiponectin, a large protein that is released into blood from fat and affects feeding, is thought to influence feeding behaviors by working on receptors on the luminal surface. BEC monolayers exposed to adiponectin led to significant increases in IL-6 from the abluminal surface of the monolayer. Taken together, these studies speak to the emerging role of the BEC in the BBB as a signaling/communication interface between the peripheral and central compartments (Banks 2006). In addition to these studies, it is now well accepted that parenchymal inflammatory mediators signal the BEC to increase expression of selectins to signal peripheral monocytes to attach and enter brain (see below). As we begin to evaluate the role of the BBB as a signaling interface, it is likely that changes in this capability will aid in our understanding of the pathogenesis of neurodegenerative disease. Given the recent characterization of the BBB as a dynamic structure, its emerging signaling function will likely play an important role in both normal and pathological function that will undoubtedly impact the way we look at neurodegenerative disorders.
VI: Entry of Peripheral Cells into Brain Parenchyma
The historical notion that the brain is an immune privileged area is not true. There is strong consensus that the normal brain undergoes limited, but routine immune surveillance and adaptive immunity likely plays a role in all neurodegenerative diseases. Unlike the periphery where peripheral mononuclear cells migrate readily across capillary beds, immune cells enter brain primarily across post-capillary venules (Owens et al. 2008b). Two additional sites of immune cell entry have been identified thus far. Immune cells enter cerebrospinal fluid (CSF) through the choroid plexus as well as through post-capillary venules at the pial surface that invade parenchyma to form Virchow-Robbins spaces. Immune cells cluster in Virchow-Robbins spaces to produce a continuous supply of immune cells as documented in the EAE model of MS. Regardless of location, extravasation (transmigration) of monocytes across the BBB is a complex process involving activation of the monocytes and BECs as well as migration mediated by gradients of chemokines. This process was recently reviewed in detail by Man and colleagues (Man et al. 2007).
Pro-inflammatory mediators including cytokines, activate BECs and lead to increased expression of CAM of the E-selectin subfamily on the luminal surface of BECs (another example of the BEC as a signaling interface). These selectins bind to glycosylated ligands (integrins) on the monocytes in a process called tethering (Figure 4). Such tethering may not overcome the sheer forces created by blood flow, but will cause monocytes to slow and roll along the luminal surface of the vessel. Normally, there is little adherence of the monocyte to the luminal wall because of low expression of selectins explaining the very low level of normal immune surveillance. However, immune cell activation peripherally, or the presence of immobilized chemokines on the luminal wall in particular, leads to increased selectin expression that results in tethering. Tethering does not lead to transmigration, but provides adequate attachment so that the normal flow of blood through the venules creates a rolling behavior of the monocytes. The binding of the monocyte integrin to BEC selectins signals the BECs to increase expression of VCAM, ICAM and fibronectin connecting segments (FN-CS-1), which then form into clusters increasing binding avidity for the monocytes. Similarly, this binding causes conformational changes in other integrins on the monocytes allowing for greater adherence. Slowed down by tethering, the monocytes can then bind with greater avidity to clustered proteins on the luminal wall causing movement arrest.
If the monocyte integrins are in a high affinity state due to stimulation, and BEC receptors have become clustered as a result of inflammatory effects on the BECs, the monocyte will slowly roll down the luminal wall to paracellular junctions (although transcellular transmigration through a BEC has also been described (Carman and Springer 2004). Here the monocyte will form firm adhesion via integrin receptors to endothelial counter-receptors. In conjunction with release of MMPs that digest the basement membrane, the monocyte will extend processes rich in chemokine receptors into the perivascular space “sniffing” the microenvironment for chemokines. If detected, the monocyte will completely enter the perivascular space. Provided an adequate gradient of chemokines is present, the monocyte will then traverse the glia limitans; a step that similarly involves the MMPs.
Clearly, the multiple steps and proteins involved with transmigration of peripheral monocytes into brain offer numerous opportunities for therapeutic intervention. Since MS likely involves an autoimmune component, interfering with T-cell entry into brain should reduce the relapses many MS patients experience following their initial presentation. Natalizumab is a neutralizing antibody to α4 integrin, which mediates transmigration of activated T-cells. Patients treated with natalizumab experience significantly reduced numbers of flare ups in association with fewer white matter lesions (see review by (Rommer et al. 2008)). This suggests that interfering with monocyte transmigration can provide therapeutic benefit. There are likely other means of interfering with this process suggesting that neuroinflammation-mediated immune cell invasion can potentially be reduced in numerous neurodegenerative diseases including PD and AD, provided that the immune system plays a role in disease progression like it appears to do in MS (see below). In addition, Beers et al. have demonstrated in an animal model of ALS casued by dominant mutations in Cu2+/Sn2+ superoxide dismutase (SOD1) that wild type donor derived microglia transplanted into mSOD1G93A/Pu.1-/-transgenic animals slowed the loss of motor neurons and prolonged survival (Beers et al. 2006). This suggests that peripheral myeloid cells do transmigrate across the BBB in response to injury. To further support this notion, Giri et al. demonstrated that Aβ-mediated migration of monocytes can be inibitited by antibody to the Aβ-receptor (RAGE) and platelet endothelial cell adhesion molecule (PECAM-1(Giri et al. 2000). However, it is also clear that in addition to increased immune cell entry in CNS disease, the neuroinflammation that mediates the BEC activation responsible for the transmigration process in the first place, also appears to compromise barrier integrity. If the barrier is compromised, the need for the transmigration process described above may be less critical. Moreover, if the barrier is indeed compromised, the chemokines that guide peripheral monocytes into brain will be able to enter the vascular lumen and not only more readily activate monocytes, but provide a stronger tropic signal to guide migration. Whether or not this is the case in neurodegenerative diseases is totally unexplored and represents a new set of potential therapeutic interventions. Thus, would chemokine antagonists reduce immune cell entry into brain across a breached barrier?
We recently reviewed the literature regarding the involvement of adaptive immunity in neurodegenerative disease, which revealed scattered historical references to immune cell involvement, and recent evidence strongly implicating peripheral immune cell entry in PD and other neurodegenerative diseases (Monahan AJ and Carvey PM 2008) (See Figure 6.). When mouse chimeras with green fluorescent protein bone marrow were treated with MPTP, bone marrow derived cells migrated into the SN and adopted the phenotype of activated microglia (Rodriguez et al. 2007). These results suggest that MPTP treatment induces extravasation of peripheral cells into the brain as part of an innate immune response. There is also evidence for ongoing immune surveillance in the brain mediated by naive T cells (Cose 2007;Cose et al. 2006). In addition, infiltration of CD 4 and CD 8 T cells have been observed following MPTP treatment (Kurkowska-Jastrzebska et al. 1999). Kurkowska et al., found that MPTP induced DA neuron damage could be reduced by dexamethasone treatment implicating immune involvement in DA neuron loss (Kurkowska-Jastrzebska et al. 1999). More recently, Brochard and colleagues showed that the MPTP induced DA loss was reduced in mice lacking CD4 helper T-cells suggesting their involvement in DA neurodegeneration (Brochard et al. 2009).
Figure 6.
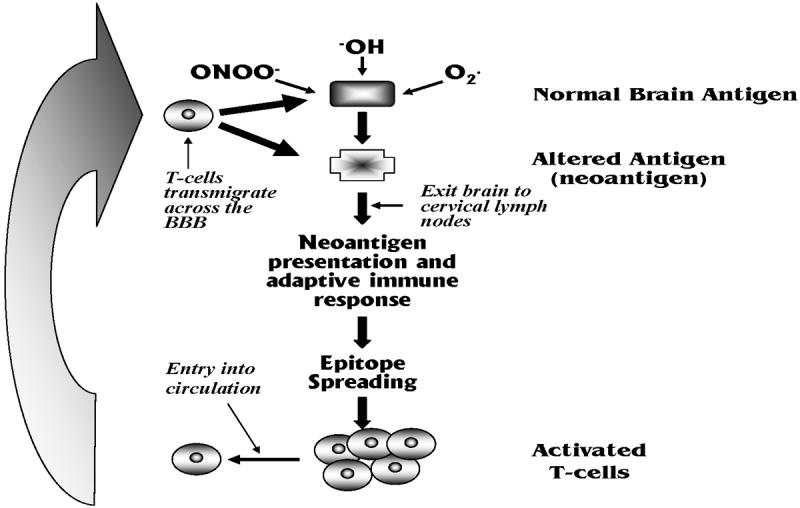
Depiction of positive feedback mechanism that could potentially contribute to neurodegenerative disease progression. Neuroinflammation leads to generation of reactive oxygen species (ROS) that attack normal brain antigens to form neoantigens. These neoantigens leave brain and collect in the cervical lymph nodes where they induce and adaptive immune response. T-cells of multiple specificieties are generated through epitope spreading, that bind to the original neoantigen and potentially normal brain antigens. These cells circulate and transmigrate across the BBB facilitated by changes in the BBB induced by neuroinflammation where they attack cells and further increase neuroinflammation contributing to progression.
Benner et al. provided a possible mechanism for an adaptive immune response in PD (Benner et al. 2008). In this study, they showed that CNS antigens exited the brain following MPTP treatment and they isolated a neo-antigen (nitrated α-synuclein (N-α-syn)) from the cervical lymph nodes. As has been shown in MS, antigen does indeed leak from the CNS and enters cervical lymph nodes where it can induce an adaptive immune response (Weller et al. 2009). Benner’s group then adoptively transferred cells from mice exposed to N-α-syn, into mice that had been treated with MPTP. The mice receiving the cells exposed to N-α-syn had greater losses of DA neurons indicating that the introduction of immune cells previously exposed to an antigen associated with MPTP treatment exacerbated the MPTP induced DA neuron loss. Moreover, adoptive transfer from immune depleted SCID mice treated in the same fashion did not show DA neuron losses. This study suggests that self-antigens attacked by reactive oxygen species form neo-antigens that become antigenic (Figure 5). One can envision some initial insult that causes a regional neuroinflammatory response that leads to neuron loss, which then sheds antigen that is attacked by hydroxyl radicals or peroxynitrite for instance, producing neoantigens that initiate a secondary immune response. Entry of T-cells, as well as antigen spreading, leads to further attack in an area of inflammation that evolves into a self-sustaining cycle of neuron loss and further immune activation that produces progression of disease. Early in life, the system may be able to maintain control of this cycle. However, as the system reduces its anti-inflammatory capacity with age, is less able to detoxify free radicals, or has a compromised BBB, the antigenic capacity reaches a tipping point where cell loss begins and continues. This is clearly a simplistic depiction of a complex process. Regardless, the neurocentric view often precludes consideration of such a hypothesis, yet has not been able to provide a sound mechanism for the progressive nature of most neurodegenerative diseases, with the notable exception of MS where an immune component to the disease as well as its progression is widely accepted. Do such phenomena occur in PD, AD, and/or ALS? If this is the case, would interfering with immune cell entry or altering transmigration of cells as shown to be successful in MS, slow disease progression? As has been true of most of the rhetorical questions in this review, they are intriguing questions that have not been asked by those who study neurodegenerative disease.
