

Abstract
Aldo-Keto Reductases (AKRs) are a major superfamily of monomeric NADPH-dependent carbonyl oxidoreductases. They are characterized by an (α/β)8-barrel structure, which at its base contains a conserved catalytic tetrad of Tyr, Lys, His and Asp. Two AKR subfamilies contain other residues substituted for the catalytic His and perform different functions. First, the steroid 5β-reductase (AKR1D1), which reduces C=C double bonds instead of carbonyl groups, has a Glu substituted for His. Second, the Kvβ subunits (AKR6A3, AKR6A5 and AKR6A9) which modulate opening of the voltage-gated potassium channel (Kv1) by oxidizing NADPH, have an Asn substituted for the His. Previously, we noted that conserved catalytic residues in AKRs perform similar functions in the short-chain dehydrogenases (SDRs). With the availability of crystal structures of AKR1D1 and two SDRs that catalyze double-bond reduction reactions, Digitalis steroid 5β-reductase and 2,4-dienoyl-CoA reductase, we have compared their active sites to outline the features that govern whether 1,2- 1,4- or 1,6-hydride transfer occurs.
Keywords: short-chain dehydrogenase-reductase/steroid 5β-reductase, potassium channel β-subunit, cardiac glycoside, oxidative stress
1. Introduction
Two of the most prominent NAD(P)(H)-dependent oxidoreductases are the short-chain dehydrogenase reductases (SDRs) and the aldo-keto reductases (AKRs) [1–5]. These two enzyme superfamilies adopt different protein folds but share common endogenous substrates, e.g., steroid hormones, prostaglandins, and lipid aldehydes, as well as exogenous substrates such as xenobiotics, including aldehydes, ketones, and carbonyl containing drugs. Despite the differing structures of SDRs and AKRs, these enzyme superfamilies share numerous features of catalytic mechanism. The SDR active site contains a YXX(S)K motif, while the AKR active site contains conserved residues D50, Y55, K84, and H117 [4–11]. Superposition of the active site residues of the SDR 3α(20β)-hydroxysteroid dehydrogenase (HSD) from Streptomyces hydrogenas [7] and rat 3α-hydroxysteroid dehydrogenase (AKR1C9) [11] shows that the general positions of the conserved tyrosine and lysine residues are generally similar relative to the si and re face of the nicotinamide ring of the cofactor, which donates either the 4-pro-S or 4-pro-R hydride in the SDR or AKR reactions, respectively (Fig. 1). We also noted that the Ser in 3α(20β)-HSD and the Nε2 atom of His in AKR1C9 are both within hydrogen bonding distance of a water molecule that is displaced by the substrate carbonyl, suggesting that these residues play a facilitatory role in catalysis [7,12]. It is apparent that the two enzyme superfamilies have convergently evolved to a common catalytic mechanism in which the tyrosine residue serves as a general acid-base [11]. In the SDRs the pKa of the Tyr is likely lowered by interaction with the nearby lysine residue, and the conserved Ser donate a hydrogen bond to the substrate carbonyl group (Fig. 2). In the AKRs, the Tyr acts a general base due to deprotonation by the lysine, but acts as a general acid by participating in a proton relay with the conserved His (Fig. 2) [13].
Fig. 1. Superposition of the active site residues of rat 3α-HSD (AKR1C9) and 3α,20β-HSD [11].
