

Abstract
Background
IgM natural antibodies bind oxidatively modified low-density lipoprotein (LDL) and apoptotic cells and have been implicated as important for protection from atherosclerosis. We have directly investigated the requirement for IgM by studying the effects of IgM deficiency in LDL receptor-deficient mice (Ldlr−/−).
Methods and Results
Mice deficient in serum IgM (sIgM) or complement C1q were crossed with Ldlr−/− mice and studied on both low (LF) and high fat (HF) semi-synthetic diets. On both diets, en face and aortic root atherosclerotic lesions in sIgM.Ldlr−/− mice were substantially larger and more complex, with accelerated cholesterol crystal formation and increased smooth muscle cell content in aortic root lesions. Combined C1q and IgM deficiency had the same effect as IgM deficiency alone. Increased apoptosis was observed in aortic root lesions of both sIgM.Ldlr−/− and C1qa.Ldlr−/− mice. Since lesions were significantly larger in IgM deficient mice than in the absence of C1q, IgM protective mechanisms therefore appear to be partially independent of classical pathway activation and apoptotic cell clearance. Levels of IgG antibodies against copper-oxidised LDL were lower in high fat fed sIgM.Ldlr−/− mice, suggesting compensatory consumption of IgG in the absence of IgM.
Conclusions
This study provides direct evidence that IgM antibodies play a central role in protection against atherosclerosis. The mechanism appears to be at least partly independent of classical pathway complement activation by C1q.
Keywords: atherosclerosis, IgM, complement, apoptosis
Introduction
Oxidative modifications of low-density lipoprotein (LDL) induce inflammatory responses that are key initial steps in the pathogenesis of atherosclerosis.1 Both innate and adaptive immune responses are thought to contribute to lesion development, with involvement of antibodies which recognise epitopes on oxidised LDL (oxLDL) among other targets.2 Both IgG and IgM autoantibodies to oxLDL can be readily detected in both humans and animal models of atherosclerosis, and in some studies antibody titres correlated with disease severity.3-5 IgM monoclonal antibodies reacting with oxLDL have been cloned from apolipoprotein E deficient (ApoE−/−) mice and some have been found to bind the phosphorylcholine (PC) headgroup of oxidatively modified phospholipids.6-8 The prototypic and best characterised of these, EO6, is identical in sequence to T15, a classical natural antibody known to recognise PC expressed as a capsular epitope on Streptococcus pneumoniae.8
While initial studies focused on the detrimental effects of antibodies in atherosclerosis, evidence has accumulated suggesting that they may also have protective roles.9 Numerous studies have shown that immunisation with malondialdehyde-modified LDL (MDA-LDL) reduces atherosclerosis and is associated with robust T cell-dependent IgG anti-oxLDL responses.10-12 Immunisation of LDL receptor deficient (Ldlr−/−) or ApoE−/− mice with Streptococcus pneumoniae or phosphorylcholine (PC)-conjugated keyhole limpet hemocyanin is also atheroprotective, in association with a rise in IgM anti-PC antibody titre.13,14 However, the precise roles of antibodies in active immunization experiments is difficult to interpret conclusively in view of the greater antibody titres achieved by active immunisation compared with those obtained spontaneously,15 as well as the use of adjuvants and the effects of immunisation on other immunological parameters, such as regulatory T cells.16
One mechanism by which IgM antibodies may contribute to protection against atherosclerosis is by facilitating the clearance of apoptotic material from developing lesions. Opsonisation of apoptotic cells with IgM is known to accelerate their non-inflammatory clearance by macrophages, and this process requires cooperation with complement C1q.17 We have recently shown that C1q deficiency in C1qa.Ldlr−/− mice accelerates atherosclerosis.18 Increased numbers of apoptotic cells were observed in atherosclerotic lesions, consistent with the concept that defective waste disposal leads to accumulation of cellular debris within plaques.19-21
In the present study we sought to examine directly the contribution of IgM antibodies in atherogenesis by crossing Ldlr−/− mice with mice deficient in serum IgM (sIgM−/−) through gene-targeted disruption of the secretory tail of immunoglobulin μ chain.22 Although unable to secrete IgM, sIgM−/− B cells maintain membrane IgM expression, and have intact class switching and IgG production.22 We compared sIgM.Ldlr−/− mice with C1qa.Ldlr−/− mice and with triple deficient C1qa.sIgM.Ldlr−/− mice to contrast the effect of IgM on atherogenesis with selective disruption of the classical pathway.
Methods
Mice
sIgM−/− mice were generated as described previously,22 and crossed with Ldlr−/− mice (both backcrossed 10 times on C57BL/6 background) to generate sIgM.Ldlr−/− mice. sIgM.Ldlr−/− mice were further crossed with C1qa.Ldlr−/− mice18 to create “triple knockout” C1qa.sIgM.Ldlr−/− mice. From 10 to 22 weeks of age, experimental groups of female mice received either a high fat (HF) diet or a low fat (LF) semisynthetic reference diet (see Supplementary Methods for details). The following numbers of mice were used for LF and HF diets respectively: Ldlr−/− (n=12, n=12), C1qa.Ldlr−/− (n=12, n=15), sIgM.Ldlr−/− (n=14, n=15) and C1qa.sIgM.Ldlr−/− (n=10, n=9). The aortic root of one LF-fed C1qa.sIgM.Ldlr−/− mouse was damaged in processing and was not used for analysis. Animal care and procedures were conducted according to institutional guidelines, and mice were kept under specific pathogen-free conditions. Total serum cholesterol and triglycerides were measured using colorimetric enzymatic assays (Infinity, Alpha Labs, Eastleigh, UK). Lipoprotein profiles were generated by fast performance liquid chromatography (FPLC) on a Superose 6 size-exclusion column.
Atherosclerotic lesion analysis
Mice were killed by CO2 inhalation and blood removed from the inferior vena cava. Using a cannula inserted in the left ventricle, hearts were perfused sequentially with Krebs-Henseleit buffer at 37°C for 5 minutes, 2% formalin for 5min, and 2mL of Sudan IV solution by direct slow injection over 5 minutes. Each aorta was microdissected to remove adventitial fat, cut open longitudinally, destained briefly in 80% ethanol and photographed. En face plaque quantification was performed using ImagePro software (Media Cybernetics, MD) by a single operator blinded to group allocation. Aortic root cryosection, Oil red O staining and lesion quantification were performed as previously described.18
Immunohistochemistry
Aortic root frozen sections were stained using standard immunohistochemistry to identify the following cell types: macrophages (MOMA-2 rat mAb, Serotec), VSMC (alkaline phosphatase (AP)-conjugated anti-α-smooth muscle actin Ab, Sigma-Aldrich, Poole, UK), T cells (goat anti-mouse CD3ε Ab, Santa Cruz Biotechnology, Santa Cruz, CA). The presence of lesional deposition of C5b-9 (rabbit anti-human C5b-9 Ab, Calbiochem, Merck Biosciences, Darmstadt, Germany) was identified using the same technique, and quantified as percentage lesion area staining positive using ImagePro. Lesional C3 (FITC-conjugated goat anti-mouse C3 Ab, MP Biomedicals, Cambridge, UK) and IgG (FITC-conjugated goat anti-mouse IgG Ab, Sigma-Aldrich) were identified using immunofluorescence and quantified as mean fluorescence intensity per pixel.