Figure 5.
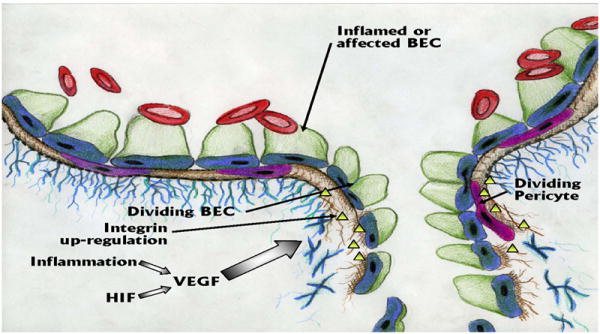
Angiogenesis within the vasculature of the brain is precipitated by Hypoxia Inducing Factor (HIF) or inflammation that induce increased production of Vascular Endothelial Growth Factor (VEGF). VEGF induces the brain endothelial cells (BECs) to divide, or alternatively, new BECs develop following de-differentiation of pericytes that act as stem cells in the angiogenesis process. As would be expected, matrix metalloproteinases (MMPs) are also needed to digest the basement membranes. As depicted, angiogenic vessels are leaky.
VII: Angiogenesis and BBB integrity
Angiogenesis is the formation of new blood vessels and is a critical component in a number of normal physiological functions including development, follicle cycles in women, and wound healing. In cancer, tumors must induce angiogenesis in order to grow and anti-angiogenic drugs are well characterized and effective in treating some tumors. Angiogenesis also occurs in the brain. Periods of hypoxia lead to increased production of the transcription factor hypoxia inducing factor (HIF) that has been shown to up-regulate production of vascular endothelial growth factor (VEGF). VEGF is increased in AD (Kalaria et al. 1998);(Thirumangalakudi et al. 2006), stroke (Kaur and Ling 2008), PD (Wada et al. 2006) and perhaps in ALS (Sathasivam, 2009). Although VEGF may have additional properties in terms of direct effects on neurons and neurogenesis (Udo et al. 2008), there is ample evidence of its increased production in PD implicating growth of new vessels. Faucheux et al. showed an increase in vascular density in the SN, but not the VTA of PD patients (Faucheux et al. 1999) and Barcia et al. found evidence of microangiogenesis in the PD brain, which is often associated with barrier dysfunction (Barcia et al. 2004). Barcia et al. also noted an increase in the number of blood vessels in close proximity to degenerating DA neurons in non-human primates that was highly correlated with increases in VEGF expression (Barcia et al. 2005). VEGF is also elevated in the SNpc and in the striatum of PD patients (Wada et al. 2006;Yasuda et al. 2007). In fact, a single intra-nigral injection of 1μg VEGF led to BBB compromise that peaked at 4 days (Rite et al. 2008) although Tian et al. did not demonostrate angiogenesis or BBB dysfunction after administering the VEGF gene via an adeno- associated viral vector (AAV-VEGF) to the striatum (Tian et al. 2007). More importantly, the leakage correlated with the degeneration of DA neurons adjacent to the injection site. In addition, we have observed a co-localization of β3 immunoreactivity, a marker for angiogenesis, with BBB permeability in an animal model of PD (Carvey et al. 2005). Taken together, these studies suggest that angiogenesis might actually participate in the DA degenerative process.
It is important to recognize that angiogenic vesicles are leaky and may explain the BBB dysfunction observed in all neurodegenerative diseases (see above). But, angiogenesis is generally viewed as transient. Thus, neuroinflammatory events or hypoxia lead to angiogenic changes that lead to new vessel formation with subsequent return to normal. However, what are the consequences of chronic neuroinflammation? Does this lead to continuing angiogenesis as the neuroinflammation spreads from an initial area of injury? Indeed, there is evidence of chronic angiogenesis as well in neurodegenerative diseases.
In a mouse model of hypoxic retinopathy, a hypoxic event results in the expression of VEGF for 2 days. After the first day, the integrin αvβ3, a marker for angiogenic vessels, is expressed on vessel buds (Chavakis et al. 2002b). Angiogenesis ensues and the maximum number of vessels are present between 6-8 days but by 23 days the number of vessels has returned to baseline (Chavakis et al. 2002a). Folkman hypothesized a balance of angiogenic and anti-angiogenic factors that constitutes an angiogenic switch (Hanahan et al. 1996). The change in the expression of pro or anti-angiogenic factors then determines whether angiogenesis continues or regresses. However, the chronic exposure to angiogenic factors, such as VEGF, as might occur during continued neuroinflammation as has been proposed in PD, produces the pathological angiogenic vessels found in many tumors (Nagy et al. 2008). These vessels do not mature and are constantly leaky (Nagy et al. 2008). Interestingly, chronic angiogenesis has been implicated in other progressive diseases including inflammatory bowel disease (Pousa et al. 2008), diabetic retinopathy (Penn et al. 2008), psoriasis (Heidenreich et al. 2008), and rheumatoid arthritis (Yoo et al. 2008) Thus, do the neuroinflammatory changes that occur in neurodegenerative disease alter a pro/anti-angiogenic balance that contributes to a chronic state of BBB dysfunction and further degeneration? Since, VEGF can produce DA neuron loss in a PD animal model, does blocking VEGF prevent DA neuron loss and more importantly, the progressive loss of DA neurons? Chronic angiogenesis and disease progression in neurodegenerative diseases is certainly worthy of further study and could provide new therapeutic targets.
VIII: Do changes in the BBB cause, participate or result from degenerative changes?
Alterations in the BBB are likely involved in all neurodegenerative diseases. The question then becomes do these changes cause the disease, participate in its pathogenesis, or are they simply a consequence of degeneration? The answer to this question has significant implications. In disease models, such as reperfusion injury, a model for stroke, there is a biphasic opening of the BBB in which the onset is due to the activation of MMP-2 while the later opening is due to activation of MMP-9 and MMP-3 (Rosenberg and Yang 2007);(Gasche et al. 2006). Vascular changes that lead to BBB compromise clearly play a role in the development of neuron loss in stroke. Notwithstanding stroke, there are several studies that argue that dysfunction in barrier function precipitates in AD. According to the neurovascular hypothesis of AD, faulty BBB clearance of A-beta via LRP-1/RAGE-mediated transport, may be the mechanism responsible for A-beta (Aβ) accumulation in the brain. Soluble forms of Aβ are transported from the blood to the brain via the RAGE receptor and from brain to blood via the LRP receptor (Zlokovic et al. 2005;Deane et al. 2004). Homeostasis of Aβ in the brain is controlled by binding of Aβ to transport proteins such as apolipoprotein E4 and apolipoprotein J (Fagan et al. 2002). Martel et al. demonstrated in guinea pig brain that apoE4 forms a stable complex with soluble Aβ, which reduces degradation of the peptide and in turn may enhance transport across the BBB and therefore amyloid formation in brain whereas ApoE2 and E3 prevented transport of soluble Aβ (Martel et al. 1997). Additional features of AD such as angiogenesis and arterial dysfunction may also initiate neurovascular uncoupling leading to Aβ accumulation and neurovascular inflammation resulting in synaptic and neuronal dysfunction (Zlokovic 2005;Deane and Zlokovic 2007). This hypothesis suggests that neurovascular dysfunction precedes the neuronal degeneration implicating BBB dysfunction as causal in AD. In support of this hypothesis, brain imaging studies using 2-[18F] fluoro-2-deoxy-D-glucose (FDG)-PET, a measure of cerebral glucose transport across the BBB, show a reduction in cerebral glucose uptake in patients with mild cognitive decline and possible AD prior to progression to AD (Drzezga et al. 2003) (Hunt et al. 2007;Bell and Zlokovic 2009b). It is also likely that barrier dysfunction participates in the progression of AD (Jaeger L.B. 2009).
Recent studies document various vascular anatomical defects in AD patients including arteriole and capillary atrophy and irregularities, swelling and increased numbers of pinocytic vesicles in BECs, disruption of the basement membrane, and reduced total microvascular density (Bell and Zlokovic 2009a). In addition, Wu and colleagues (2005) reported that the low expression of vascular-restricted mesenchyme homeobox 2 gene leads to angiogenesis and premature pruning of capillary networks in AD patients. Indeed, we recently showed increased αvβ3 immunoreactivity in AD brains suggesting angiogenesis (Desai et al. 2009). Since angiogenic vessels are notoriously leaky, the possibility exists that elements of the peripheral vasculature may participate in AD progression, and as argued above, may even be involved with its development. Regardless of timing, it is likely that BBB dysfunction plays an important role in AD pathogenesis.
In multiple sclerosis, the early development of the disease is associated with increases in MMPs 1,2,3,7,9 and 12 activity, which are known to disrupt the BBB and lead to an increased entry of leukocytes as well as degradation of myelin (Kurzepa et al. 2005;Lindberg et al. 2001). In addition, Kirk and colleagues (2004) argue that angiogenic changes are well established in MS pointing to a link between BEC inflammation, barrier abnormalities, and angiogenic changes that could contribute to disease progression (Kirk et al. 2004). Although it is clear that barrier dysfunction contributes to disease progression, whether or not it is causative remains a question. However, it must be acknowledged that since MS is considered an autoimmune disease and passage of immune mediators across the BBB is critical step in its development, it can be argued that barrier dysfunction and not compromise may also be causative.