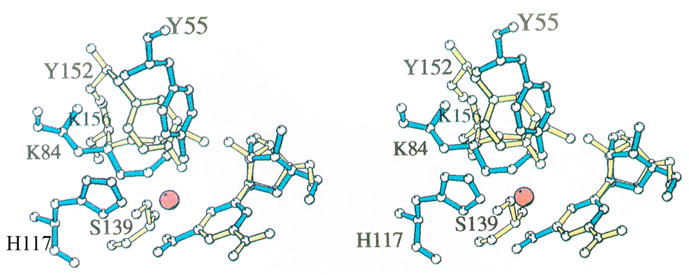
Comparison of AKR and SDR enzymes in which the protein folds differ but the active site residues superimpose. Active site residues in 3α-HSD (blue) superimposed with those in 3α,20β–HSD, a member of the SDR superfamily (yellow) [7]. All non-hydrogen atoms in Tyr 55, Lys 84, His 117, and nicotinamide ribose in the 3α-HSD-NADP+ binary complex and in Ser 139, Tyr 152, Lys 156, and nicotinamide ribose in the 3α,20β-HSD-NAD+ binary complex are shown. The water molecule in 3α-HSD that may mimic the carbonyl oxygen in a 3-ketosteroid substrate is represented by a red sphere. The superposition was based on the nicotinamide ring position, excluding the carboxamide substituent at the C3 position. The reason for excluding this substituent was that 3α,20β-HSD transfers the pro-S hydrogen, while 3α-HSD transfers the pro-R hydrogen, so that although the nicotinamide rings lie in the same plane, they are flipped 180° relative to one another and the carboxamide groups do not superimpose. This figure was prepared using MOLSCRIPT. Reproduced with permission from Biochemistry 1996 35, 10702–10711. Copyright 1996 the American Chemical Society.
Fig. 2.

Carbonyl reduction and alcohol oxidation by AKRs and SDRs is evolutionary conserved.
In the AKR superfamily, there are two subfamilies in which the conserved catalytic histidine residue is substituted [5]. In the AKR1D subfamily, which contains the steroid 5β-reductases, H117 is substituted by glutamic acid. In the AKR6A family, which contains the β-subunit Kvβ of the potassium channel Kv1, H117 is substituted by asparagine. Recently determined crystal structures of these AKRs have led to mechanistic proposals concerning the respective functions of these substituted residues [14]. These proposals are supported by comparisons to SDRs.
2. Comparison of the steroid double-bond reductases AKR1D1 and progesterone 5β-reductase
There are two forms of steroid hormone double bond reductases that reduce Δ4-3-ketosteroids to the corresponding 5α- and 5β-reduced dihydrosteroids (Fig. 3). The steroid 5α-reductases are annotated as SRD5 genes and belong to neither the SDRs or the AKRs. In humans both type 1 and type 2 5α-reductases are found [15]. These enzymes generate steroid products containing a trans-A/B ring configuration. In contrast, the steroid 5β-reductases catalyze a reaction that is unique in steroid enzymology since the resultant product contains a cis-A/B ring configuration and accordingly contains a 90° bend. The cis A/B ring configuration is an essential characteristic of cardiac glycosides, e.g., digioxin and bile acids and their precursors. Steroid 5β-reduction is almost impossible to perform using chemical reductants unless a superacid is used and unless there is a directing group to facilitate the correct facial attack of the reductant [16–17]. To gain insight on these reactions it is useful to compare the crystal structure of human 5β-reductase (AKR1D1) with that of the SDR progesterone 5β-reductase (5β-POR) from Digitalis lanata (foxglove), both of which have been recently reported [14,18].
Fig. 3. Reduction of the carbon-carbon double bond at C-5 of a Δ4-3-ketosteroid to form 5β-dihydrosteroid as catalyzed by SDR or AKR family members.

Steroid rings and selected carbon atoms are labeled according to standard practice.