Quantification of lesional apoptosis
Apoptotic cells were detected using TUNEL (Roche, Welwyn Garden City, UK) on aortic root cryosections, following the manufacturer's instructions. Randomised slides were quantified by a single operator blinded to group allocation, and assessed for number of TUNEL positive cells fitting morphological criteria for apoptosis, including cell shrinkage, nuclear condensation or fragmentation and expressed as percentage of lesional cells.
Confocal microscopy
For confocal microscopy,18 aortic root cryosections were double-immunostained for CD68 (Alexafluor 488-conjugated anti-CD68 Ab, Molecular Probes, Invitrogen, Paisley, UK) and IgM (biotin-conjugated mouse anti-mouse IgMb mAb and biotin-conjugated mouse anti-mouse IgMa, BD Pharmingen, Oxford, UK) secondarily labelled with Alexa 568-conjugated streptavidin (Molecular Probes), counterstained with TOPRO-3.
Lipoprotein isolation and modification
Human LDL (density 1.019-1.063g/mL) was isolated from plasma of healthy donors after overnight fasting by differential density ultracentrifugation,23 and modified with either freshly synthesized malondialdehyde (MDA) or CuSO4 to generate MDA-LDL and copper-oxidised LDL (CuOxLDL) (see Supplementary Methods).24
Serum autoantibody measurement and assessment of autoimmunity
For the detection of anti-MDA-LDL24 and anti-CuOxLDL antibodies, non-irradiated microtiter plates (Greiner Bio-One, Stonehouse, UK) were coated overnight at 4°C with either 15μg/mL MDA-LDL, CuOxLDL or native LDL in a coating buffer of 100mM NaHCO3 and 1mg/mL Na2EDTA or with coating buffer alone to assess non-specific binding. After washing with PBS with 0.5% Tween-20, plates were blocked with 5% BSA. Sera diluted in PBS-Tween were applied. AP-conjugated anti-mouse IgG, IgM or IgG1 Abs (Southern Biotech, Birmingham AL) were used for detection and plates were developed with p-nitrophenol phosphate. As Ldlr−/− mice on the C57BL/6 background produce IgG2c (equivalent to IgG2ab) antibodies, whereas sIgM.Ldlr−/− mice (carrying a129-derived interval over the Ig gene region) produce IgG2a (equivalent to IgG2aa) (unpublished, M.Botto), to detect IgG2a and IgG2c anti-MDA-LDL or anti-CuOxLDL antibodies, we used an AP-conjugated polyclonal goat anti-mouse IgG2a (Southern Biotech) with equal specificity for both mouse IgG2a and IgG2c over the detection range used (see Supplementary data). Anti-single-stranded DNA, anti-dsDNA, anti-chromatin and anti-histone antibodies were assayed by ELISA.25 For anti-cardiolipin and anti-β2glycoprotein I antibody ELISA assays and scoring of renal histology for glomerulonephritis see Supplementary Methods.
Statistical analysis
Results were analysed using Graphpad Prism version 3.0 (Graphpad Software, San Diego CA). Where data were not normally distributed non-parametric statistical tests were used and results are expressed as median (interquartile range): to compare two groups Mann-Whitney test was used; to compare three or more groups Kruskal-Wallis test followed by Dunn's post-test was used. Normally distributed data was compared using unpaired two-tailed Student's t-test, with Bonferroni correction for multiple comparisons. Probability values were considered significant at P<0.05.
The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
Results
Localisation of IgM in plaques of Ldlr−/− mice
Confocal microscopy was used to examine IgM deposition within atherosclerotic plaques and localisation relative to macrophages. IgM was found within lesions following either low fat (LF) or high fat (HF) diets, particularly in the acellular core and with a reciprocal relationship to CD68-positive macrophages (Figure 1A). No IgM was detected within atherosclerotic lesions of sIgM.Ldlr−/− mice, indicating detection specificity (Figure 1B).
Figure 1. Deposition of IgM in atherosclerotic lesions.

Confocal images of aortic root sections double-immunostained for CD68 (green) and IgM (red) and counterstained with TOPRO nuclear dye (blue). (A) Low power view of aortic root lesion from LF-fed Ldlr−/− mouse showing IgM deposition in the acellular base of the lesion and its relationship to lesional macrophages. (B) Low power view of similar lesion from LF-fed sIgM.Ldlr−/− mouse showing complete absence of IgM.
Accelerated atherosclerosis in serum IgM deficient mice
No significant differences between all four mouse strains on either diet were noted in final body weight, total serum cholesterol or triglycerides (Supplementary Table 1), or lipoprotein FPLC profiles (not shown). Atherosclerotic plaque burden was quantified in sIgM.Ldlr−/− mice and compared with Ldlr−/− control mice and with C1qa.Ldlr−/− mice. On the LF diet, a hierarchy was apparent such that C1qa.Ldlr−/− mice showed a moderate increase (2.8-fold) in atherosclerotic lesion area fraction at the aortic root (C1qa.Ldlr−/−, median 4.58% (interquartile range 1.42-8.44%), n=12 vs. Ldlr−/−, 1.64% (1.11-2.72%), n=12; P<0.05, using Kruskall-Wallis test with Dunn's post-test), whereas both sIgM.Ldlr−/− and C1qa.sIgM.Ldlr−/− mice demonstrated substantially increased lesion size of 6.1-fold and 4.1-fold respectively (sIgM.Ldlr−/− 10.0% (4.96-12.9%), n=14; C1qa.sIgM.Ldlr−/− 6.76% (5.11-10.4%), n=9; both P<0.001 vs. Ldlr−/−) (Figure 2A and 2B). On the HF diet (Figure 2A and 2C), aortic root lesions in sIgM.Ldlr−/− mice were 29% increased compared to Ldlr−/− mice (sIgM.Ldlr−/− 35.8% (31.1-39.1%), n=15; Ldlr−/− 27.8% (24.0-30.5%), n=12; P<0.05) and 33% increased in C1qa.sIgM.Ldlr−/− mice (36.9% (33.5-39.9%), n=9; P<0.01). There was no difference between C1qa.Ldlr−/− (26.4% (20.7-31.8%), n=15, P=1.00) and Ldlr−/− mice following the HF diet confirming our previously published data.18
Figure 2. Accelerated atherosclerotic lesion formation in aortic roots of sIgM deficient mice.