Disruption of the BBB has also been implicated as causative in HIV associated dementia (HAD). With the advent of new drug therapies, HAD is becoming more prevalent and patients exhibit cognitive deficits from sub-clinical and mild cognitive motor deficits to dementia. HAD generally develops late in disease and patients deteriorate into a vegetative state. Studies by Kanmogne and colleagues (2002) revealed that the HIV protein gp120 was toxic to BECs implicating barrier compromise as an early step in the CNS infection process, suggesting that BBB dysfunction or compromise is causative in HAD (Kanmogne et al. 2002). Since BECs express a number of different receptors or membrane-bound cytokines that affect TJ integrity, it is quite possible that circulating factors initiate TJ alterations that lead to barrier compromise to initiate disease.
Westerlund and colleagues (2009) identified polymorphinsms in P-gp as a risk factor for PD suggesting the possibility of reduced capacity to extrude DA neurotoxins (Westerlund et al. 2009). In a similar vein, Menegon et al., (1998) demonstrated polymorphisms in glutathione transferase in the BBB suggesting reduced capacity to detoxify xenobiotics at the BBB (Menegon et al. 1998). Although these studies suggest that environmental toxins are the initiating factor, they also argue for the involvement of the BBB in causality. Interestingly, there has been a long-standing inverse relationship between caffeine consumption and PD. Chen and colleagues (2008) demonstrated that caffeine prevented the breakdown in the BBB normally produced by the DA neurotoxin MPTP. Thus, caffeine administration not only prevented MPTP-induced barrier compromise, it prevented the increases in MMP9 and losses of ZO-1 normally associated with DA neuron loss (Chen et al. 2008). Although these effects could result from the effects of caffeine on adenosine receptors, they could also reflect its inhibition of phosphodiesterase thereby increasing cAMP which is known to enhance TJs (Ishizaki et al. 2003). Indeed, Rite and colleagues (2007) demonstrated that injection of VEGF alone caused DA neuron loss and neuroinflammation typical of PD suggesting that BBB dysfunction may be a very early event in the PD process (Rite et al. 2007).
The discussion above alludes to a significant role for a causal role or involvement of the BBB in the initial degenerative process. However, the commonality of BBB compromise in all neurodegenerative diseases studied to date suggests at least, that barrier dysfunction results from the pathogenic process. Indeed, numerous inflammogens present in these disease states have all been repeatedly shown to alter barrier function suggesting in the very least that barrier function results from the degenerative process. This has significant implications for understanding disease progression as well as therapy.
IX: Consequences of barrier dysfunction in neurodegenerative disease
As reviewed above, there is emerging evidence for the involvement of adaptive immunity in the disease process. Activation of BECs facilitates entry of immune cells that would assist the neurodegenerative process creating more neuroinflammation in a self-sustaining process leading to disease progression that characterizes these disease states (Figure 7). This is likely facilitated by advanced age and its associated impact on BBB function, detoxification capacity, and anti-inflammatory capability. BBB dysfunction and/or barrier compromise resulting from, and adaptations to the degenerative process, would allow other elements of the peripheral vasculature including complement, toxins, and metals normally excluded from the central compartment to enter brain and potentially contribute to the progression of disease. BBB dysfunction would likely also alter ion balance, disrupt transport systems, and potentially alter constituents of the enzymatic barrier from being effective. Finally, chronic BEC inflammation leads to chronic angiogenesis that is often aberrant leading to altered perfusion and its potential consequences. Thus, consideration of the involvement of the peripheral compartment is likely critical to our understanding of the progression of neurodegenerative disease.
Figure 7.
Summary of consequences of a dysfunctional/compromised BBB. Dysfunction of the BBB, angiogenic leak, or TJ separation mediated by neuroinflammation leads to disruption of the normal boundaries between the peripheral blood and the parenchymal compartments. As a result, peripheral factors including toxins, monocytes, and complement etc., more readily enter the brain. In addition, chemokines, metabolites, and neurotransmitters etc., more readily leave brain. The mingling of these compartments that are usually separated is likely to have significant effects on our perception of how neurodegenerative diseases develop and progress.
Barrier compromise in neurodegenerative diseases also has significant implications for drug therapy. Studies described above demonstrate increased entry of drugs normally excluded from brain in animals with barrier compromise. As we considered the implications of this, we came to realize that it is likely that numerous drugs that could be used in neurodegenerative diseases are never developed for therapy because preliminary studies suggest they do not cross the BBB. But, what if the barrier is not intact? Indeed, conventional wisdom assumes an intact BBB and water-soluble drugs or larger molecules that could be effective in these diseases, are never tried in animal models of the disease where BBB compromise is likely present. Would systemically administered peptide, protein, or glycoprotein drugs be effective in an animal model of AD or PD if they were tried in actual animal models? Interestingly, Banks and colleagues (2007) recently demonstrated increased anti-amyloid beta protein entry across the blood-brain barrier in the SAMP8 mouse model of AD suggesting that barrier compromise or BBB dysfunction may indeed facilitate entry of substances that normally do not cross the BBB (Banks et al. 2007).
It must also be appreciated that the BBB alterations we are alluding to above are not system-wide, but rather punctate (Figure 8). Thus, barrier compromise does not occur throughout an affected structure in which neurodegeneration is occurring, but rather in small localized areas within a structure. Thus, we have exposed rats and mice to various known DA neurotoxins and demonstrated barrier compromise by injecting FITC-labeled albumin into the carotid artery just prior to sacrifice and then scanned the brains using confocal microscopy. In the hundreds of animals we have studied, we always observe punctate areas of FITC-labeled albumin leakage in DA rich areas, but not in other areas (Zhao et al. 2007). We hypothesized that these punctate areas of barrier compromise reflect regions undergoing active neuroinflammation as evidenced by finding activated microglia co-localized to areas of FITC-labeled albumin penetration. Indeed, areas of barrier compromise in MS are also punctate. This raises the intriguing possibility of directed delivery following systemic administration. Thus, systemic administration of drugs that normally do not penetrate the BBB would enter only in areas of active neuroinflammation. This strategy may be particularly useful in slowing or even arresting the self-perpetuating cycles of neuroinflammation thought to underlie progression of neurodegenerative disease. Such punctate patterns of leakage may also underlie drug side effects as has been argued by Westin and colleagues (Westin et al. 2006). Thus, the dyskinesias that attend chronic treatment of PD with levodopa/carbidopa could result in hot spots of increased DA activity in the brain due to inhomogeneous delivery of levodopa or carbidopa due to barrier compromise.
Figure 8.
Section of rat and mouse mesencephalon at approximately that same level (accessory optic tract) demonstrating punctate areas of BBB compromise in animals treated with various neurotoxins. Just prior to sacrifice, animals were injected with FITC-labeled albumin which normally does not cross the BBB. Three minutes later the animals were sacrificed and brain sections were scanned using confocal microscopy. a: section from the mesencephalon from a control rat revealing typical FITC-labeled albumin confined to the vessels; b: punctate areas of FITC-labeled albumin leakage from vessels in rats that had received 6-hydroxydopamine (6OHDA) 30 days prior to sacrifice; c: same as b, but rats had received paraquat 14 days prior to sacrifice; d: 16 month old rats that had received rotenone one month prior to sacrifice; e: mesencephalon of a mouse exposed to MPTP 72 hours prior to sacrifice; f: 17 month old rats that were exposed to lipopolysaccharide in utero at E10.5 similarly show punctate areas of leakage in the mesencephalon in the vicinity of DA neurons (redish orange cells).
Finally, do therapeutic strategies designed to enhance barrier integrity reduce progression? The study by (Chen et al. 2008) suggests this might be the case. Although caffeine has a number of effects that could prevent the MPTP induced DA neuron loss, the possibility of increasing cAMP levels that, in turn, stabilize TJs may be involved. Are patients on chronic β-adrenergic agonists or phosphodiesterase inhibitors, both of which increase cAMP, less likely to develop PD or AD or do they exhibit slowed progression after they go on these drugs? We recently showed that animals treated with cyclic RGD peptide, that binds αvβ3 and prevents angiogenesis, prevented MPTP-induced leakage as well as DA neuron loss suggesting the involvement of barrier compromise in the acute effects of this neurotoxin as well (unpublished observation). Given that there are currently several angiogenic inhibitors on the market or in late phases of clinical trials for cancer treatment, would these drugs slow or stop the progression of neurodegenerative diseases? For those of us who study neurodegenerative disease, it is time for us to begin considering the role of BBB dysfunction, barrier compromise and the potential involvement of the peripheral compartment in our neurocentric fields.
Acknowledgments
The authors would like to thank William A. Banks for his critical read of the manuscript and excellent suggestions on its content. Thanks also go to Chris Carvey for the artwork. This work was supported by a grant from the Kenneth Douglass Foundation.