AKR1D1 catalyzes the stereospecific reduction of the Δ4-double bond of circulating steroid hormones that contain the Δ4-3-ketosteroid functionality, e.g. the reduction of testosterone, progesterone and cortisol to yield the corresponding 5β-dihydrosteroid (Fig. 3). The crystal structures of AKR1D1 reveals the characteristic (α/β)8-barrel fold of AKRs with three large loops (A, B and C) that form the steroid binding site [14]. The structures of the AKR1D1-NADP+-cortisone and AKR1D1-NADP+-progesterone complexes show that the NADP+ cofactor lies across the lip of the barrel with the nicotinamide ring oriented into the active site, perpendicular to the steroid substrates. Presuming that the binding conformation of NADP+ is similar to that of NADPH, the 4-pro-R hydride would be oriented towards the β-face of the steroid, adjacent to the reactive C5 atom of the steroid substrate. The position of the catalytic tetrad supports the proposed role of Y58 as a general acid [14]. Importantly, E120 appears to be a fully protonated, anti-oriented carboxylic acid based on its intermolecular interactions presumed to be hydrogen bonds in the enzyme-substrate complexes [14]. The side chain of E120 is proposed to serve two roles in the mechanism by replacing the histidine residue found at this position in other AKR enzymes. First, the histidine→glutamate substitution removes steric bulk from this portion of the active site and thereby permits the steroid to bind more deeply in the active site cavity [14]. This creates the optimal reaction trajectory for hydride transfer to the substrate C5 atom. In addition, both E120 and Y58 donate hydrogen bonds to the C3-ketone and contribute to a superacidic environment that can stabilize the enolate intermediate in catalysis, which facilitates hydride transfer (Fig. 4a) [14,19].
Fig. 4. a) Proposed catalytic mechanism for steroid double bond reduction [14].

Reproduced with permission of the American Society of Biochemistry and Molecular Biology. b) Proposed catalytic mechanism for 2,4-dienoyl-CoA reductase [21].
5β-POR catalyzes the stereospecific reduction of the Δ4-double bond of progesterone to form 5β-pregnane-3,20-dione using NADPH as cofactor in cardenolide biosynthesis by Digitalis (Fig. 5). The crystal structure of the 5β-POR-NADP+ binary complex reveals that 5β-POR exhibits a characteristic SDR fold with an N-terminal domain consisting of a double Rossmann fold for cofactor binding and an insertional domain between strands βF and βG of ~100 residues for substrate binding (Fig. 5b) [18]. Additionally, the N-terminal domain of 5β-POR contains the NFY179YDLED motif with residues separating the catalytic tyrosine and lysine residues (this is a variation of the YXX(S)K motif). Notably, for the majority of SDRs in which the YXX(S)K motif is found, the lysine residue assists in the protonation of the tyrosine residue during catalysis and stabilizes the binding of the cofactor through a hydrogen bond interaction with the 2′-hydroxyl group of the cofactor ribose group. The side chain of Y179 plays a central role in the catalytic activity of 5β-POR. In fact, when this residue is mutated to either alanine or phenylalanine, the enzyme completely loses activity [18]. This suggests that the hydroxyl group of Y179 in the NFY179YXXED segment plays a similar role in catalysis as the corresponding tyrosine in the YXX(S)K motif of SDRs [18].
Fig. 5. Evolution of SDRs to accommodate different reduction mechanisms.

(a) Human 17β-hydroxysteroid dehydrogenase-NADP+-estradiol ternary complex (PDB accession code 1FDT, [20]) which catalyzes a 1,2-addition (b) 5β-POR-NADP+ (D. lanata) binary complex (PDB code 2V6G [18]) which catalyzes a 1,4-addition, (c) human 2,4-dienoyl-CoA reductase-NADP+-2,4-hexadienoyl-CoA ternary complex (PDB code 1W6U, [21]) which catalyzes a 1,6-addition. The catalytic residues and NADP+ are shown in sticks and are magenta; the other ligands are yellow. The cofactor binding domain is essentially conserved among these different enzymes at the N-terminal domain. The insertional domain for binding of progesterone in 5β-POR is indicated in cyan. The position of the catalytic tyrosine is shifted among these enzymes to accommodate different addition mechanisms. In enzymes catalyzing 1,2, 1,4, and 1,6 addition reactions, the catalytic tyrosine OH---NADP+ C4 separation is 5.2 Å, 6.0 Å, and 7.1 Å, respectively.
