

Developmentally, podocytes evolve from columnar epithelial cells linked together by an apical junction complex containing both tight junctions (TJ) and adherent junctions (AJ). As epithelial cells develop into podocytes, the apical TJ and AJ migrate basally and ultimately morph into a single slit diaphragm (SD) that links the foot processes from adjacent podocytes (1). Mature podocytes develop spindle like foot processes, re-organize their actin cytoskeleton and express vimentin and synaptopodin. The mature SD retains features of both the TJ and the AJ. Immunostaining for the TJ-associated protein zonula occludens-1 (ZO-1) in adult rat kidney reveals the greatest intensity at the insertion sites of the SD into the foot processes (2). Also, like the TJ, the SD functions as a barrier that imposes selective permeability between the blood and the urinary space and serves as a signaling platform (3).
Podocyte injury and reduced podocyte density have been documented in patients with diabetic kidney disease and are among the strongest predictors of its progression (4). Once a critical proportion of the total podocyte population is lost, the remaining cells are unable to compensate and glomerulosclerosis develops (5). Although the mechanism of podocyte loss is still being debated; detachment and apoptosis are both thought to contribute to the decrease in podocyte density (6). The diabetic milieu, including hyperglycemia, oxidative stress, and advanced glycation end-products (AGE) decreases podocyte survival via multiple pathways, including decreased phosphorylation of survival factor AKT and activation of proapoptotic P38 mitogen activated protein kinases (MAPK) (7, 8). Activation of Notch in diabetic nephropathy alters cell cycle signaling and induces apoptosis via the p53 pathway (9–12). While apoptosis can be identified in experimental diabetic nephropathy, it has been challenging to identify apoptotic podocytes in human diabetic nephropathy (13). Several recent studies described urinary podocytes in both experimental and clinical diabetic nephropathy suggesting that podocyte dropout might be caused by decreased podocyte adhesion (6, 14). In support of this hypothesis, failure of attachment of podocytes to the glomerular basement membrane (GBM) by integrin receptors, and increased expression of anti-adhesive proteins have both been identified in diabetic nephropathy (15, 16). Many recent studies, however, question whether these cells are truly glomerular epithelial cells or they are of parietal epithelial origin (17). In addition, the majority (approx 90%) of podocytes that can be detected in the urine are apoptotic (18). Therefore, the possibility arises that apoptosis and detachment contribute to the disease development together; they might even be linked as cells that do not attach to a basement membrane die and apoptotic cells may detach from the GBM.
In this issue of the American Journal of Kidney Diseases, Yamaguchi et al. propose a new mechanism for podocyte loss via an epithelial to mesenchymal transition (EMT). EMT is believed to signify a reversal of the developmental transition from metanephric mesenchyme to the epithelial phenotype characteristic of differentiated podocytes and tubular cells. EMT has been proposed to contribute to tubulointerstitial fibrosis and to the progression of kidney disease (10, 19). Transforming growth factor beta (TGFβ) is a strong inducer of EMT of tubular epithelial cells cultured in vitro. Diabetic kidney disease is associated with increased expression of TGFβ in glomerular and tubular epithelial cells (20). In 2008, Li et al. proposed that TGFβ induces the expression of snail, a transcriptional inducer of EMT. Downregulation of nephrin, ZO-1 and P-cadherin in cultured podocytes might be dependent on snail (21). Li et al. also described the expression of fibroblast specific protein-1 (FSP-1), a marker of EMT, in human diabetic nephropathy(22).
What is EMT? EMT is loosely defined by three major changes in cellular phenotype (23, 24): (1) morphological changes from a cobblestone-like monolayer of epithelial cells with an apical basal polarity to dispersed, spindle-shaped mesenchymal cells with migratory protrusions; (2) changes of differentiation markers from cell-cell junction proteins and cytokeratin intermediate filaments to vimentin filaments and fibronectin and (3) the functional changes associated with the conversion of stationary cells to motile cells that can invade through the extracellular matrix (ECM) (Figure 1). Not all three changes are invariably observed during an EMT; however, acquisition of the ability to migrate and invade ECM as single cells is considered a functional hallmark of the EMT program.
Figure 1.