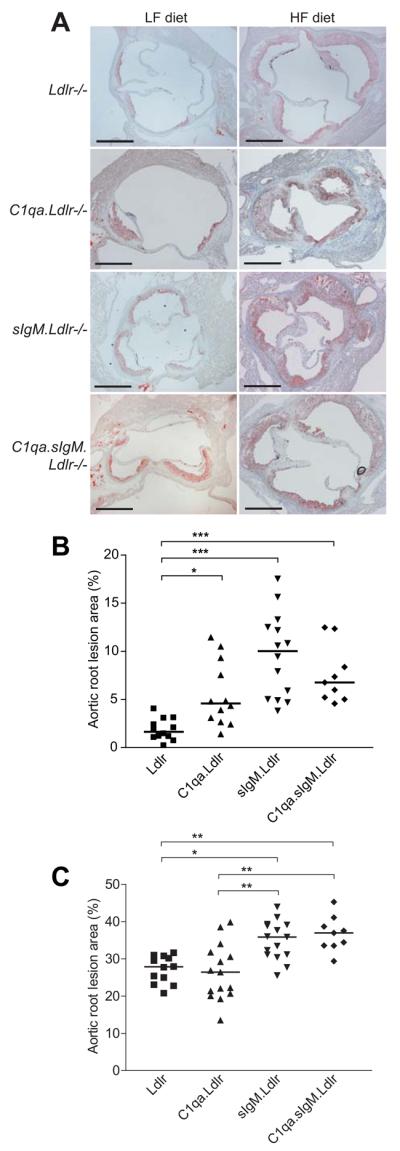
(A) Representative sections from Ldlr−/−, C1qa.Ldlr−/−, sIgM.Ldlr−/− and C1qa.sIgM.Ldlr−/− mice stained with Oil Red O and haematoxylin. Scale bars 500μm. (B, C) Cross-sectional aortic root lesion area fraction (%) on (B) LF or (C) HF diet. Each point represents the mean of 5 sections per mouse, and bars show overall median. *P<0.05, **P<0.01, ***P<0.001 by Kruskal-Wallis test with Dunn's post-test.
Whole aorta en face atherosclerotic lesion area mirrored the aortic root lesion data, showing a comparable hierarchy of effects of C1q and sIgM deficiency. On the LF diet C1qa.Ldlr−/− mice showed a moderate (although not statistically significant) increase of approximately 2.1-fold in en face atherosclerotic lesion area compared to Ldlr−/− mice (C1qa.Ldlr−/− 2.87% (1.71-3.56%), n=12 vs. Ldlr−/− 1.36% (0.73-1.72%), n=12; P=0.26) (Figure 3A and 3B). A substantially greater increase (~7-fold) in en face staining was evident in the sIgM.Ldlr−/− group compared to Ldlr−/− controls, with an equal increase seen in C1qa.sIgM.Ldlr−/− triple knockouts (sIgM.Ldlr−/−, 9.48% (5.36-10.9%), n=14; C1qa.sIgM.Ldlr−/−, 7.18% (4.79-11.4%), n=10; both P<0.001 vs. Ldlr−/−). On the HF diet, a 66% increase in Sudan IV staining was still seen in the sIgM.Ldlr−/− group compared to Ldlr−/− controls (12.4% (8.48-17.2%), n=15 vs. 7.49% (6.13-8.77%), n=12; P<0.05) (Figure 3C and 3D). C1qa.sIgM.Ldlr−/− showed a similar level of en face atherosclerotic lesions (10.1% (7.88-14.1%), n=9) as sIgM.Ldlr−/− mice, while C1qa.Ldlr−/− (5.23% (4.77-6.70%), n=15) were not statistically different to Ldlr−/− mice (P=0.16).
Figure 3. Accelerated lesion formation in en face aorta preparations of sIgM deficient mice.
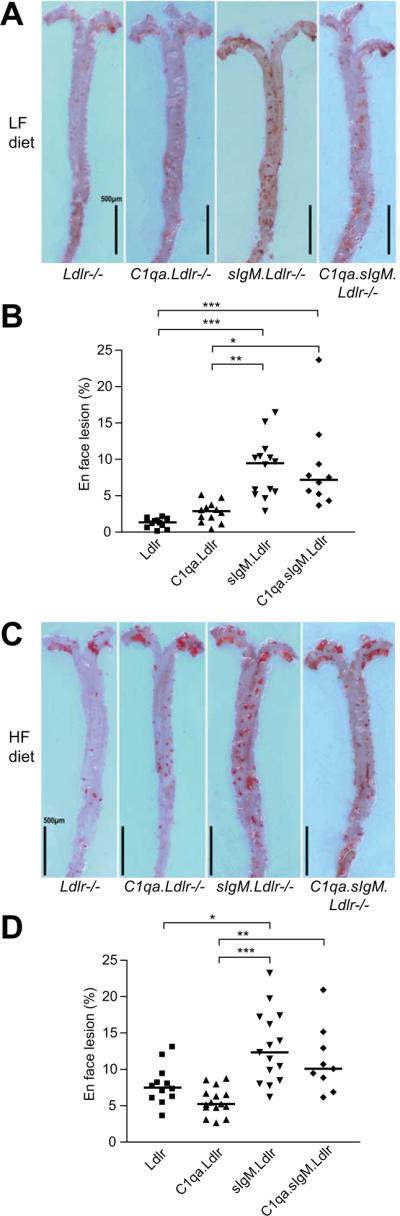
(A, C) Representative Sudan IV-stained en face aorta preparations from Ldlr−/−, C1qa.Ldlr−/−, sIgM.Ldlr−/− and C1qa.sIgM.Ldlr−/− mice following 12 weeks of (A) LF or (C) HF diet. Scale bars 0.5mm. (B, D) Quantification of en face aorta lesion area expressed as % lesion area fraction on (B) LF or (D) HF diet, with bars showing the median. *P<0.05, **P<0.01, ***P<0.001 by Kruskal-Wallis test with Dunn's post-test.
Increased lesion complexity in the absence of serum IgM
Aortic root lesions in Ldlr−/− mice fed the LF diet were largely fatty streaks, composed virtually entirely of macrophages. On both LF (Figure 4A) and HF (Figure 4B) diets, sIgM.Ldlr−/− lesions showed a significant increase in population by vascular smooth muscle cells (VSMC), identified by positive staining for α-smooth muscle actin (LF diet: sIgM.Ldlr−/− 4.95% (1.80-7.87%), n=14 vs. Ldlr−/− 0.0% (0.0-2.29%), n=12; P=0.008; HF diet: sIgM.Ldlr−/− 19.7% (11.6-33.9%), n=15 vs. Ldlr−/− 6.73% (3.85-10.4%), n=12; P=0.0004), with a reciprocal drop in macrophage content. On the HF diet, substantial numbers of sIgM.Ldlr−/− aortic root sections demonstrated fibrous caps (Figure 4C). Even on the LF diet, cholesterol crystal deposition was noted in the aortic root lesions of sIgM.Ldlr−/− mice (Figure 4C), whereas there was no evidence of cholesterol crystal deposition in the LF fed Ldlr−/− mice. There was no difference between sIgM.Ldlr−/− and Ldlr−/− in the proportion of lesional T lymphocytes (assessed with anti-CD3ε) on either diet (Figure 4A & B). Using immunofluorescence for IgG and C3, mean fluorescent intensity per pixel was quantified as an estimate of the density of lesional IgG and C3 deposition. There was no difference in the mean fluorescent intensity for IgG or C3 deposition in aortic root lesions between Ldlr−/− and sIgM.Ldlr−/− mice on either diet (Figure 4A, B and C), nor any difference in the level of lesional C5b-9 (not shown).