The crystal structure of the 5β-POR-NADP+ binary complex reveals that the side chain of K147 is found in a similar position to the lysine residue of the YXX(S)K motif in standard SDRs (Fig. 5a–b) [18]. The terminal amino group of K147 is ~4.4 Å from the hydroxyl group of Y179, which is longer than the hydrogen bond separation of 2.7 Å between K87 and Y58 in the crystal structure of AKR1D1 (Fig. 5b). In turn, Y179 donates a hydrogen bond to the 2′-hydroxyl of the cofactor ribose group in the crystal structure of 5β-POR-NADP+ binary complex (Fig. 5b).
The mechanism for the reduction of the double bond in common with other SDRs follows two steps: first, the 4-pro-S-hydride transfer occurs from the NAD(P)H to the substrate, while in the second step protonation neutralizes the enolate intermediate. For the majority of SDRs that catalyze the reduction of a carbonyl bond, the hydrogenation occurs as a 1,2 addition mechanism. The crystal structure of human 17β-hydroxysteroid dehydrogenase type 1 in complex with 17β-estradiol and NADP+ reveals that the O17 hydroxyl group of 17β-estradiol hydrogen bonds with Y155 and S142 (Fig. 5a) [20]. In this way the substrate carbonyl is further polarized to facilitate the nucleophilic addition of the hydride ion.
Significantly, the carbon-carbon double bond is often part of an α,β-unsaturated ketone, and the enzymes AKR1D1 and 5β-POR evolved to promote a 1,4 addition mechanism. While AKR1D1 evolved to have the polarization of the carbon-carbon double bond assisted by the extra acidic residue E120 [14], Thorn and colleagues observe that the catalytic residue Y179 in 5β-POR is shifted by a complete helical turn with respect to common SDRs and it assumes a geometry that would favor the 1,4 addition [18].
It is also informative to extend the comparison of AKR1D1 to another SDR family member for which the crystal structure is available, 2,4-dienoyl-CoA reductase [21]. This enzyme catalyzes the 1,6 addition mechanism for the reduction of trans-2, trans-4-hexadienoyl-CoA to form trans-3-hexenoyl-CoA, in which a γ,δ-carbon-carbon double bond is conjugated with an α,β-unsaturated thioester [21]. Thorn and colleagues note that for this enzyme the catalytic tyrosine (Y199) is moved to a completely different region to donate a hydrogen bond to the carbonyl of the substrate (Fig. 5c) [18,20,21]. The location of the catalytic tyrosine is even further removed than that of 5β-POR to accommodate the 1,6 addition mechanism of 2,4-dienoyl-CoA reductase versus the 1,4 addition mechanism of 5β-POR (Figs. 4b, 5b–c). In other words, the further away the carbonyl group is from the reactive double bond of the unsaturated ketone, the further away the catalytic tyrosine is from the reactive C4-H hydride of NAD(P)H; in enzymes catalyzing 1,2, 1,4, and 1,6 addition reactions shown in Fig. 5, the catalytic tyrosine OH---NADP+ C4 separation is 5.2 Å, 6.0 Å, and 7.1 Å, respectively.
Finally, we note that the crystal structure of the ternary complex 2,4-dienoyl-CoA reductase-trans-2, trans-4-hexadienoyl-CoA-NADP+ reveals that the carbonyl oxygen of the substrate accepts a hydrogen bond from general acid Y199 and also N148 [21] (Fig. 5c). The substitution of a smaller asparagine side chain for the larger histidine side chain found in AKR enzymes catalyzing 1,2 addition reactions permits the substrate to bind more deeply in the active site to accommodate the 1,6-addition reaction.
3. Comparison of AKR1D1 and the Kvβ subunit of potassium channel Kv1
Voltage-gated potassium-selective channels are found in virtually all living organisms and form transmembrane pores. These pores are found in many cells types and tissues where they function to regulate electrical signaling and other physiological processes [22–24]. The pores are formed by the association of two subunits: the voltage-dependent potassium channel (Kv1) and a β-subunit (Kvβ) [25–27]. The crystal structure of the potassium channel associated with the β-subunit shows that 4 Kv1:Kvβ heterodimers assemble to form a heterooctamer with C4 symmetry, and the central ion conducting pore defines the axis of symmetry [28–30].