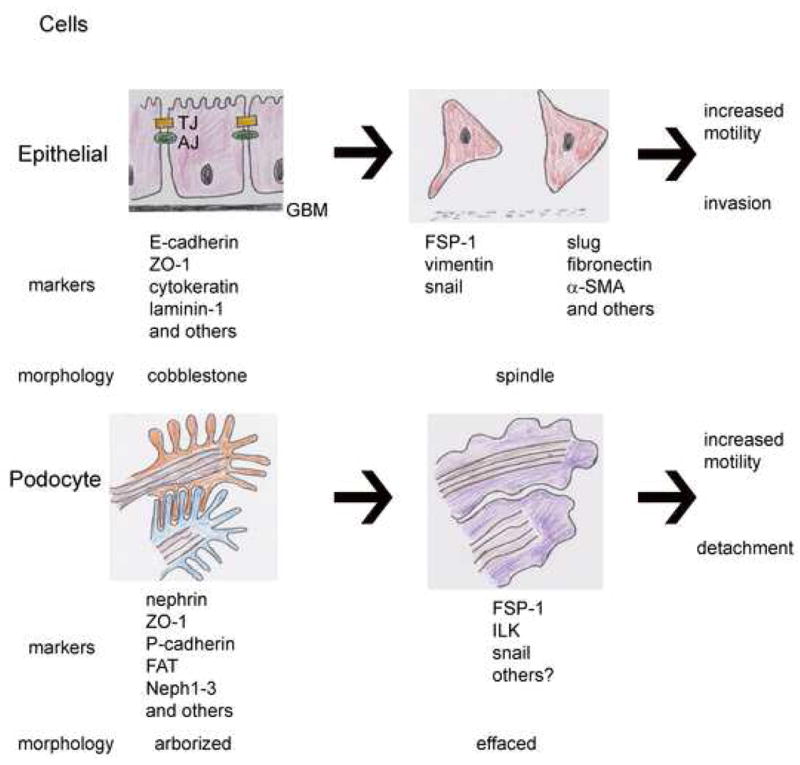
Top panel depicts an arrow demarking the epithelial-mesenchymal transition (EMT) in columnar epithelial cells that are characterized by the presence of tight junctions (TJ) and adherens junctions (AJ) at their apical aspect and adherence to the glomerular basement membrane (GBM) at the basal aspect. The columnar epithelial cells lose cell-cell junctions and their cobblestone morphology and develop a spindle shape, permitting increased motility and promoting migration into and through a degenerating GBM. Bottom panel depicts the phenotype change in podocytes (arrow): arborized podocytes have interdigitating foot processes linked by slit diaphragms (SD), but retain more mesenchymal characteristics at baseline, with spindle shaped processes and expression of vimentin. In diabetic nephrophathy, podocytes become effaced, and have increased expression of mesenchymal markers. They do not invade the GBM, but rather increased motility results in detachment and loss of podocytes.
Is there a podocyte EMT? When podocytes are grown in cell culture, a cobblestone like morphology resembling that of epithelial cells can be observed; however, differentiated podocytes have many spindle like protrusions. Similarly, podocytes in vivo have primary, secondary and tertiary foot processes and express vimentin giving them a more mesenchymal appearance at baseline (25). Therefore podocytes cannot necessarily fulfill the general morphological change premise of EMT. Increased podocyte motility recently has been considered an important aspect of proteinuria. In addition, injured or diseased podocytes de novo express different, more mesenchymal like markers. Most importantly, however, podocytes do not fulfill the last and maybe the most important marker of EMT, the ECM invasion. For these reasons, podocyte EMT remains a controversial issue.
What is unique about this current study is that it also links EMT to podocyte loss in human diabetic kidney disease. The authors identify expression of FSP1 in the majority of urinary podocytes from patients with diabetic nephropathy, and these podocytes did not have apoptotic nuclei, although no direct assay for apoptosis was performed. Notably, urinary FSP1+ podocytes correlated with severity of diabetic lesions, and increased glomerular expression of FSP1 and urinary loss of FSP1+ podocytes was specific to diabetic lesions and not identified in patients with minimal change disease and proteinuria. This finding is important as it has long remained unclear why patients with proteinuria and minimal change disease do not develop decreased glomerular filtration rate (GFR), while in other kidney diseases, proteinuria correlates with progressive decline in GFR. The authors also show increased expression of snail1, and downregulation of ZO-1 in human diabetic glomeruli (21). In addition, the authors identify upregulation of integrin linked kinase (ILK) in the FSP1+ podocytes of patients with diabetic nephropathy; ILK has been shown to mediate EMT in tubular cells and functions in adhesion of podocytes to the GBM (26, 27). Taken together, the data indicate that, in vitro under the influence of TGF-β, podocytes might take on mesenchymal characteristics, and that this mesenchymal phenotype may contribute to podocyte dehiscence and loss.