Figure 4. Increased complexity of aortic root lesions in sIgM deficient mice.
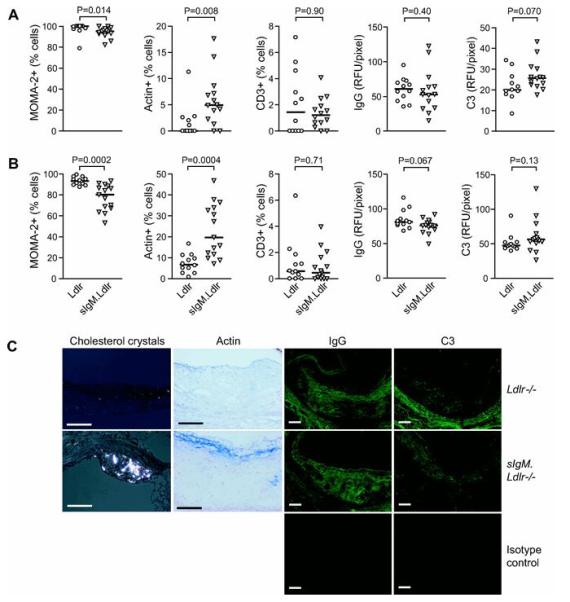
MOMA-2 positive macrophages, α-smooth muscle actin positive VSMC and CD3+ T cells expressed as % of lesional cells on (A) low fat (Ldlr−/− n=12, sIgM.Ldlr−/− n=14) and (B) high fat diet (Ldlr−/− n=12, sIgM.Ldlr−/− n=15). Density of aortic root lesion IgG and C3 deposition measured as mean fluorescence intensity per pixel on (A) low fat and (B) high fat diet (n numbers as before), expressed as relative fluorescence units (RFU) per pixel. Bars represent median, and statistical analysis was by Mann-Whitney test. (C) Representative photomicrographs of aortic root lesions showing cholesterol crystals under polarizing microscopy (age 22 wk, LF diet), smooth muscle fibrous cap formation (blue), lesional IgG and C3 deposition using fluorescence microscopy on HF diet in sIgM.Ldlr−/− compared to Ldlr−/− mice. Autofluorescence using a FITC-conjugated isotype control is shown below. Scale bars 100μm.
Clearance of apoptotic cell debris in atherosclerotic lesions in the absence of IgM
Lesional apoptosis in sIgM.Ldlr−/− mice was at a level similar to that found in C1qa.Ldlr−/−, which were previously demonstrated to have impaired lesional apoptotic cell clearance.18 Thus, using a combination of TUNEL staining and morphological identification, aortic root lesions of sIgM.Ldlr−/− mice fed the LF diet contained increased numbers of apoptotic cells compared to Ldlr−/− controls, reaching statistical significance when analysed by Mann-Whitney test (P=0.047). However, using more stringent Kruskal-Wallis post-tests only C1qa.Ldlr−/− mice showed increased numbers of apoptotic cells (P<0.01), and neither sIgM.Ldlr−/− nor C1qa.sIgM.Ldlr−/− mice reached significance, compared to Ldlr−/− controls (Figure 5A and 5B). More apoptotic cells were detectable in Ldlr−/− controls following the HF diet, such that differences between HF-fed groups were not significant (Figure 5C).
Figure 5. Increased apoptosis in atherosclerotic lesions of LF-fed C1qa.Ldlr−/−, sIgM.Ldlr−/− and C1qa.sIgM.Ldlr−/− mice.
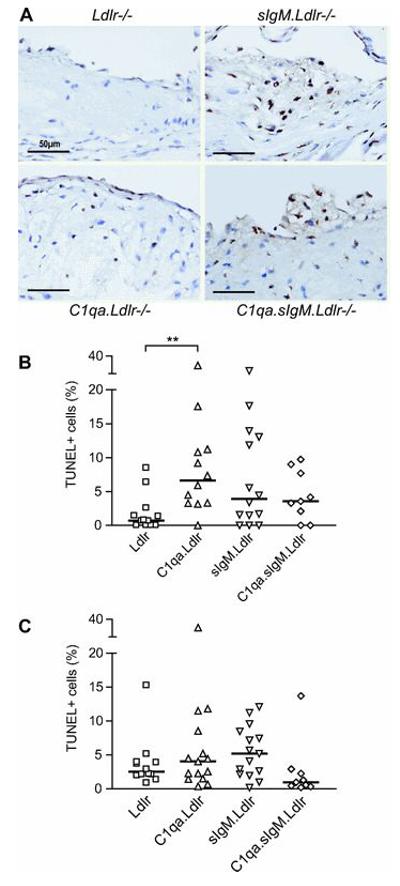
(A) Representative photomicrographs of TUNEL staining (brown) from LF-fed mice, counterstained with haematoxylin, showing few apoptotic cells in Ldlr−/− mice, but increased lesional apoptotic cells in C1qa.Ldlr−/−, sIgM.Ldlr−/− and C1qa.sIgM.Ldlr−/− mice. Scale bars 50μm. Quantification of lesional apoptotic cells on basis of TUNEL positive staining and morphological nuclear changes consistent with apoptosis on (B) LF diet and (C) HF diet, expressed as % of total lesional nuclei. Bars represent median. **P<0.01 by Kruskall-Wallis post-test.
Autoantibody production in sIgM.Ldlr−/− mice
sIgM−/− mice on a mixed C57BL/6×129 background have previously been reported to produce anti-double-stranded DNA (dsDNA) and aCL antibodies.26 However sIgM−/− mice backcrossed for 10 generations on the C57BL/6 background do not produce autoantibodies (unpublished data, M. Botto). Serological tests were conducted to check for evidence of autoimmunity in sIgM.Ldlr−/− mice. LF-fed sIgM.Ldlr−/− mice showed low levels of IgG anti-single-stranded DNA (ssDNA) antibodies, which were not statistically different to levels in Ldlr−/− mice (Table 1). However a significant increase in IgG anti-ssDNA antibodies was noted when sIgM.Ldlr−/− mice were fed the HF diet compared to HF-fed Ldlr−/− controls (P<0.001, Kruskal-Wallis post-test). Although raised, levels of anti-ssDNA antibodies in HF-fed sIgM.Ldlr−/− mice were low compared with 6 month old MRL/lpr mouse serum that was used as a standard (1000 ELISA units). Only HF-fed Ldlr−/− mice showed modest titres of IgG anticardiolipin antibodies (aCL), while all other groups showed non-significant levels. Other autoantibodies, including antinuclear antibodies, anti-dsDNA, antichromatin and anti-β2glycoprotein I antibodies were essentially undetectable in all experimental groups. There was no evidence of proteinuria or glomerulonephritis in any group (not shown).