Three human Kvβ proteins (Kvβ1-3) have been identified that belong to the AKR6A subfamily, which also contains the voltage-dependent Shaker potassium channels [5,25]. The Kvβ structures adopt the (α/β)8-barrel fold typical of AKRs despite sharing low amino acid sequence identity with other members of the AKR superfamily [5,25]. Additionally, three residues of the catalytic tetrad, Y55, K84, and D50 (numbering scheme based on AKR1C9, rat 3α-HSD), are conserved with respect to other members of the AKR superfamily, but a neutral asparagine residue (N158) replaces the conserved H117 [5,25]. The imidazole group of H117 plays a crucial role in substrate binding and catalysis in AKR1C9, where the role of H117 is to facilitate the protonation of Y55, which serves as the general acid by donating a proton to the steroid carbonyl [13].
Several crystal structures of the Kvβ proteins or the (Kv1:Kvβ)4 channel include a bound NADP+/NADPH cofactor [28–30]. Recently, it has been demonstrated that Kvβ is a functional AKR capable of reducing aromatic aldehydes at the expense of oxidizing NADPH, which is accompanied by an increase in channel current. An identical effect is seen if H2O2 is used to oxidize the bound NADPH [31,32]. Interestingly, typical AKR1D1 substrates such as cortisone or AKR1C9 substrates such as 5α-androstane-3α,17β-diol show no activity [33]. Instead, aldehydes having an aromatic ring with an electron withdrawing group in the para position, or substrates with carbonyl groups polarized by α,β-unsaturation, are favored. Additionally, the reduction of 4-oxo-2-noneal produces only 4-oxo-2-nonenol (corresponding to the reduction of the aldehyde group at C1), while the C=C double bond between C2 and C3, and the keto group at C4, are not reduced [33]. When the catalytic tyrosine of Kvβ, Y90, is mutated to phenylalanine, catalytic activity is mostly obliterated [32]. It is not immediately clear why the residue corresponding to H117 in AKRs is a neutral asparagine in Kvβ.
The comparison of the crystal structure of AKRs having different catalytic residues in place of H117 support the concept that this difference determines the substrate specificity of these AKRs. Even though the root-mean-square (r.m.s.) deviation between the cytoplasmic β subunit (Kvβ) in complex with the cofactor NADPH complex (PDB accession code 1EXB [29]) and the AKR1D1-NADP+-progesterone complex (PDB accession code 3COT [14]), or the rat AKR1C9-NADP+-testosterone complex (PDB accession code 1AFS [12]), is 1.60 Å and 1.50 Å, respectively, larger variations involving loop segments A, B and C occur (Fig. 6a). Specifically, loops A (I119-L147) and C (L302-Y326) in AKR1D1 are much longer than the respective loops A (A157-T165) and C (G356-S361, 6 residues are missing from the X-ray model) in the Kvβ and their role in AKR1D1 is to accommodate substrate binding (Fig. 6). In contrast, loop B (Y219-L238) of AKR1D1 is less structured than the corresponding region of Kvβ. Comparison of the Kvβ-NADPH complex and the AKR1C9-NADP+-testosterone complex yields similar observations. However, a closer inspection of the Kvβ active site reveals ~1 Å deeper binding of the NADPH cofactor with respect to the cofactor in AKR1D1 and AKR1C9. The side chain of N158 in the Kvβ is coplanar with both the side chain of H117 of AKR1C9 and the side chain of E120 of AKR1D1. We speculate that N158 in the Kvβ provides more room for the optimal positioning of bulky lipid and phospholipid aldehyde substrates. We note that the conversion of NADPH to NADP+ provides a mechanism by which potassium channel opening is redox-regulated. Lipid and phospholipid aldehydes are formed as products of lipid peroxidation and oxidative stress. The optimal positioning of these substrates may contribute to the redox-regulation of channel opening. It is also interesting to note that N167 in rat 3α-HSD and the equivalent residue N170 in AKR1D1 is found as R189 in Kvβ; the crystal structure of the Kvβ-NADPH complex reveals that this residue is very close to the position of progesterone in the structure of the AKR1D1-NADP+-progesterone complex (Fig. 6) and may contribute to substrate specificity. The recent crystal structure of the ternary complex Kvβ-NADPH-cortisone reveals cortisone binding sites on the surface of the protein in addition to the enzyme active site, where the cortisone molecule is bound backwards with respect to the productive binding of cortisone and progesterone in the active site of AKR1D1, and it causes the side chain of R189 to flip (Fig. 6c) [34]. The crystal structure reveals that the binding of cortisone to the protein surface has an important role in dissociating the β subunits from the Shaker protein channel [34].