Abbreviations
- AD
Alzheimer’s disease
- ABC
ATP-Binding Cassette
- BBB
blood brain barrier
- BEC
brain endothelial cell
- BCRP
breast cancer resistance protein
- CAM
cell adhesion molecule
- DA
dopamine
- EC
endothelial cell
- EAAT
excitatory amino acid transporters
- EAE
experimental allergic encephalomyelitis
- FN-CS-1
fibronectin connecting segments
- FITC
fluorescein isothiocyanate
- GAT/BGT
gamma aminobutryic acid transporter/big gamma aminobutryic acid transporter
- HAD
HIV- associated dementia
- HIF
hypoxia inducing factor
- HVA
homovanillic acid
- IL-6
interleukin -6
- ICAM
inter-cellular adhesion molecule
- JAM
junctional adhesion molecule
- LNAA
large neutral amino acid transporters
- LPS
lipopolysaccharide
- LPR
low-density lipoprotein receptor-related protein
- MDR
multidrug resistant protein
- MMP
matrix metalloproteinase
- MRPs
MDR associated proteins
- MS
multiple sclerosis
- NE
norepinephrine
- OAT
organic anion transport
- OAT/SLCO
organic anion transport/SoLute Carrier Family
- PD
Parkinson’s Disease
- P-gp
P-glycoprotein
- PECAM
platelet endothelial cell adhesion molecule
- RAGE
receptor for advanced glycation end products
- RMT
receptor mediated transport
- RSP
renal-specific transporter
- 5-HT
serotonin
- SLCO
SoLute Carrier Family
- TLE
temporal lobe epilepsy
- TJ
tight junction
- TNF-α
tumor necrosis factor – alpha
- ZO
zona occludin
- VCAM
vascular cell adhesion molecule
- VTA
ventral tegmental area
Reference List
- 1.Abbott N. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. 2002a;200:527. doi: 10.1046/j.1469-7580.2002.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. 2002b;200:629–638. doi: 10.1046/j.1469-7580.2002.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al Anizy M, Horley NJ, Kuo CW, Gillett LC, Laughton CA, Kendall D, Barrett DA, Parker T, Bell DR. Cytochrome P450 Cyp4x1 is a major P450 protein in mouse brain. FEBS J. 2006;273:936–947. doi: 10.1111/j.1742-4658.2006.05119.x. [DOI] [PubMed] [Google Scholar]
- 4.Balabanov R, Dore-Duffy P. Role of the CNS microvascular pericyte in the blood-brain barrier. J Neurosci Res. 1998;53:637–644. doi: 10.1002/(SICI)1097-4547(19980915)53:6<637::AID-JNR1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 5.Banks WA. The blood-brain barrier as a regulatory interface in the gut-brain axes. Physiol Behav. 2006;89:472–476. doi: 10.1016/j.physbeh.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 6.Banks WA, Farr SA, Morley JE, Wolf KM, Geylis V, Steinitz M. Anti-amyloid beta protein antibody passage across the blood-brain barrier in the SAMP8 mouse model of Alzheimer’s disease: an age-related selective uptake with reversal of learning impairment. Exp Neurol. 2007;206:248–256. doi: 10.1016/j.expneurol.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bar T. The vascular system of the cerebral cortex. Adv Anat Embryol Cell Biol. 1980;59:I–62. doi: 10.1007/978-3-642-67432-7. [DOI] [PubMed] [Google Scholar]
- 8.Barcia C, Bautista V, Sanchez-Bahillo A, Fernandez-Villalba E, Faucheux B, Poza M, Fernandez BA, Hirsch EC, Herrero MT. Changes in vascularization in substantia nigra pars compacta of monkeys rendered parkinsonian. J Neural Transm. 2005;112:1237–1248. doi: 10.1007/s00702-004-0256-2. [DOI] [PubMed] [Google Scholar]
- 9.Barcia C, Emborg ME, Hirsch EC, Herrero MT. Blood vessels and parkinsonism. Front Biosci. 2004;9:277–282. doi: 10.2741/1145. [DOI] [PubMed] [Google Scholar]
- 10.Barlow BK, Cory-Slechta DA, Richfield EK, Thiruchelvam M. The gestational environment and Parkinson’s disease: evidence for neurodevelopmental origins of a neurodegenerative disorder. Reprod Toxicol. 2007;23:457–470. doi: 10.1016/j.reprotox.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 11.Bartels AL, Kortekaas R, Bart J, Willemsen AT, de Klerk OL, de Vries JJ, van Oostrom JC, Leenders KL. Blood-brain barrier P-glycoprotein function decreases in specific brain regions with aging: A possible role in progressive neurodegeneration. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.02.002. in press. [DOI] [PubMed] [Google Scholar]
- 12.Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, Siklos L, McKercher SR, Appel SH. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103:16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Begley DJ. ABC transporters and the blood-brain barrier. Curr Pharm Des. 2004;10:1295–1312. doi: 10.2174/1381612043384844. [DOI] [PubMed] [Google Scholar]
- 14.Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009a doi: 10.1007/s00401-009-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009b doi: 10.1007/s00401-009-0522-3. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bendayan R, Ronaldson PT, Gingras D, Bendayan M. In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J Histochem Cytochem. 2006;54:1159–1167. doi: 10.1369/jhc.5A6870.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, Nemachek C, Ciborowski P, Przedborski S, Mosley RL, Gendelman HE. Nitrated alpha-Synuclein Immunity Accelerates Degeneration of Nigral Dopaminergic Neurons. PLoS ONE. 2008;3:e1376. doi: 10.1371/journal.pone.0001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beretta S, Begni B, Ferrarese C. Pharmacological manipulation of glutamate transport. Drug News Perspect. 2003;16:435–445. doi: 10.1358/dnp.2003.16.7.829355. [DOI] [PubMed] [Google Scholar]
- 19.Bernacki J, Dobrowolska A, Nierwinska K, Malecki A. Physiology and pharmacological role of the blood-brain barrier. Pharmacol Rep. 2008;60:600–622. [PubMed] [Google Scholar]
- 20.Betz AL. Transport of ions across the blood-brain barrier. Fed Proc. 1986;45:2050–2054. [PubMed] [Google Scholar]
- 21.Betz AL, Goldstein GW. Polarity of the blood-brain barrier: neutral amino acid transport into isolated brain capillaries. Science. 1978;202:225–227. doi: 10.1126/science.211586. [DOI] [PubMed] [Google Scholar]
- 22.Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, Duyckaerts C, Flavell RA, Hirsch EC, Hunot S. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119:182–192. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butt AM, Jones HC, Abbott NJ. Electrical resistance across the blood-brain barrier in anaesthetized rats: a developmental study. J Physiol. 1990;429:47–62. doi: 10.1113/jphysiol.1990.sp018243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campbell SJ, Carare-Nnadi RO, Losey PH, Anthony DC. Loss of the atypical inflammatory response in juvenile and aged rats. Neuropathol Appl Neurobiol. 2007;33:108–120. doi: 10.1111/j.1365-2990.2006.00773.x. [DOI] [PubMed] [Google Scholar]
- 25.Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. 2004;167:377–388. doi: 10.1083/jcb.200404129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carvey PM, Chang Q, Lipton JW, Ling Z. Prenatal exposure to the bacteriotoxin lipopolysaccharide leads to long-term losses of dopamine neurons in offspring: a potential, new model of Parkinson’s disease. Front Biosci. 2003;8:s826–s837. doi: 10.2741/1158. [DOI] [PubMed] [Google Scholar]
- 27.Carvey PM, Zhao CH, Hendey B, Lum H, Trachtenberg J, Desai BS, Snyder J, Zhu YG, Ling ZD. 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur J Neurosci. 2005;22:1158–1168. doi: 10.1111/j.1460-9568.2005.04281.x. [DOI] [PubMed] [Google Scholar]
- 28.Chan-Ling T, Hughes S, Baxter L, Rosinova E, McGregor I, Morcos Y, van Nieuwenhuyzen P, Hu P. Inflammation and breakdown of the blood-retinal barrier during “physiological aging” in the rat retina: a model for CNS aging. Microcirculation. 2007;14:63–76. doi: 10.1080/10739680601073451. [DOI] [PubMed] [Google Scholar]
- 29.Chat M, Bayol-Denizot C, Suleman G, Roux F, Minn A. Drug metabolizing enzyme activities and superoxide formation in primary and immortalized rat brain endothelial cells. Life Sci. 1998;62:151–163. doi: 10.1016/s0024-3205(97)01061-8. [DOI] [PubMed] [Google Scholar]
- 30.Chavakis E, Riecke B, Lin J, Linn T, Bretzel RG, Preissner KT, Brownlee M, Hammes HP. Kinetics of integrin expression in the mouse model of proliferative retinopathy and success of secondary intervention with cyclic RGD peptides. Diabetologia. 2002a;45:262–267. doi: 10.1007/s00125-001-0727-z. [DOI] [PubMed] [Google Scholar]
- 31.Chavakis E, Riecke B, Lin J, Linn T, Bretzel RG, Preissner KT, Brownlee M, Hammes HP. Kinetics of integrin expression in the mouse model of proliferative retinopathy and success of secondary intervention with cyclic RGD peptides. Diabetologia. 2002b;45:262–267. doi: 10.1007/s00125-001-0727-z. [DOI] [PubMed] [Google Scholar]
- 32.Chen X, Lan X, Roche I, Liu R, Geiger JD. Caffeine protects against MPTP-induced blood-brain barrier dysfunction in mouse striatum. J Neurochem. 2008;107:1147–1157. doi: 10.1111/j.1471-4159.2008.05697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cose S. T-cell migration: a naive paradigm? Immunology. 2007;120:1–7. doi: 10.1111/j.1365-2567.2006.02511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cose S, Brammer C, Khanna KM, Masopust D, Lefrancois L. Evidence that a significant number of naive T cells enter non-lymphoid organs as part of a normal migratory pathway. Eur J Immunol. 2006;36:1423–1433. doi: 10.1002/eji.200535539. [DOI] [PubMed] [Google Scholar]