Table 1. Autoantibody profiles in Ldlr−/− and sIgM.Ldlr−/− mice at 22 weeks of age.
| LF diet |
HF diet |
|||
|---|---|---|---|---|
|
Ldlr−/− n=12 |
sIgM.Ldlr−/− n=14 |
Ldlr−/− n=12 |
sIgM.Ldlr−/− n=15 |
|
| ssDNA | 14.2 (10.5-24.66) | 22.0 (16.2-37.8) | 14.3 (4.70-29.3) | 49.6 (26.7-87.9)* |
| dsDNA | 2.16 (1.67-4.22) | 1.46 (1.10-5.35) | 2.50 (1.62-3.44) | 5.27 (3.08-9.12) |
| Chromatin | 9.98 (6.67-15.2) | 11.5 (8.25-26.5) | 17.1 (13.7-25.5) | 16.1 (9.82-22.3) |
| aCL | 7.43 (0-43.7) | 14.2 (0-44.7) | 38.4 (0-75.1) | 0 (0-18.3) |
| β2GPI | 4.84 (2.43-7.27) | 1.96 (0.26-10.5) | 3.74 (2.42-7.43) | 4.21 (1.22-8.64) |
Serum IgG anti-single-stranded DNA (ssDNA), anti-dsDNA, anti-chromatin, anticardiolipin (aCL) and anti-β2glycoprotein I (β2GPI) antibody levels expressed as median (interquartile range) in arbitrary ELISA units, compared against a reference standard of pooled MRL/lpr mouse sera.
P<0.001 vs. HF-fed Ldlr−/− (post-test following Kruskal-Wallis test)
Changes in levels of IgG antibodies against MDA-LDL and CuOxLDL
To examine whether IgM deficiency influences IgG antibody responses against oxidised LDL (oxLDL), titres of anti-MDA-LDL and anti-CuOxLDL antibodies were assayed in Ldlr−/− and sIgM.Ldlr−/− mice at 22 weeks of age by ELISA. There was a significant increase in IgM and IgG antibodies to CuOxLDL, but not to MDA-LDL, in HF-fed versus LF-fed Ldlr−/− mice, and, as expected, IgM antibodies were not detected in sIgM.Ldlr−/− sera (Figure 6). There were no significant differences in IgG anti-MDA-LDL or anti-CuOxLDL antibody titre in sIgM.Ldlr−/− mice compared with Ldlr−/− mice on the LF diet. However, there was a significant reduction in IgG anti-CuOxLDL antibodies in sIgM.Ldlr−/− mice fed a HF diet compared with HF-fed Ldlr−/− mice.
Figure 6. IgG and IgM anti-oxLDL antibodies in Ldlr−/− and sIgM.Ldlr−/− mice.

Serial dilution curves showing titres of (A) anti-MDA-LDL and (B) anti-CuOxLDL antibodies in Ldlr−/− and sIgM.Ldlr−/− mice on low and high fat diets at 22 weeks of age. Non-specific binding to native LDL is shown at a single dilution. Error bars show SEM. †P<0.05 sIgM.Ldlr−/− (LF) vs. Ldlr−/− (LF). **P<0.01 sIgM.Ldlr−/− (HF) vs. Ldlr−/− (HF). §P<0.05, §§P<0.01, §§§P<0.001 Ldlr−/− (LF) vs. Ldlr−/− (HF). Statistical analysis by unpaired t-test with Bonferroni correction for multiple comparisons.
Hypercholesterolaemia in ApoE−/− mice has been shown to cause a Th1 to Th2 switch resulting in increased IgG1 anti-MDA-LDL antibody formation.24 To investigate whether increased atherosclerosis in sIgM.Ldlr−/− mice was associated with an increased Th2 response, IgG1 and IgG2a/2c isotypes were also assayed. For both isotypes generally no differences were seen between sIgM.Ldlr−/− and Ldlr−/− mice, with the exception of slightly lower IgG2a/2c anti-CuOxLDL antibodies observed in LF-fed sIgM.Ldlr−/− mice (significant only at the lowest dilution).
Discussion
In this paper, we have shown that Ldlr−/− mice deficient in serum IgM display a substantial acceleration of atherosclerosis on both a LF and HF diet, with larger lesions, an increase in lesional cholesterol crystal formation and an increase in VSMC content consistent with fibrous cap formation. Overall, our data provide the first direct evidence that endogenous IgM natural antibodies are necessary for atheroprotection, and build on previous experimental studies on humoral immunity in mice that have shown reduction of atherosclerosis by (i) B cell rescue of splenectomised mice;27 (ii) administration of polyclonal human IgG;28,29 (iii) administration of a monoclonal human IgG1 antibody against MDA-LDL;30,31 and (iv) raising IgM anti-PC antibody levels by vaccination with pneumococci13 or PC conjugated to keyhole limpet hemocyanin.14
One of the primary functions of IgM is to activate complement. Previous studies in mouse models have shown increased arterial lipid deposition and macrophage infiltration in mice with deficiency of C1q or C3, suggesting that complement has atheroprotective effects.18,32,33 However, whilst C1q deficiency was associated with increased VSMC content in lesions,18 the opposite was reported in C3 deficient mice,32 raising the possibility that different modes of complement activation may have distinct effects on atherogenesis. The direct comparison of sIgM.Ldlr−/− mice with C1qa.Ldlr−/− and triple “knockout” C1qa.sIgM.Ldlr−/− mice allowed us to gauge how much of the IgM-related atheroprotection was mechanistically linked to downstream classical pathway complement activation via C1q. As previously published, C1qa.Ldlr−/− mice on the LF diet showed a modest increase in aortic root lesion size, but C1q deficiency had no detectable effect in mice fed a HF diet.18 The differences in this paper between sIgM.Ldlr−/− and C1qa.Ldlr−/− mice now demonstrate a hierarchy, with IgM playing a dominant role. Consistent with this, combined deficiency of C1q and sIgM showed no difference in the level of atherosclerosis compared to IgM deficiency alone. It is possible that in the absence of C1q, IgM may bypass the classical pathway and instead activate the lectin pathway,34 as has been shown in ischaemia-reperfusion injury.35 However, in the sIgM.Ldlr−/− mice we found no difference in the density of lesional C3 or C5b-9 staining compared with the Ldlr−/− mice, suggesting that at least part of the mechanism by which IgM exerts its protective effect in atherosclerosis is independent of complement activation in the arterial wall.