Fig. 6. Comparison of AKRs crystal structures in which the conserved catalytic histidine is substituted.

(a) Least-squares superposition of the AKR1D1-NADP+-progesterone complex (magenta, PDB code 3COT [14]), the Kvβ-NADPH complex (light blue, PDB code 1EXB [29]; for clarity, the Kv1.2 subunit is omitted) and the AKR1C9-NADP+-testosterone complex (green, PDB code 1AFS [11]). (b) Superposition of selected active site residues, cofactor and ligand of AKR1D1-NADP+-progesterone complex, Kvβ-NADPH complex, and AKR1C9-NADP+-testosterone complex. The side chains of His 117, E120, and N148 from the different enzymes are coplanar. (c) Superposition of AKR1D1-NADP+-progesterone complex, Kvβ-NADPH complex, and Kvβ-NADPH-cortisone complex (carbon atoms are yellow, PDB code 3EAU [34]). Cofactor, progesterone, testosterone and catalytic tetrad are color-coded as follow: oxygen, red; nitrogen, blue; phosphorus, orange.
Acknowledgments
Supported by grants R01-DK40715 to T.M.P and R01-GM56838 to D.W.C. We thank Dr. Ming Zhou for sharing the coordinates of the ternary complex Kvβ-NADPH-cortisone.
Footnotes
Conflict of Interest Statement: The authors declare that they have no conflicts of interest.
References
- 1.Wermuth B, Omar A, Forster A, Di Francesco C, Wolf M, Wartburg JP. Primary structure of aldehyde reductase from human liver. Prog Clin Biol Res. 1987;232:297–307. [PubMed] [Google Scholar]
- 2.Jörnvall H, Persson B, Krook M, Atrian S, Gonzàlez-Duarte R, Jeffery J, Ghosh D. Short-chain dehydrogenases/reductases (SDR) Biochemistry. 1995;34:6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- 3.Persson B, Kallberg Y, Oppermann U, Jörnvall H. Coenzyme-based functional assignments of short-chain dehydrogenases/reductases (SDRs) Chem Biol Interact. 2003;143–144:271–278. doi: 10.1016/s0009-2797(02)00223-5. [DOI] [PubMed] [Google Scholar]
- 4.Jez JM, Bennett MJ, Schlegel BP, Lewis M, Penning TM. Comparative anatomy of the aldo-keto reductase superfamily. Biochem J. 1997;325:625–636. doi: 10.1042/bj3260625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jez JM, Flynn G, Penning TM. A new nomenclature for the aldo-keto reductase superfamily. Biochem Pharmacol. 1997;54:639–647. doi: 10.1016/s0006-2952(97)84253-0. [DOI] [PubMed] [Google Scholar]
- 6.Varughese KI, Skinner MM, Whiteley JM, Matthews DA, Xuong NH. Crystal structure of rat liver dihydropteridine reductase. Proc Natl Ac Sci. 1992;89:6080–6084. doi: 10.1073/pnas.89.13.6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghosh D, Wawrzak Z, Weeks CM, Duax WL, Erman M. The refined three-dimensional structure of 3α,20β-hydroxysteroid dehydrogenase and possible roles of the residues conserved in short-chain dehydrogenases. Structure. 1994;2:629–640. doi: 10.1016/s0969-2126(00)00064-2. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh D, Pletnev VZ, Zhu DW, Wawrzak Z, Duax WL, Pangborn W, Labrie F, Lin SX. Structure of human estrogenic 17β-hydroxysteroid dehydrogenase at 2.20 Å resolution. Structure. 1995;3:503–513. doi: 10.1016/s0969-2126(01)00183-6. [DOI] [PubMed] [Google Scholar]