- 35.Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke. 2004;35:2628–2631. doi: 10.1161/01.STR.0000143452.85382.d1. [DOI] [PubMed] [Google Scholar]
- 36.Deane R, Zlokovic BV. Role of the blood-brain barrier in the pathogenesis of Alzheimer’s disease. Curr Alzheimer Res. 2007;4:191–197. doi: 10.2174/156720507780362245. [DOI] [PubMed] [Google Scholar]
- 37.Desai BS, Schneider JA, Li JL, Carvey PM, Hendey B. Evidence of angiogenic vessels in Alzheimer’s disease. J Neural Transm. 2009 doi: 10.1007/s00702-009-0226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dey A, Jones JE, Nebert DW. Tissue- and cell type-specific expression of cytochrome P450 1A1 and cytochrome P450 1A2 mRNA in the mouse localized in situ hybridization. Biochem Pharmacol. 1999;58:525–537. doi: 10.1016/s0006-2952(99)00110-0. [DOI] [PubMed] [Google Scholar]
- 39.Diaz-Flores L, Gutierrez R, Varela H, Rancel N, Valladares F. Microvascular pericytes: a review of their morphological and functional characteristics. Histol Histopathol. 1991;6:269–286. [PubMed] [Google Scholar]
- 40.DiCarlo LA, Jr, Botvinick EH, Canhasi BS, Schwartz AS, Chatterjee K. Value of noninvasive assessment of patients with atypical chest pain and suspected coronary spasm using ergonovine infusion and thallium-201 scintigraphy. Am J Cardiol. 1984;54:744–748. doi: 10.1016/s0002-9149(84)80201-5. [DOI] [PubMed] [Google Scholar]
- 41.Diffley JM, Wu M, Sohn M, Song W, Hammad SM, Lyons TJ. Apoptosis induction by oxidized glycated LDL in human retinal capillary pericytes is independent of activation of MAPK signaling pathways. Mol Vis. 2009;15:135–145. [PMC free article] [PubMed] [Google Scholar]
- 42.DiNapoli VA, Huber JD, Houser K, Li X, Rosen CL. Early disruptions of the blood-brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol Aging. 2008;29:753–764. doi: 10.1016/j.neurobiolaging.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dombrowski SM, Desai SY, Marroni M, Cucullo L, Goodrich K, Bingaman W, Mayberg MR, Bengez L, Janigro D. Overexpression of multiple drug resistance genes in endothelial cells from patients with refractory epilepsy. Epilepsia. 2001;42:1501–1506. doi: 10.1046/j.1528-1157.2001.12301.x. [DOI] [PubMed] [Google Scholar]
- 44.Dore-Duffy P. Pericytes: pluripotent cells of the blood brain barrier. Curr Pharm Des. 2008;14:1581–1593. doi: 10.2174/138161208784705469. [DOI] [PubMed] [Google Scholar]
- 45.Drzezga A, Lautenschlager N, Siebner H, Riemenschneider M, Willoch F, Minoshima S, Schwaiger M, Kurz A. Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer’s disease: a PET follow-up study. Eur J Nucl Med Mol Imaging. 2003;30:1104–1113. doi: 10.1007/s00259-003-1194-1. [DOI] [PubMed] [Google Scholar]
- 46.Dvorska I, Brust P, Hrbas P, Ruhle HJ, Barth T, Ermisch A. On the blood-brain barrier to peptides: effects of immobilization stress on regional blood supply and accumulation of labelled peptides in the rat brain. Endocr Regul. 1992;26:77–82. [PubMed] [Google Scholar]
- 47.Ehrlich P. Ueber die beziehungen von chemischer constitution, verteilung und pharmakologischer wirkung. Gesammelte Arbeiten zur Immunitaetsforschung. 1904;574 [Google Scholar]
- 48.El Masry EM, Abou-Donia MB. Interaction of pyridostigmine bromide and N,N-diethyl-m-toluamide alone and in combination with P-glycoprotein expressed in Escherichia coli leaky mutant. J Toxicol Environ Health A. 2006;69:919–933. doi: 10.1080/15287390500360588. [DOI] [PubMed] [Google Scholar]
- 49.Engelhardt B. Development of the blood-brain barrier. Cell Tissue Res. 2003;314:119–129. doi: 10.1007/s00441-003-0751-z. [DOI] [PubMed] [Google Scholar]
- 50.Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, Hosoyamada M, Takeda M, Sekine T, Igarashi T, Matsuo H, Kikuchi Y, Oda T, Ichida K, Hosoya T, Shimokata K, Niwa T, Kanai Y, Endou H. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002a;417:447–452. doi: 10.1038/nature742. [DOI] [PubMed] [Google Scholar]
- 51.Enomoto A, Wempe MF, Tsuchida H, Shin HJ, Cha SH, Anzai N, Goto A, Sakamoto A, Niwa T, Kanai Y, Anders MW, Endou H. Molecular identification of a novel carnitine transporter specific to human testis. Insights into the mechanism of carnitine recognition. J Biol Chem. 2002b;277:36262–36271. doi: 10.1074/jbc.M203883200. [DOI] [PubMed] [Google Scholar]
- 52.Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM. Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2002;9:305–318. doi: 10.1006/nbdi.2002.0483. [DOI] [PubMed] [Google Scholar]
- 53.Faucheux BA, Bonnet AM, Agid Y, Hirsch EC. Blood vessels change in the mesencephalon of patients with Parkinson’s disease. Lancet. 1999;353:981–982. doi: 10.1016/S0140-6736(99)00641-8. [DOI] [PubMed] [Google Scholar]
- 54.Fenstermacher J, Kaye T. Drug “diffusion” within the brain. Ann N Y Acad Sci. 1988;531:29–39. doi: 10.1111/j.1749-6632.1988.tb31809.x. [DOI] [PubMed] [Google Scholar]
- 55.Fisher M. Pericyte signaling in the neurovascular unit. Stroke. 2009;40:S13–S15. doi: 10.1161/STROKEAHA.108.533117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Friedman A, Kaufer D, Shemer J, Hendler I, Soreq H, Tur-Kaspa I. Pyridostigmine brain penetration under stress enhances neuronal excitability and induces early immediate transcriptional response. Nat Med. 1996;2:1382–1385. doi: 10.1038/nm1296-1382. [DOI] [PubMed] [Google Scholar]
- 57.Garlanda C, Dejana E. Heterogeneity of endothelial cells. Specific markers. Arterioscler Thromb Vasc Biol. 1997;17:1193–1202. doi: 10.1161/01.atv.17.7.1193. [DOI] [PubMed] [Google Scholar]
- 58.Gasche Y, Soccal PM, Kanemitsu M, Copin JC. Matrix metalloproteinases and diseases of the central nervous system with a special emphasis on ischemic brain. Front Biosci. 2006;11:1289–1301. doi: 10.2741/1883. [DOI] [PubMed] [Google Scholar]
- 59.Ghersi-Egea JF, Leninger-Muller B, Suleman G, Siest G, Minn A. Localization of drug-metabolizing enzyme activities to blood-brain interfaces and circumventricular organs. J Neurochem. 1994;62:1089–1096. doi: 10.1046/j.1471-4159.1994.62031089.x. [DOI] [PubMed] [Google Scholar]
- 60.Giri R, Shen Y, Stins M, Du YS, Schmidt AM, Stern D, Kim KS, Zlokovic B, Kalra VK. beta-amyloid-induced migration of monocytes across human brain endothelial cells involves RAGE and PECAM-1. Am J Physiol Cell Physiol. 2000;279:C1772–C1781. doi: 10.1152/ajpcell.2000.279.6.C1772. [DOI] [PubMed] [Google Scholar]
- 61.Goldmann E. Vitalfarbung am zentralnervensystem. Abhandl Konigl preuss Akad Wiss. 1913;1:1–60. [Google Scholar]
- 62.Gosselin D, Rivest S. MyD88 signaling in brain endothelial cells is essential for the neuronal activity and glucocorticoid release during systemic inflammation. Mol Psychiatry. 2008;13:480–497. doi: 10.1038/sj.mp.4002122. [DOI] [PubMed] [Google Scholar]
- 63.Granberg L, Ostergren A, Brandt I, Brittebo EB. CYP1A1 and CYP1B1 in blood-brain interfaces: CYP1A1-dependent bioactivation of 7,12-dimethylbenz(a)anthracene in endothelial cells. Drug Metab Dispos. 2003;31:259–265. doi: 10.1124/dmd.31.3.259. [DOI] [PubMed] [Google Scholar]
- 64.Hanahan D, Christofori G, Naik P, Arbeit J. Transgenic mouse models of tumour angiogenesis: the angiogenic switch, its molecular controls, and prospects for preclinical therapeutic models. Eur J Cancer. 1996;32A:2386–2393. doi: 10.1016/s0959-8049(96)00401-7. [DOI] [PubMed] [Google Scholar]
- 65.Hardebo JE, Emson PC, Falck B, Owman C, Rosengren E. Enzymes related to monoamine transmitter metabolism in brain microvessels. J Neurochem. 1980;35:1388–1393. doi: 10.1111/j.1471-4159.1980.tb09014.x. [DOI] [PubMed] [Google Scholar]
- 66.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 67.Hediger MA, Romero MF, Peng JB, Rolfs A, Takanaga H, Bruford EA. The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteinsIntroduction. Pflugers Arch. 2004;447:465–468. doi: 10.1007/s00424-003-1192-y. [DOI] [PubMed] [Google Scholar]
- 68.Hedlund E, Gustafsson JA, Warner M. Cytochrome P450 in the brain; a review. Curr Drug Metab. 2001;2:245–263. doi: 10.2174/1389200013338513. [DOI] [PubMed] [Google Scholar]
- 69.Heidenreich R, Rocken M, Ghoreschi K. Angiogenesis: the new potential target for the therapy of psoriasis? Drug News Perspect. 2008;21:97–105. doi: 10.1358/dnp.2008.21.2.1188196. [DOI] [PubMed] [Google Scholar]
- 70.Hennessy M, Spiers JP. A primer on the mechanics of P-glycoprotein the multidrug transporter. Pharmacol Res. 2007;55:1–15. doi: 10.1016/j.phrs.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 71.Ho KL. Ultrastructure of cerebellar capillary hemangioblastoma. IV. Pericytes and their relationship to endothelial cells. Acta Neuropathol. 1985;67:254–264. doi: 10.1007/BF00687810. [DOI] [PubMed] [Google Scholar]
- 72.Huang P, Rannug A, Ahlbom E, Hakansson H, Ceccatelli S. Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the expression of cytochrome P450 1A1, the aryl hydrocarbon receptor, and the aryl hydrocarbon receptor nuclear translocator in rat brain and pituitary. Toxicol Appl Pharmacol. 2000;169:159–167. doi: 10.1006/taap.2000.9064. [DOI] [PubMed] [Google Scholar]