Both C1q and IgM have been implicated as important recognition molecules mediating clearance of dying cells.17,36 We observed increased levels of apoptotic cells and TUNEL positive cellular debris within aortic root atherosclerotic plaques of sIgM.Ldlr−/− mice, similar to those seen in C1qa.Ldlr−/−.18 Increased lesional apoptosis was not observed in C1qa.sIgM.Ldlr−/− mice, raising the possibility of compensatory mechanisms occurring within the triple knock-outs. There was no difference in apoptotic cell numbers between the groups on the HF diet, primarily due to increased lesional apoptosis in Ldlr−/− controls. It is important to note that although defective clearance of cellular debris within atherosclerotic plaques may be a significant contributing factor to the increase in lesion size noted in sIgM.Ldlr−/− and C1qa.Ldlr−/− mice on the LF diet, increased numbers of lesional apoptotic cells may also be a function of lesion size. Overall, the data on lesional apoptotic cells do not support a failure of apoptotic cell clearance being the only or even the major mechanism by which IgM deficiency accelerates atherosclerosis, and certainly do not account for the greater lesion size in sIgM.Ldlr−/− compared to C1qa.Ldlr−/− mice.
Although deficiency of serum IgM has been shown to predispose to the development of anti-dsDNA and aCL antibodies on certain murine genetic backgrounds,26,37 sIgM−/− mice do not develop autoantibodies when fully backcrossed onto the C57BL/6 background (unpublished, M.Botto). Unexpectedly we observed that sIgM.Ldlr−/− mice on the HF diet developed anti-ssDNA autoantibodies, supporting previous observations that there is an interaction between hypercholesterolaemia and predisposition to lupus-like autoimmunity.19 Hence IgM deficiency enhances autoimmunity both in a strain-dependent and lipid-dependent manner. It should be noted however that the levels of anti-ssDNA in sIgM.Ldlr−/− mice on the HF diet were low compared to those seen in classical lupus models (e.g. MRL/lpr), and no mice developed renal disease. It therefore seems unlikely that lupus-like autoimmunity contributed significantly to the accelerated arterial disease we observed.
The main purpose of measuring levels of IgG antibodies against MDA- and CuOxLDL was to check whether removal of IgM would result in a change in predominant IgG isotype or a compensatory increase in levels of IgG antibodies to these antigens. There was no evidence in sIgM.Ldlr−/− mice of a change in IgG isotype reflecting a Th1 to Th2 switch.24 Paradoxically, there was a significant reduction in IgG antibodies against CuOxLDL in sIgM.Ldlr−/− mice fed a HF diet compared with Ldlr−/− mice. Whilst theoretically this may be due to an idiosyncratic reduction in IgG synthesis related to IgM deficiency, class switching is thought to be relatively normal in sIgM−/− mice.22 One plausible explanation is that the lower IgG CuOxLDL antibody level in HF-fed sIgM.Ldlr−/− mice is due to depletion of IgG by oxLDL particles that would have bound IgM in Ldlr−/− mice. This raises the question as to what extent the protective effect of IgM is related to actions such as inhibition of uptake of oxLDL by macrophages within lesions,7 and how much is due to facilitating safe clearance of oxLDL from the circulation or other tissues. For example, IgM may well be important for opsonising circulating oxLDL particles and targeting them to the liver, which is known to be the major organ for circulating oxLDL uptake.38
Although mechanistic conclusions cannot be drawn from clinical studies relating antibody levels with disease, it is relevant that there have been reports showing inverse correlations between serum levels of IgM anti-oxLDL or anti-phosphorylcholine antibodies and progression of carotid atherosclerosis,39-41 as well as angiographically determined coronary artery disease.42 Taken together with the emerging experimental evidence in this and previous studies that IgM and IgG antibodies can be atheroprotective, there is therefore increasing rationale for therapeutic strategies aimed at boosting humoral immunity. Conversely, B cell depletion therapy using the anti-CD20 monoclonal antibody rituximab is increasingly used to treat autoimmune conditions including systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA). Up to 20% of patients with RA treated with repeated courses of B cell depletion develop abnormally low levels of sIgM, with less effect on serum IgG levels.43 It seems possible that repeated B cell depletion might increase cardiovascular risk due to preferential depletion of serum IgM, in a population already susceptible to cardiovascular complications.44
Short Commentary of Clinical Impact.
Previous immunization experiments in animals and clinical association studies have suggested that IgM antibodies may have protective effects against the development of atherosclerosis. In this study we have addressed the role of IgM in atherogenesis by crossing the low density lipoprotein receptor-deficient mouse model of atherosclerosis (Ldlr−/−) with mice that have no IgM in their serum due to defective release of IgM from B lymphocytes (sIgM−/−). sIgM.Ldlr−/− mice showed marked acceleration of atherosclerosis compared with Ldlr−/− with larger lesions, early lesional cholesterol crystal formation, and an increase in VSMC content consistent with fibrous cap formation. Although IgM is a potent activator of complement, and thus has an important function in disposing of apoptotic cells, atheroprotection by IgM, in part, is not mediated through complement activation and consequently is at least partially independent of apoptotic cell clearance. Overall, our data provide the first direct evidence that endogenous IgM natural antibodies are necessary for arterial homeostasis and corroborate clinical studies that have observed an inverse correlation between IgM anti-oxidized LDL antibodies and progression of carotid atherosclerosis or angiographically determined coronary artery disease. This work lends support for developing immunization strategies to boost natural antibody protection and potentially limit atherosclerosis.
Supplementary Material
Acknowledgments
We are grateful for assistance from H.T. Cook (renal pathology), D.P. Patel (LDL purification), N. Navaratnam (FPLC), D. Carassiti (immunofluorescence), M. Lewis (histology processing) and the Biological Services Unit staff for the welfare of the animals.
Sources of Funding
This study was funded by programme grants from the British Heart Foundation and the Wellcome Trust. Dr Lewis is the recipient of a Wellcome Trust Clinical Research Fellowship.
Footnotes
Disclosures
None.