- 9.Ensor CM, Tai H-H. Site-directed mutagenesis of the conserved tyrosine 151 of human placental NAD+-dependent 15-hydroxyprostaglandin dehydrogenase yields a catalytically inactive enzyme. Biochem Biophys Res Commun. 1991;172:840–845. doi: 10.1016/s0006-291x(05)80262-1. [DOI] [PubMed] [Google Scholar]
- 10.Obeid J, White PC. Tyr-179 and Lys-183 are essential for enzymatic activity of 11β-hydroxysteroid dehydrogenase. Biochem Biophys Res Commun. 1992;188:222–227. doi: 10.1016/0006-291x(92)92373-6. [DOI] [PubMed] [Google Scholar]
- 11.Bennett MJ, Schlegel BP, Penning TM, Lewis M. Structure of 3α-hydroxysteroid/dihydrodiol dehydrogenase complexed with NADP+ Biochemistry. 1996;35:10702–10711. doi: 10.1021/bi9604688. [DOI] [PubMed] [Google Scholar]
- 12.Bennett MJ, Albert RH, Jez JM, Ma H, Penning TM, Lewis M. Steroid recognition and regulation of hormone action: crystal structure of testosterone and NADP+ bound to 3α-hydroxysteroid/dihydrodiol dehydrogenase. Structure. 1997;5:799–812. doi: 10.1016/s0969-2126(97)00234-7. [DOI] [PubMed] [Google Scholar]
- 13.Schlegel BP, Jez JM, Penning TM. Mutagenesis of 3α-hydroxysteroid dehydrogenase reveals a “push-pull” mechanism for proton transfer in aldo-keto reductases. Biochemistry. 1998;37:3538–3548. doi: 10.1021/bi9723055. [DOI] [PubMed] [Google Scholar]
- 14.Di Costanzo L, Drury JE, Penning TM, Christianson DW. Crystal structure of human liver Δ4-3-ketosteroid 5β-reductase (AKR1D1) and implications for substrate binding and catalysis. J Biol Chem. 2008;283:16830–16839. doi: 10.1074/jbc.M801778200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russell DW, Wilson JD. Steroid 5α-reductase: two genes/two enzymes. Annu Rev Biochem. 1994;63:25–61. doi: 10.1146/annurev.bi.63.070194.000325. [DOI] [PubMed] [Google Scholar]
- 16.Norymberski JK, Woods GF. Partial reduction of hormones and related substances. J Chem Soc (Lond) 1955:3426–3430. [Google Scholar]
- 17.March J. Advanced Organic Chemistry, Reactions, Mechanisms and Structure. John Wiley & Sons, Inc; New York: pp. 694–695. [Google Scholar]
- 18.Thorn A, Egerer-Sieber C, Jäger CM, Herl V, Müller-Uri F, Kreis W, Muller YA. The crystal structure of progesterone 5β-reductase from Digitalis lanata defines a novel class of short chain dehydrogenases/reductases. J Biol Chem. 2008;283:17260–1726. doi: 10.1074/jbc.M706185200. [DOI] [PubMed] [Google Scholar]
- 19.Di Costanzo L, Drury JE, Christianson DW. Structure and catalytic mechanism of human steroid 5β-reductase (AKR1D1) Mol Cell Endocrinol. 2009 doi: 10.1016/j.mce.2008.09.013. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breton R, Housset D, Mazza C, Fontecilla-Camps JC. The structure of a complex of human 17β-hydroxysteroid dehydrogenase with estradiol and NADP+ identifies two principal targets for the design of inhibitors. Structure. 1996;4:905–915. doi: 10.1016/s0969-2126(96)00098-6. [DOI] [PubMed] [Google Scholar]