- 73.Huber JD, Campos CR, Mark KS, Davis TP. Alterations in blood-brain barrier ICAM-1 expression and brain microglial activation after lambda-carrageenan-induced inflammatory pain. Am J Physiol Heart Circ Physiol. 2006;290:H732–H740. doi: 10.1152/ajpheart.00747.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hunt A, Schonknecht P, Henze M, Seidl U, Haberkorn U, Schroder J. Reduced cerebral glucose metabolism in patients at risk for Alzheimer’s disease. Psychiatry Res. 2007;155:147–154. doi: 10.1016/j.pscychresns.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 75.Ishizaki T, Chiba H, Kojima T, Fujibe M, Soma T, Miyajima H, Nagasawa K, Wada I, Sawada N. Cyclic AMP induces phosphorylation of claudin-5 immunoprecipitates and expression of claudin-5 gene in blood-brain-barrier endothelial cells via protein kinase A-dependent and -independent pathways. Exp Cell Res. 2003;290:275–288. doi: 10.1016/s0014-4827(03)00354-9. [DOI] [PubMed] [Google Scholar]
- 76.Jaeger LB, Dohgu S, Hwang MC, Farr SA, Murphy MP, Fleegal-Demotta MA, Lynch JL, Robinson SM, Niehoff ML, Johnson SN, Kumar VB, Banks WA. Testing the neurovascular hypothesis of Alzheimer’s disease: LRP-1 antisense reduces blood-brainbarrier clearance, increases brain levels of amyloid beta protein, and impairs cognition. J Alzheimer’s Disease. 2009 doi: 10.3233/JAD-2009-1074. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Janzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 1987;325:253–257. doi: 10.1038/325253a0. [DOI] [PubMed] [Google Scholar]
- 78.Jones AR, Shusta EV. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm Res. 2007;24:1759–1771. doi: 10.1007/s11095-007-9379-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 80.Kalaria RN, Cohen DL, Premkumar DR, Nag S, LaManna JC, Lust WD. Vascular endothelial growth factor in Alzheimer’s disease and experimental cerebral ischemia. Brain Res Mol Brain Res. 1998;62:101–105. doi: 10.1016/s0169-328x(98)00190-9. [DOI] [PubMed] [Google Scholar]
- 81.Kalaria RN, Harik SI. Blood-brain barrier monoamine oxidase: enzyme characterization in cerebral microvessels and other tissues from six mammalian species, including human. J Neurochem. 1987;49:856–864. doi: 10.1111/j.1471-4159.1987.tb00973.x. [DOI] [PubMed] [Google Scholar]
- 82.Kalinin S, Feinstein DL, Xu HL, Huesa G, Pelligrino DA, Galea E. Degeneration of noradrenergic fibres from the locus coeruleus causes tight-junction disorganisation in the rat brain. Eur J Neurosci. 2006;24:3393–3400. doi: 10.1111/j.1460-9568.2006.05223.x. [DOI] [PubMed] [Google Scholar]
- 83.Kamiie J, Ohtsuki S, Iwase R, Ohmine K, Katsukura Y, Yanai K, Sekine Y, Uchida Y, Ito S, Terasaki T. Quantitative atlas of membrane transporter proteins: development and application of a highly sensitive simultaneous LC/MS/MS method combined with novel in-silico peptide selection criteria. Pharm Res. 2008;25:1469–1483. doi: 10.1007/s11095-008-9532-4. [DOI] [PubMed] [Google Scholar]
- 84.Kanmogne GD, Kennedy RC, Grammas P. HIV-1 gp120 proteins and gp160 peptides are toxic to brain endothelial cells and neurons: possible pathway for HIV entry into the brain and HIV-associated dementia. J Neuropathol Exp Neurol. 2002;61:992–1000. doi: 10.1093/jnen/61.11.992. [DOI] [PubMed] [Google Scholar]
- 85.Kapadia SE, de Lanerolle NC. Immunohistochemical and electron microscopic demonstration of vascular innervation in the mammalian brainstem. Brain Res. 1984;292:33–39. doi: 10.1016/0006-8993(84)90887-4. [DOI] [PubMed] [Google Scholar]
- 86.Kaur C, Ling EA. Blood brain barrier in hypoxic-ischemic conditions. Curr Neurovasc Res. 2008;5:71–81. doi: 10.2174/156720208783565645. [DOI] [PubMed] [Google Scholar]
- 87.Keep RF, Ennis SR, Beer ME, Betz AL. Developmental changes in blood-brain barrier potassium permeability in the rat: relation to brain growth. J Physiol. 1995;488(Pt 2):439–448. doi: 10.1113/jphysiol.1995.sp020978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kim JH, Kim JH, Yu YS, Kim DH, Kim KW. Recruitment of pericytes and astrocytes is closely related to the formation of tight junction in developing retinal vessels. J Neurosci Res. 2009;87:653–659. doi: 10.1002/jnr.21884. [DOI] [PubMed] [Google Scholar]
- 89.Kirk S, Frank JA, Karlik S. Angiogenesis in multiple sclerosis: is it good, bad or an epiphenomenon? J Neurol Sci. 2004;217:125–130. doi: 10.1016/j.jns.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 90.Kniesel U, Wolburg H. Tight junctions of the blood-brain barrier. Cell Mol Neurobiol. 2000;20:57–76. doi: 10.1023/A:1006995910836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Konig J, Nies AT, Cui Y, Leier I, Keppler D. Conjugate export pumps of the multidrug resistance protein (MRP) family: localization, substrate specificity, and MRP2-mediated drug resistance. Biochim Biophys Acta. 1999;1461:377–394. doi: 10.1016/s0005-2736(99)00169-8. [DOI] [PubMed] [Google Scholar]
- 92.Krueger M, Bechmann I. CNS pericytes: Concepts, misconceptions, and a way out. Glia. 2009 doi: 10.1002/glia.20898. in press. [DOI] [PubMed] [Google Scholar]
- 93.Kuhnke D, Jedlitschky G, Grube M, Krohn M, Jucker M, Mosyagin I, Cascorbi I, Walker LC, Kroemer HK, Warzok RW, Vogelgesang S. MDR1-P-Glycoprotein (ABCB1) Mediates Transport of Alzheimer’s amyloid-beta peptides--implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain Pathol. 2007;17:347–353. doi: 10.1111/j.1750-3639.2007.00075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kurkowska-Jastrzebska I, Wronska A, Kohutnicka M, Czlonkowski A, Czlonkowska A. The inflammatory reaction following 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine intoxication in mouse. Exp Neurol. 1999;156:50–61. doi: 10.1006/exnr.1998.6993. [DOI] [PubMed] [Google Scholar]
- 95.Kurzepa J, Bartosik-Psujek H, Suchozebrska-Jesionek D, Rejdak K, Stryjecka-Zimmer M, Stelmasiak Z. Role of matrix metalloproteinases in the pathogenesis of multiple sclerosis. Neurol Neurochir Pol. 2005;39:63–67. [PubMed] [Google Scholar]
- 96.Langhans W. Signals generating anorexia during acute illness. Proc Nutr Soc. 2007;66:321–330. doi: 10.1017/S0029665107005587. [DOI] [PubMed] [Google Scholar]
- 97.Li Q, Zhang Q, Wang C, Liu X, Li N, Li J. Disruption of tight junctions during polymicrobial sepsis in vivo. J Pathol. 2009 doi: 10.1002/path.2525. [DOI] [PubMed] [Google Scholar]
- 98.Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, Reis M, Felici A, Wolburg H, Fruttiger M, Taketo MM, von Melchner H, Plate KH, Gerhardt H, Dejana E. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol. 2008;183:409–417. doi: 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liebner S, Fischmann A, Rascher G, Duffner F, Grote EH, Kalbacher H, Wolburg H. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol (Berl) 2000;100:323–331. doi: 10.1007/s004010000180. [DOI] [PubMed] [Google Scholar]
- 100.Lindberg RL, De Groot CJ, Montagne L, Freitag P, van d V, Kappos L, Leppert D. The expression profile of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in lesions and normal appearing white matter of multiple sclerosis. Brain. 2001;124:1743–1753. doi: 10.1093/brain/124.9.1743. [DOI] [PubMed] [Google Scholar]
- 101.Ling Z, Zhu Y, Tong CW, Snyder JA, Lipton JW, Carvey PM. Prenatal lipopolysaccharide does not accelerate progressive dopamine neuron loss in the rat as a result of normal aging. Exp Neurol. 2009;216:312–320. doi: 10.1016/j.expneurol.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 102.Loscher W. How to explain multidrug resistance in epilepsy? Epilepsy Curr. 2005;5:107–112. doi: 10.1111/j.1535-7511.2005.05311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Man S, Ubogu EE, Ransohoff RM. Inflammatory cell migration into the central nervous system: a few new twists on an old tale. Brain Pathol. 2007;17:243–250. doi: 10.1111/j.1750-3639.2007.00067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martel CL, Mackic JB, Matsubara E, Governale S, Miguel C, Miao W, McComb JG, Frangione B, Ghiso J, Zlokovic BV. Isoform-specific effects of apolipoproteins E2, E3, and E4 on cerebral capillary sequestration and blood-brain barrier transport of circulating Alzheimer’s amyloid beta. J Neurochem. 1997;69:1995–2004. doi: 10.1046/j.1471-4159.1997.69051995.x. [DOI] [PubMed] [Google Scholar]
- 105.Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, Simmons D, Dejana E. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McCaffrey G, Seelbach MJ, Staatz WD, Nametz N, Quigley C, Campos CR, Brooks TA, Davis TP. Occludin oligomeric assembly at tight junctions of the blood-brain barrier is disrupted by peripheral inflammatory hyperalgesia. J Neurochem. 2008;106:2395–2409. doi: 10.1111/j.1471-4159.2008.05582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McFadyen MC, Melvin WT, Murray GI. Cytochrome P450 in normal human brain and brain tumours. Biochem Soc Trans. 1997;25:S577. doi: 10.1042/bst025s577. [DOI] [PubMed] [Google Scholar]