It is unclear as yet how expression of mesenchymal genes results in detachment of podocytes; however it is clear that podocytes that detach undergo a change in their transcription profile. It is interesting to note that while both snail and ILK have been implicated in EMT, they are also regulators of apical-basal polarity and cell motility. As apical-basal polarity is required to properly localize ZO-1 and adherens junction proteins at cell-cell interfaces and for adhesion to matrix components, these finding suggest the possibility that there may be in a defect in podocyte polarity in diabetic nephropathy. Podocyte polarity has recently been a topic of interest, with demonstration that apical basal polarity factors Crumbs and Par3/Par6/aPKC are required to localize nephrin to the podocyte slit diaphragm, with loss of function inducing proteinuria and glomerulosclerosis (1, 28).
From a therapeutic standpoint, the important role of TGF-β in EMT provides yet another mechanism by which TGFβ inhibitors could be protective in progressive proteinuric kidney disease. It also suggests that ongoing trials using antiTGFβ agents in the treatment of progressive proteinuric kidney diseases should examine urinary podocyte loss as an outcome measure.
In sum, phenotypic changes of podocytes associated with an increase in podocyte motility and expression of new markers is documented in human diabetic kidney disease and many different animal models and appears to be a common theme associated with proteinuria. The functional role of these changes and whether we can call these changes EMT or not remains to be determined. Nevertheless Yamaguchi at al provides a new paradigm of podocyte detachment, with EMT contributing to podocyte loss in diabetic nephropathy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Huber TB, Hartleben B, Winkelmann K, et al. Loss of podocyte aPKClambda/iota causes polarity defects and nephrotic syndrome. J Am Soc Nephrol. 2009;20:798–806. doi: 10.1681/ASN.2008080871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reiser J, Kriz W, Kretzler M, Mundel P. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol. 2000;11:1–8. doi: 10.1681/ASN.V1111. [DOI] [PubMed] [Google Scholar]
- 3.Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- 4.Pagtalunan ME, Miller PL, Jumping Eagle S, et al. Podocyte loss and progressive glomerular injury in type II diabetes. The Journal of clinical investigation. 1997;99:342–348. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim YH, Goyal M, Kurnit D, et al. Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN treated rat. Kidney international. 2001;60:957–968. doi: 10.1046/j.1523-1755.2001.060003957.x. [DOI] [PubMed] [Google Scholar]
- 6.Petermann AT, Pippin J, Krofft R, et al. Viable podocytes detach in experimental diabetic nephropathy: potential mechanism underlying glomerulosclerosis. Nephron. 2004;98:e114–123. doi: 10.1159/000081555. [DOI] [PubMed] [Google Scholar]
- 7.Li JH, Huang XR, Zhu HJ, et al. Advanced glycation end products activate Smad signaling via TGF beta dependent and independent mechanisms: implications for diabeticrenal and vascular disease. Faseb J. 2004;18:176–178. doi: 10.1096/fj.02-1117fje. [DOI] [PubMed] [Google Scholar]
- 8.Szabo C, Biser A, Benko R, Bottinger E, Susztak K. Poly(ADP ribose) polymerase inhibitors ameliorate nephropathy of type 2 diabetic Leprdb/db mice. Diabetes. 2006;55:3004–3012. doi: 10.2337/db06-0147. [DOI] [PubMed] [Google Scholar]
- 9.Niranjan T, Bielesz B, Gruenwald A, et al. The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat Med. 2008;14:290–298. doi: 10.1038/nm1731. [DOI] [PubMed] [Google Scholar]
- 10.Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes. 2005;54:1626–1634. doi: 10.2337/diabetes.54.6.1626. [DOI] [PubMed] [Google Scholar]