References
- 1.Glass CK, Witztum JL. Atherosclerosis. The road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 2.Binder CJ, Chang MK, Shaw PX, Miller YI, Hartvigsen K, Dewan A, Witztum JL. Innate and acquired immunity in atherogenesis. Nat Med. 2002;8:1218–1226. doi: 10.1038/nm1102-1218. [DOI] [PubMed] [Google Scholar]
- 3.Palinski W, Rosenfeld ME, Yla-Herttuala S, Gurtner GC, Socher SS, Butler SW, Parthasarathy S, Carew TE, Steinberg D, Witztum JL. Low density lipoprotein undergoes oxidative modification in vivo. Proc Natl Acad Sci USA. 1989;86:1372–1376. doi: 10.1073/pnas.86.4.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palinski W, Tangirala RK, Miller E, Young SG, Witztum JL. Increased autoantibody titers against epitopes of oxidized LDL in LDL receptor-deficient mice with increased atherosclerosis. Arterioscler Thromb Vasc Biol. 1995;15:1569–1576. doi: 10.1161/01.atv.15.10.1569. [DOI] [PubMed] [Google Scholar]
- 5.Shoenfeld Y, Wu R, Dearing LD, Matsuura E. Are anti-oxidized low-density lipoprotein antibodies pathogenic or protective? Circulation. 2004;110:2552–2558. doi: 10.1161/01.CIR.0000143225.07377.EA. [DOI] [PubMed] [Google Scholar]
- 6.Palinski W, Horkko S, Miller E, Steinbrecher UP, Powell HC, Curtiss LK, Witztum JL. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest. 1996;98:800–814. doi: 10.1172/JCI118853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horkko S, Bird DA, Miller E, Itabe H, Leitinger N, Subbanagounder G, Berliner JA, Friedman P, Dennis EA, Curtiss LK, Palinski W, Witztum JL. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J Clin Invest. 1999;103:117–128. doi: 10.1172/JCI4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaw PX, Horkko S, Chang MK, Curtiss LK, Palinski W, Silverman GJ, Witztum JL. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J Clin Invest. 2000;105:1731–1740. doi: 10.1172/JCI8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Binder CJ, Chou MY, Fogelstrand L, Hartvigsen K, Shaw PX, Boullier A, Witztum JL. Natural antibodies in murine atherosclerosis. Current Drug Targets. 2008;9:190–195. doi: 10.2174/138945008783755520. [DOI] [PubMed] [Google Scholar]
- 10.Palinski W, Miller E, Witztum JL. Immunization of low density lipoprotein (LDL) receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduces atherogenesis. Proc Natl Acad Sci USA. 1995;92:821–825. doi: 10.1073/pnas.92.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ameli S, Hultgardh-Nilsson A, Regnstrom J, Calara F, Yano J, Cercek B, Shah PK, Nilsson J. Effect of immunization with homologous LDL and oxidized LDL on early atherosclerosis in hypercholesterolemic rabbits. Arterioscler Thromb Vasc Biol. 1996;16:1074–1079. doi: 10.1161/01.atv.16.8.1074. [DOI] [PubMed] [Google Scholar]
- 12.Freigang S, Horkko S, Miller E, Witztum JL, Palinski W. Immunization of LDL receptor-deficient mice with homologous malondialdehyde-modified and native LDL reduces progression of atherosclerosis by mechanisms other than induction of high titers of antibodies to oxidative neoepitopes. Arterioscler Thromb Vasc Biol. 1998;18:1972–1982. doi: 10.1161/01.atv.18.12.1972. [DOI] [PubMed] [Google Scholar]
- 13.Binder CJ, Horkko S, Dewan A, Chang MK, Kieu EP, Goodyear CS, Shaw PX, Palinski W, Witztum JL, Silverman GJ. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med. 2003;9:736–743. doi: 10.1038/nm876. [DOI] [PubMed] [Google Scholar]
- 14.Caligiuri G, Khallou-Laschet J, Vandaele M, Gaston AT, Delignat S, Mandet C, Kohler HV, Kaveri SV, Nicoletti A. Phosphorylcholine-targeting immunization reduces atherosclerosis. J Am Coll Cardiol. 2007;50:540–546. doi: 10.1016/j.jacc.2006.11.054. [DOI] [PubMed] [Google Scholar]
- 15.Mironova M, Virella G, Lopes-Virella MF. Isolation and characterization of human antioxidized LDL autoantibodies. Arterioscler Thromb Vasc Biol. 1996;16:222–229. doi: 10.1161/01.atv.16.2.222. [DOI] [PubMed] [Google Scholar]
- 16.Mallat Z, Taleb S, Ait-Oufella H, Tedgui A. The role of adaptive T cell immunity in atherosclerosis. J Lipid Res. 2009;50:S364–S369. doi: 10.1194/jlr.R800092-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quartier P, Potter PK, Ehrenstein MR, Walport MJ, Botto M. Predominant role of IgM-dependent activation of the classical pathway in the clearance of dying cells by murine bone marrow-derived macrophages in vitro. Eur J Immunol. 2005;35:252–260. doi: 10.1002/eji.200425497. [DOI] [PubMed] [Google Scholar]
- 18.Bhatia VK, Yun S, Leung V, Grimsditch DC, Benson GM, Botto MB, Boyle JJ, Haskard DO. Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am J Pathol. 2007;170:416–426. doi: 10.2353/ajpath.2007.060406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aprahamian T, Rifkin I, Bonegio R, Hugel B, Freyssinet JM, Sato K, Castellot JJ, Jr., Walsh K. Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. J Exp Med. 2004;199:1121–1131. doi: 10.1084/jem.20031557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boisvert WA, Rose DM, Boullier A, Quehenberger O, Sydlaske A, Johnson KA, Curtiss LK, Terkeltaub R. Leukocyte transglutaminase 2 expression limits atherosclerotic lesion size. Arterioscler Thromb Vasc Biol. 2006;26:563–569. doi: 10.1161/01.ATV.0000203503.82693.c1. [DOI] [PubMed] [Google Scholar]
- 21.Ait-Oufella H, Kinugawa K, Zoll J, Simon T, Boddaert J, Heeneman S, Blanc-Brude O, Barateau V, Potteaux S, Merval R, Esposito B, Teissier E, Daemen MJ, Leseche G, Boulanger C, Tedgui A, Mallat Z. Lactadherin deficiency leads to apoptotic cell accumulation and accelerated atherosclerosis in mice. Circulation. 2007;115:2168–2177. doi: 10.1161/CIRCULATIONAHA.106.662080. [DOI] [PubMed] [Google Scholar]