- 108.Menegon A, Board PG, Blackburn AC, Mellick GD, Le Couteur DG. Parkinson’s disease, pesticides, and glutathione transferase polymorphisms. Lancet. 1998;352:1344–1346. doi: 10.1016/s0140-6736(98)03453-9. [DOI] [PubMed] [Google Scholar]
- 109.Meyer RP, Gehlhaus M, Knoth R, Volk B. Expression and function of cytochrome p450 in brain drug metabolism. Curr Drug Metab. 2007;8:297–306. doi: 10.2174/138920007780655478. [DOI] [PubMed] [Google Scholar]
- 110.Mollgard K, Dziegielewska KM, Saunders NR, Zakut H, Soreq H. Synthesis and localization of plasma proteins in the developing human brain. Integrity of the fetal blood-brain barrier to endogenous proteins of hepatic origin. Dev Biol. 1988;128:207–221. doi: 10.1016/0012-1606(88)90283-7. [DOI] [PubMed] [Google Scholar]
- 111.Monahan AJ, Carvey PM. Neuroinflammation and Peripheral Immune Infiltration in Parkinson’s disease: An autoimmune hypothesis. Cell Transplantation. 2008;17:1–10. [PubMed] [Google Scholar]
- 112.Moos T. Brain iron homeostasis. Dan Med Bull. 2002;49:279–301. [PubMed] [Google Scholar]
- 113.Mori S, Takanaga H, Ohtsuki S, Deguchi T, Kang YS, Hosoya K, Terasaki T. Rat organic anion transporter 3 (rOAT3) is responsible for brain-to-blood efflux of homovanillic acid at the abluminal membrane of brain capillary endothelial cells. J Cereb Blood Flow Metab. 2003;23:432–440. doi: 10.1097/01.WCB.0000050062.57184.75. [DOI] [PubMed] [Google Scholar]
- 114.Morse DC, Stein AP, Thomas PE, Lowndes HE. Distribution and induction of cytochrome P450 1A1 and 1A2 in rat brain. Toxicol Appl Pharmacol. 1998;152:232–239. doi: 10.1006/taap.1998.8477. [DOI] [PubMed] [Google Scholar]
- 115.Nagy JA, Benjamin L, Zeng H, Dvorak AM, Dvorak HF. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis. 2008;11:109–119. doi: 10.1007/s10456-008-9099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nakagawa S, Deli MA, Nakao S, Honda M, Hayashi K, Nakaoke R, Kataoka Y, Niwa M. Pericytes from brain microvessels strengthen the barrier integrity in primary cultures of rat brain endothelial cells. Cell Mol Neurobiol. 2007;27:687–694. doi: 10.1007/s10571-007-9195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nakaoke R, Verma S, Deli M, Shirabe S, Niwa M, Banks WA. Glucose-regulated blood-brain barrier transport of insulin: Pericyte-astrocyte-endothelial cell cross talk. International Journal of Neuroprotection and Neuroregeneration. 2007;3:195–200. [Google Scholar]
- 118.Nalivaeva NN, Fisk LR, Belyaev ND, Turner AJ. Amyloid-degrading enzymes as therapeutic targets in Alzheimer’s disease. Curr Alzheimer Res. 2008;5:212–224. doi: 10.2174/156720508783954785. [DOI] [PubMed] [Google Scholar]
- 119.Nico B, Frigeri A, Nicchia GP, Corsi P, Ribatti D, Quondamatteo F, Herken R, Girolamo F, Marzullo A, Svelto M, Roncali L. Severe alterations of endothelial and glial cells in the blood-brain barrier of dystrophic mdx mice. Glia. 2003;42:235–251. doi: 10.1002/glia.10216. [DOI] [PubMed] [Google Scholar]
- 120.O’Kane RL, Hawkins RA. Na+-dependent transport of large neutral amino acids occurs at the abluminal membrane of the blood-brain barrier. Am J Physiol Endocrinol Metab. 2003;285:E1167–E1173. doi: 10.1152/ajpendo.00193.2003. [DOI] [PubMed] [Google Scholar]
- 121.Ohtsuki S. Physiological function of blood-brain barrier transporters as the CNS supporting and protecting system. Yakugaku Zasshi. 2004;124:791–802. doi: 10.1248/yakushi.124.791. [DOI] [PubMed] [Google Scholar]
- 122.Ohtsuki S, Asaba H, Takanaga H, Deguchi T, Hosoya K, Otagiri M, Terasaki T. Role of blood-brain barrier organic anion transporter 3 (OAT3) in the efflux of indoxyl sulfate, a uremic toxin: its involvement in neurotransmitter metabolite clearance from the brain. J Neurochem. 2002;83:57–66. doi: 10.1046/j.1471-4159.2002.01108.x. [DOI] [PubMed] [Google Scholar]
- 123.Oldendorf WH, Cornford ME, Brown WJ. The large appent work capability of the blood-brain barrier: A study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann Neurol. 1977;1:409–417. doi: 10.1002/ana.410010502. [DOI] [PubMed] [Google Scholar]
- 124.Onyszchuk G, He YY, Berman NE, Brooks WM. Detrimental effects of aging on outcome from traumatic brain injury: a behavioral, magnetic resonance imaging, and histological study in mice. J Neurotrauma. 2008;25:153–171. doi: 10.1089/neu.2007.0430. [DOI] [PubMed] [Google Scholar]
- 125.Owens T, Bechmann I, Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol. 2008b;67:1113–1121. doi: 10.1097/NEN.0b013e31818f9ca8. [DOI] [PubMed] [Google Scholar]
- 126.Owens T, Bechmann I, Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol. 2008a;67:1113–1121. doi: 10.1097/NEN.0b013e31818f9ca8. [DOI] [PubMed] [Google Scholar]
- 127.Pelegri C, Canudas AM, del Valle J, Casadesus G, Smith MA, Camins A, Pallas M, Vilaplana J. Increased permeability of blood-brain barrier on the hippocampus of a murine model of senescence. Mech Ageing Dev. 2007;128:522–528. doi: 10.1016/j.mad.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 128.Penn JS, Madan A, Caldwell RB, Bartoli M, Caldwell RW, Hartnett ME. Vascular endothelial growth factor in eye disease. Prog Retin Eye Res. 2008;27:331–371. doi: 10.1016/j.preteyeres.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Peppiatt CM, Howarth C, Mobbs P, Attwell D. Bidirectional control of CNS capillary diameter by pericytes. Nature. 2006;443:700–704. doi: 10.1038/nature05193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Perloff ES, Duan SX, Skolnik PR, Greenblatt DJ, von Moltke LL. Atazanavir: effects on P-glycoprotein transport and CYP3A metabolism in vitro. Drug Metab Dispos. 2005;33:764–770. doi: 10.1124/dmd.104.002931. [DOI] [PubMed] [Google Scholar]
- 131.Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. 2006;1:223–236. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- 132.Pousa ID, Mate J, Gisbert JP. Angiogenesis in inflammatory bowel disease. Eur J Clin Invest. 2008;38:73–81. doi: 10.1111/j.1365-2362.2007.01914.x. [DOI] [PubMed] [Google Scholar]
- 133.Rappa G, Finch RA, Sartorelli AC, Lorico A. New insights into the biology and pharmacology of the multidrug resistance protein (MRP) from gene knockout models. Biochem Pharmacol. 1999;58:557–562. doi: 10.1016/s0006-2952(99)00074-x. [DOI] [PubMed] [Google Scholar]
- 134.Redzic ZB, Preston JE, Duncan JA, Chodobski A, Szmydynger-Chodobska J. The choroid plexus-cerebrospinal fluid system: from development to aging. Curr Top Dev Biol. 2005;71:1–52. doi: 10.1016/S0070-2153(05)71001-2. [DOI] [PubMed] [Google Scholar]
- 135.Rennels ML, Nelson E. Capillary innervation in the mammalian central nervous system: an electron microscopic demonstration. Am J Anat. 1975;144:233–241. doi: 10.1002/aja.1001440208. [DOI] [PubMed] [Google Scholar]
- 136.Risau W. Embryonic angiogenesis factors. Pharmacol Ther. 1991;51:371–376. doi: 10.1016/0163-7258(91)90066-u. [DOI] [PubMed] [Google Scholar]
- 137.Rite I, Machado A, Cano J, Venero JL. Blood-brain barrier disruption induces in vivo degeneration of nigral dopaminergic neurons. J Neurochem. 2007;101:1567–1582. doi: 10.1111/j.1471-4159.2007.04567.x. [DOI] [PubMed] [Google Scholar]
- 138.Rite I, Machado A, Cano J, Venero JL. Intracerebral VEGF injection highly upregulates AQP4 mRNA and protein in the perivascular space and glia limitans externa. Neurochem Int. 2008;52:897–903. doi: 10.1016/j.neuint.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 139.Robertson PL, Du BM, Bowman PD, Goldstein GW. Angiogenesis in developing rat brain: an in vivo and in vitro study. Brain Res. 1985;355:219–223. doi: 10.1016/0165-3806(85)90044-6. [DOI] [PubMed] [Google Scholar]
- 140.Rodriguez M, Alvarez-Erviti L, Blesa FJ, Rodriguez-Oroz MC, Arina A, Melero I, Ramos LI, Obeso JA. Bone-marrow-derived cell differentiation into microglia: a study in a progressive mouse model of Parkinson’s disease. Neurobiol Dis. 2007;28:316–325. doi: 10.1016/j.nbd.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 141.Rommer PS, Stuve O, Goertsches R, Mix E, Zettl UK. Monoclonal antibodies in the therapy of multiple sclerosis: an overview. J Neurol. 2008;255(Suppl 6):28–35. doi: 10.1007/s00415-008-6006-x. [DOI] [PubMed] [Google Scholar]
- 142.Ronaldson PT, Persidsky Y, Bendayan R. Regulation of ABC membrane transporters in glial cells: relevance to the pharmacotherapy of brain HIV-1 infection. Glia. 2008;56:1711–1735. doi: 10.1002/glia.20725. [DOI] [PubMed] [Google Scholar]
- 143.Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39:279–291. doi: 10.1002/glia.10108. [DOI] [PubMed] [Google Scholar]
- 144.Rosenberg GA. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009;8:205–216. doi: 10.1016/S1474-4422(09)70016-X. [DOI] [PubMed] [Google Scholar]
- 145.Rosenberg GA, Yang Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg Focus. 2007;22:E4. doi: 10.3171/foc.2007.22.5.5. [DOI] [PubMed] [Google Scholar]