- 21.Alphey MS, Yu W, Byres E, Li D, Hunter WN. Structure and reactivity of human mitochondrial 2,4-dienoyl-CoA reductase: enzyme-ligand interactions in a distinctive short-chain reductase active site. J Biol Chem. 2005;280:3068–3077. doi: 10.1074/jbc.M411069200. [DOI] [PubMed] [Google Scholar]
- 22.Connor JA, Stevens CF. Prediction of repetitive firing behaviour from voltage clamp data on an isolated neurone soma. J Physiol (Lond) 1971;213:31–53. doi: 10.1113/jphysiol.1971.sp009366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aldrich RW, Getting PA, Thompson SH. Inactivation of delayed outward current in molluscan neurone somata. J Physiol (Lond) 1979;291:531–544. doi: 10.1113/jphysiol.1979.sp012828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isom LL, De Johngh KS, Catterall WA. Auxiliary subunits of voltage-gated ion channels. Neuron. 1994;12:1183–1194. doi: 10.1016/0896-6273(94)90436-7. [DOI] [PubMed] [Google Scholar]
- 25.McCormack K, McCormack T. Shaker K+ channel β subunits belong to an NAD(P)H-dependent oxidoreductase superfamily. Cell. 1994;79:1133–1135. doi: 10.1016/0092-8674(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 26.Shi G, Nakahira K, Hammond S, Rhodes KJ, Schechter LE, Trimmer JS. β subunits promote K+ channel surface expression through effects early in biosynthesis. Neuron. 1996;16:843–852. doi: 10.1016/s0896-6273(00)80104-x. [DOI] [PubMed] [Google Scholar]
- 27.Nagaya N, Papazian DM. Potassium channel α and β subunits assemble in the endoplasmic reticulum. J Biol Chem. 1997;272:3022–3027. doi: 10.1074/jbc.272.5.3022. [DOI] [PubMed] [Google Scholar]
- 28.Gulbis JM, Mann S, MacKinnon R. Structure of a voltage-dependent K+ channel β subunit. Cell. 1999;97:943–952. doi: 10.1016/s0092-8674(00)80805-3. [DOI] [PubMed] [Google Scholar]
- 29.Gulbis JM, Zhou M, Mann S, MacKinnon R. Structure of the cytoplasmic β subunit-T1 assembly of voltage-dependent K+ channels. Science. 2000;289:123–127. doi: 10.1126/science.289.5476.123. [DOI] [PubMed] [Google Scholar]
- 30.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 31.Weng J, Cao Y, Moss N, Zhou M. Modulation of voltage-dependent Shaker family potassium channels by an aldo-keto reductase. J Biol Chem. 2006;281:15194–15200. doi: 10.1074/jbc.M513809200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan Y, Weng J, Cao Y, Bhosle RC, Zhou M. Functional coupling between the Kv1.1 channel and aldoketoreductase Kvβ1. J Biol Chem. 2008;283:8634–8642. doi: 10.1074/jbc.M709304200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tipparaju SM, Barski OA, Srivastava S, Bhatnagar A. Catalytic mechanism and substrate specificity of the β-subunit of the voltage-gated potassium channel. Biochemistry. 2008;47:8840–8854. doi: 10.1021/bi800301b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan Y, Weng J, Kabaleeswaran V, Li H, Cao Y, Bhosle RC, Zhou M. Cortisone dissociates the Shaker family K+ channels from their β subunits. Nat Chem Biol. 2008 doi: 10.1038/nchembio.114. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]