- 22.Ehrenstein MR, O'Keefe TL, Davies SL, Neuberger MS. Targeted gene disruption reveals a role for natural secretory IgM in the maturation of the primary immune response. Proc Natl Acad Sci USA. 1998;95:10089–10093. doi: 10.1073/pnas.95.17.10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wade DP, Knight BL, Soutar AK. Detection of the low-density-lipoprotein receptor with biotin-low-density lipoprotein. A rapid new method for ligand blotting. Biochem J. 1985;229:785–790. doi: 10.1042/bj2290785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou X, Paulsson G, Stemme S, Hansson GK. Hypercholesterolemia is associated with a T helper (Th) 1/Th2 switch of the autoimmune response in atherosclerotic apo E-knockout mice. J Clin Invest. 1998;101:1717–1725. doi: 10.1172/JCI1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burlingame RW, Rubin RL. Subnucleosome structures as substrates in enzyme-linked immunosorbent assays. J Immunol Methods. 1990;134:187–199. doi: 10.1016/0022-1759(90)90380-e. [DOI] [PubMed] [Google Scholar]
- 26.Ehrenstein MR, Cook HT, Neuberger MS. Deficiency in serum immunoglobulin (Ig)M predisposes to development of IgG autoantibodies. J Exp Med. 2000;191:1253–1258. doi: 10.1084/jem.191.7.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caligiuri G, Nicoletti A, Poirier B, Hansson GK. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest. 2002;109:745–753. doi: 10.1172/JCI07272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicoletti A, Kaveri S, Caligiuri G, Bariety J, Hansson GK. Immunoglobulin treatment reduces atherosclerosis in apo E knockout mice. J Clin Invest. 1998;102:910–918. doi: 10.1172/JCI119892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan Z, Kishimoto C, Sano H, Shioji K, Xu Y, Yokode M. Immunoglobulin treatment suppresses atherosclerosis in apolipoprotein E-deficient mice via the Fc portion. Am J Physiol Heart Circ Physiol. 2003;285:H899–H906. doi: 10.1152/ajpheart.00926.2002. [DOI] [PubMed] [Google Scholar]
- 30.Schiopu A, Bengtsson J, Soderberg I, Janciauskiene S, Lindgren S, Ares MP, Shah PK, Carlsson R, Nilsson J, Fredrikson GN. Recombinant human antibodies against aldehyde-modified apolipoprotein B-100 peptide sequences inhibit atherosclerosis. Circulation. 2004;110:2047–2052. doi: 10.1161/01.CIR.0000143162.56057.B5. [DOI] [PubMed] [Google Scholar]
- 31.Schiopu A, Frendeus B, Jansson B, Soderberg I, Ljungcrantz I, Araya Z, Shah PK, Carlsson R, Nilsson J, Fredrikson GN. Recombinant antibodies to an oxidized low-density lipoprotein epitope induce rapid regression of atherosclerosis in apobec-1(−/−)/low-density lipoprotein receptor(−/−) mice. J Am Coll Cardiol. 2007;50:2313–2318. doi: 10.1016/j.jacc.2007.07.081. [DOI] [PubMed] [Google Scholar]
- 32.Buono C, Come CE, Witztum JL, Maguire GF, Connelly PW, Carroll M, Lichtman AH. Influence of C3 deficiency on atherosclerosis. Circulation. 2002;105:3025–3031. doi: 10.1161/01.cir.0000019584.04929.83. [DOI] [PubMed] [Google Scholar]
- 33.Persson L, Boren J, Robertson AK, Wallenius V, Hansson GK, Pekna M. Lack of complement factor C3, but not factor B, increases hyperlipidemia and atherosclerosis in apolipoprotein E−/− low-density lipoprotein receptor−/− mice. Arterioscler Thromb Vasc Biol. 2004;24:1062–1067. doi: 10.1161/01.ATV.0000127302.24266.40. [DOI] [PubMed] [Google Scholar]
- 34.Arnold JN, Wormald MR, Suter DM, Radcliffe CM, Harvey DJ, Dwek RA, Rudd PM, Sim RB. Human serum IgM glycosylation: identification of glycoforms that can bind to mannan-binding lectin. J Biol Chem. 2005;280:29080–29087. doi: 10.1074/jbc.M504528200. [DOI] [PubMed] [Google Scholar]
- 35.Zhang M, Takahashi K, Alicot EM, Vorup-Jensen T, Kessler B, Thiel S, Jensenius JC, Ezekowitz RA, Moore FD, Carroll MC. Activation of the lectin pathway by natural IgM in a model of ischemia/reperfusion injury. J Immunol. 2006;177:4727–4734. doi: 10.4049/jimmunol.177.7.4727. [DOI] [PubMed] [Google Scholar]
- 36.Ogden CA, Kowalewski R, Peng Y, Montenegro V, Elkon KB. IgM is required for efficient complement mediated phagocytosis of apoptotic cells in vivo. Autoimmunity. 2005;38:259–264. doi: 10.1080/08916930500124452. [DOI] [PubMed] [Google Scholar]
- 37.Boes M, Schmidt T, Linkemann K, Beaudette BC, Marshak-Rothstein A, Chen J. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc Natl Acad Sci USA. 2000;97:1184–1189. doi: 10.1073/pnas.97.3.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Berkel TJ, De Rijke YB, Kruijt JK. Different fate in vivo of oxidatively modified low density lipoprotein and acetylated low density lipoprotein in rats. Recognition by various scavenger receptors on Kupffer and endothelial liver cells. J Biol Chem. 1991;266:2282–2289. [PubMed] [Google Scholar]
- 39.Karvonen J, Paivansalo M, Kesaniemi YA, Horkko S. Immunoglobulin M type of autoantibodies to oxidized low-density lipoprotein has an inverse relation to carotid artery atherosclerosis. Circulation. 2003;108:2107–2112. doi: 10.1161/01.CIR.0000092891.55157.A7. [DOI] [PubMed] [Google Scholar]
- 40.Su J, Georgiades A, Wu R, Thulin T, de FU, Frostegard J. Antibodies of IgM subclass to phosphorylcholine and oxidized LDL are protective factors for atherosclerosis in patients with hypertension. Atherosclerosis. 2006;188:160–166. doi: 10.1016/j.atherosclerosis.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 41.Sjöberg B, Su J, Dahlbom I, Grönlund H, Wikström M, Hedblad B, Berglund G, de Faire U, Frostegard J. Low levels of IgM antibodies against phosphorylcholine--A potential risk marker for ischemic stroke in men. Atherosclerosis. 2009;203:528–532. doi: 10.1016/j.atherosclerosis.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 42.Tsimikas S, Brilakis ES, Lennon RJ, Miller ER, Witztum JL, McConnell JP, Kornman KS, Berger PB. Relationship of IgG and IgM autoantibodies to oxidized low density lipoprotein with coronary artery disease and cardiovascular events. J Lipid Res. 2007;48:425–433. doi: 10.1194/jlr.M600361-JLR200. [DOI] [PubMed] [Google Scholar]
- 43.Keystone E, Fleischmann R, Emery P, Furst DE, van VR, Bathon J, Dougados M, Baldassare A, Ferraccioli G, Chubick A, Udell J, Cravets MW, Agarwal S, Cooper S, Magrini F. Safety and efficacy of additional courses of rituximab in patients with active rheumatoid arthritis: an open-label extension analysis. Arthritis Rheum. 2007;56:3896–3908. doi: 10.1002/art.23059. [DOI] [PubMed] [Google Scholar]
- 44.Haskard DO. Accelerated atherosclerosis in inflammatory rheumatic diseases. Scand J Rheumatol. 2004;33:281–292. doi: 10.1080/03009740410010281. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.