- 11.Schiffer M, Bitzer M, Roberts IS, et al. Apoptosis in podocytes induced by TGF-beta and Smad7. The Journal of clinical investigation. 2001;108:807–816. doi: 10.1172/JCI12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doublier S, Salvidio G, Lupia E, et al. Nephrin expression is reduced in human diabetic nephropathy: evidence for a distinct role for glycated albumin and angiotensin II. Diabetes. 2003;52:1023–1030. doi: 10.2337/diabetes.52.4.1023. [DOI] [PubMed] [Google Scholar]
- 13.Susztak K, Ciccone E, McCue P, Sharma K, Bottinger EP. Multiple metabolic hits converge on CD36 as novel mediator of tubular epithelial apoptosis in diabetic nephropathy. PLoS Med. 2005;2:e45. doi: 10.1371/journal.pmed.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vogelmann SU, Nelson WJ, Myers BD, Lemley KV. Urinary excretion of viable podocytes in health and renal disease. American journal of physiology. 2003;285:F40–48. doi: 10.1152/ajprenal.00404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pozzi A, Jarad G, Moeckel GW, et al. Beta1 integrin expression by podocytes is required to maintain glomerular structural integrity. Developmental biology. 2008;316:288–301. doi: 10.1016/j.ydbio.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durvasula RV, Shankland SJ. Podocyte injury and targeting therapy: an update. Current opinion in nephrology and hypertension. 2006;15:1–7. doi: 10.1097/01.mnh.0000199012.79670.0b. [DOI] [PubMed] [Google Scholar]
- 17.Skoberne A, Konieczny A, Schiffer M. Glomerular epithelial cells in the urine: what has to be done to make them worthwhile? American journal of physiology. 2009;296:F230–241. doi: 10.1152/ajprenal.90507.2008. [DOI] [PubMed] [Google Scholar]
- 18.Shankland SJ. The podocyte’s response to injury: role in proteinuria and glomerulosclerosis. Kidney international. 2006;69:2131–2147. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- 19.Ziyadeh FN. Mediators of diabetic renal disease: the case for tgf Beta as the major mediator. J Am Soc Nephrol. 2004;15 (Suppl 1):S55–57. doi: 10.1097/01.asn.0000093460.24823.5b. [DOI] [PubMed] [Google Scholar]
- 20.Bitzer M, Sterzel RB, Bottinger EP. Transforming growth factor beta in renal disease. Kidney Blood Press Res. 1998;21:1–12. doi: 10.1159/000025837. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Kang YS, Dai C, Kiss LP, Wen X, Liu Y. Epithelial to mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. The American journal of pathology. 2008;172:299–308. doi: 10.2353/ajpath.2008.070057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Yang J, Dai C, Wu C, Liu Y. Role for integrin linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. The Journal of clinical investigation. 2003;112:503–516. doi: 10.1172/JCI17913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 24.Yang J, Weinberg RA. Epithelial mesenchymal transition: at the crossroads of development and tumor metastasis. Developmental cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Shankland SJ, Pippin JW, Reiser J, Mundel P. Podocytes in culture: past, present, and future. Kidney international. 2007;72:26–36. doi: 10.1038/sj.ki.5002291. [DOI] [PubMed] [Google Scholar]
- 26.El Aouni C, Herbach N, Blattner SM, et al. Podocyte specific deletion of integrin linked kinase results in severe glomerular basement membrane alterations and progressive glomerulosclerosis. J Am Soc Nephrol. 2006;17:1334–1344. doi: 10.1681/ASN.2005090921. [DOI] [PubMed] [Google Scholar]
- 27.Dai C, Stolz DB, Bastacky SI, et al. Essential role of integrin linked kinase in podocyte biology: Bridging the integrin and slit diaphragm signaling. J Am Soc Nephr. 2006;17:2164–2175. doi: 10.1681/ASN.2006010033. [DOI] [PubMed] [Google Scholar]
- 28.Hirose T, Satoh D, Kurihara H, et al. An essential role of the universal polarity protein, aPKClambda, on the maintenance of podocyte slit diaphragms. PLoS ONE. 2009;4:e4194. doi: 10.1371/journal.pone.0004194. [DOI] [PMC free article] [PubMed] [Google Scholar]