- 146.Salkeni MA, Lynch JL, Otamis-Price T, Banks WA. Lipopolysaccharide Impairs Blood-Brain Barrier P-glycoprotein Function in Mice Through Prostaglandin-and Nitric Oxide-Independent Pathways. J Neuroimmune Pharmacol. 2008;4:276–82. doi: 10.1007/s11481-008-9138-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sanchez del Pino MM, Peterson DR, Hawkins RA. Neutral amino acid transport characterization of isolated luminal and abluminal membranes of the blood-brain barrier. J Biol Chem. 1995;270:14913–14918. doi: 10.1074/jbc.270.25.14913. [DOI] [PubMed] [Google Scholar]
- 148.Satoh J, Tabunoki H, Yamamura T, Arima K, Konno H. Human astrocytes express aquaporin-1 and aquaporin-4 in vitro and in vivo. Neuropathology. 2007;27:245–256. doi: 10.1111/j.1440-1789.2007.00774.x. [DOI] [PubMed] [Google Scholar]
- 149.Saunders NR, Ek CJ, Habgood MD, Dziegielewska KM. Barriers in the brain: a renaissance? Trends Neurosci. 2008;31:279–286. doi: 10.1016/j.tins.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 150.Sedlakova R, Shivers RR, Del Maestro RF. Ultrastructure of the blood-brain barrier in the rabbit. J Submicrosc Cytol Pathol. 1999;31:149–161. [PubMed] [Google Scholar]
- 151.Sharabi Y, Danon YL, Berkenstadt H, Almog S, Mimouni-Bloch A, Zisman A, Dani S, Atsmon J. Survey of symptoms following intake of pyridostigmine during the Persian Gulf war. Isr J Med Sci. 1991;27:656–658. [PubMed] [Google Scholar]
- 152.Sharma HS, Kretzschmar R, Cervos-Navarro J, Ermisch A, Ruhle HJ, Dey PK. Age-related pathophysiology of the blood-brain barrier in heat stress. Prog Brain Res. 1992;91:189–196. doi: 10.1016/s0079-6123(08)62334-1. [DOI] [PubMed] [Google Scholar]
- 153.Somjen GG. Ion regulation in the brain: implications for pathophysiology. Neuroscientist. 2002;8:254–267. doi: 10.1177/1073858402008003011. [DOI] [PubMed] [Google Scholar]
- 154.Stevenson BR, Keon BH. The tight junction: morphology to molecules. Annu Rev Cell Dev Biol. 1998;14:89–109. doi: 10.1146/annurev.cellbio.14.1.89. [DOI] [PubMed] [Google Scholar]
- 155.Stewart PA, Hayakawa EM. Interendothelial junctional changes underlie the developmental ‘tightening’ of the blood-brain barrier. Brain Res. 1987;429:271–281. doi: 10.1016/0165-3806(87)90107-6. [DOI] [PubMed] [Google Scholar]
- 156.Takano T, Han X, Deane R, Zlokovic B, Nedergaard M. Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer’s disease. Ann N Y Acad Sci. 2007;1097:40–50. doi: 10.1196/annals.1379.004. [DOI] [PubMed] [Google Scholar]
- 157.Tamai I, Nezu J, Uchino H, Sai Y, Oku A, Shimane M, Tsuji A. Molecular identification and characterization of novel members of the human organic anion transporter (OATP) family. Biochem Biophys Res Commun. 2000;273:251–260. doi: 10.1006/bbrc.2000.2922. [DOI] [PubMed] [Google Scholar]
- 158.Terasaki T, Hosoya K. The blood-brain barrier efflux transporters as a detoxifying system for the brain. Adv Drug Deliv Rev. 1999;36:195–209. doi: 10.1016/s0169-409x(98)00088-x. [DOI] [PubMed] [Google Scholar]
- 159.Tetsuka K, Hosoya KI, Ohtsuki S, Takanaga H, Yanai N, Ueda M, Obinata M, Terasaki T. Acidic amino acid transport characteristics of a newly developed conditionally immortalized rat type 2 astrocyte cell line (TR-AST) Cell Struct Funct. 2001;26:197–203. doi: 10.1247/csf.26.197. [DOI] [PubMed] [Google Scholar]
- 160.Thirumangalakudi L, Samany PG, Owoso A, Wiskar B, Grammas P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J Alzheimers Dis. 2006;10:111–118. doi: 10.3233/jad-2006-10114. [DOI] [PubMed] [Google Scholar]
- 161.Tian YY, Tang CJ, Wang JN, Feng Y, Chen XW, Wang L, Qiao X, Sun SG. Favorable effects of VEGF gene transfer on a rat model of Parkinson disease using adeno-associated viral vectors. Neurosci Lett. 2007;421:239–244. doi: 10.1016/j.neulet.2007.05.033. [DOI] [PubMed] [Google Scholar]
- 162.Udo H, Yoshida Y, Kino T, Ohnuki K, Mizunoya W, Mukuda T, Sugiyama H. Enhanced adult neurogenesis and angiogenesis and altered affective behaviors in mice overexpressing vascular endothelial growth factor 120. J Neurosci. 2008;28:14522–14536. doi: 10.1523/JNEUROSCI.3673-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ueno M. Molecular anatomy of the brain endothelial barrier: an overview of the distributional features. Curr Med Chem. 2007;14:1199–1206. doi: 10.2174/092986707780597943. [DOI] [PubMed] [Google Scholar]
- 164.Uno T, Yasui-Furukori N. Effect of grapefruit juice in relation to human pharmacokinetic study. Curr Clin Pharmacol. 2006;1:157–161. doi: 10.2174/157488406776872550. [DOI] [PubMed] [Google Scholar]
- 165.Verma S, Nakaoke R, Dohgu S, Banks WA. Release of cytokines by brain endothelial cells: A polarized response to lipopolysaccharide. Brain Behav Immun. 2006;20:449–455. doi: 10.1016/j.bbi.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 166.Vorbrodt AW, Dobrogowska DH. Molecular anatomy of intercellular junctions in brain endothelial and epithelial barriers: electron microscopist’s view. Brain Res Brain Res Rev. 2003;42:221–242. doi: 10.1016/s0165-0173(03)00177-2. [DOI] [PubMed] [Google Scholar]
- 167.Wada K, Arai H, Takanashi M, Fukae J, Oizumi H, Yasuda T, Mizuno Y, Mochizuki H. Expression levels of vascular endothelial growth factor and its receptors in Parkinson’s disease. Neuroreport. 2006;17:705–709. doi: 10.1097/01.wnr.0000215769.71657.65. [DOI] [PubMed] [Google Scholar]
- 168.Wakai S, Hirokawa N. Development of the blood-brain barrier to horseradish peroxidase in the chick embryo. Cell Tissue Res. 1978;195:195–203. doi: 10.1007/BF00236719. [DOI] [PubMed] [Google Scholar]
- 169.Wakayama K, Ohtsuki S, Takanaga H, Hosoya K, Terasaki T. Localization of norepinephrine and serotonin transporter in mouse brain capillary endothelial cells. Neurosci Res. 2002;44:173–180. doi: 10.1016/s0168-0102(02)00120-7. [DOI] [PubMed] [Google Scholar]
- 170.Weller RO, Djuanda E, Yow HY, Carare RO. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol. 2009;117:1–14. doi: 10.1007/s00401-008-0457-0. [DOI] [PubMed] [Google Scholar]
- 171.Westerlund M, Belin AC, Anvret A, Hakansson A, Nissbrandt H, Lind C, Sydow O, Olson L, Galter D. Association of a polymorphism in the ABCB1 gene with Parkinson’s disease. Parkinsonism Relat Disord. 2009;15:422–4. doi: 10.1016/j.parkreldis.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 172.Westin JE, Lindgren HS, Gardi J, Nyengaard JR, Brundin P, Mohapel P, Cenci MA. Endothelial proliferation and increased blood-brain barrier permeability in the basal ganglia in a rat model of 3,4-dihydroxyphenyl-L-alanine-induced dyskinesia. J Neurosci. 2006;26:9448–9461. doi: 10.1523/JNEUROSCI.0944-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Willis CL, Davis TP. Chronic inflammatory pain and the neurovascular unit: a central role for glia in maintaining BBB integrity? Curr Pharm Des. 2008;14:1625–1643. doi: 10.2174/138161208784705414. [DOI] [PubMed] [Google Scholar]
- 174.Wittchen ES, Haskins J, Stevenson BR. Protein interactions at the tight junction. Actin has multiple binding partners, and ZO-1 forms independent complexes with ZO-2 and ZO-3. J Biol Chem. 1999;274:35179–35185. doi: 10.1074/jbc.274.49.35179. [DOI] [PubMed] [Google Scholar]
- 175.Wolburg H, Lippoldt A. Tight junctions of the blood-brain barrier: development, composition and regulation. Vascul Pharmacol. 2002;38:323–337. doi: 10.1016/s1537-1891(02)00200-8. [DOI] [PubMed] [Google Scholar]
- 176.Wolburg H, Noell S, Mack A, Wolburg-Buchholz K, Fallier-Becker P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009;335:75–96. doi: 10.1007/s00441-008-0658-9. [DOI] [PubMed] [Google Scholar]
- 177.Wood JH. Neurochemical analysis of cerebrospinal fluid. Neurology. 1980;30:645–651. doi: 10.1212/wnl.30.6.645. [DOI] [PubMed] [Google Scholar]
- 178.Yasuda T, Fukuda-Tani M, Nihira T, Wada K, Hattori N, Mizuno Y, Mochizuki H. Correlation between levels of pigment epithelium-derived factor and vascular endothelial growth factor in the striatum of patients with Parkinson’s disease. Exp Neurol. 2007;206:308–317. doi: 10.1016/j.expneurol.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 179.Yong VW, Giuliani F, Xue M, Bar-Or A, Metz LM. Experimental models of neuroprotection relevant to multiple sclerosis. Neurology. 2007;68:S32–S37. doi: 10.1212/01.wnl.0000275230.20635.72. [DOI] [PubMed] [Google Scholar]
- 180.Yoo SA, Kwok SK, Kim WU. Proinflammatory role of vascular endothelial growth factor in the pathogenesis of rheumatoid arthritis: prospects for therapeutic intervention. Mediators Inflamm. 2008;2008:129873. doi: 10.1155/2008/129873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhao C, Ling Z, Newman MB, Bhatia A, Carvey PM. TNF-alpha knockout and minocycline treatment attenuates blood-brain barrier leakage in MPTP-treated mice. Neurobiol Dis. 2007;26:36–46. doi: 10.1016/j.nbd.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zlokovic BV. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 183.Zlokovic BV, Deane R, Sallstrom J, Chow N, Miano JM. Neurovascular pathways and Alzheimer amyloid beta-peptide. Brain Pathol. 2005;15:78–83. doi: 10.1111/j.1750-3639.2005.tb00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]