

Abstract
During activation of the sympathetic nervous system, cardiac performance is increased as part of the fight-or-flight stress response. The increase in contractility with sympathetic stimulation is an orchestrated combination of intrinsic inotropic, lusitropic and chronotropic effects, mediated in part by activation of β-adrenergic receptors and protein kinase A. This causes phosphorylation of several Ca cycling proteins in cardiac myocytes (increasing Ca entry via L-type Ca channels, sarcoplasmic reticulum (SR) Ca pumping and the dissociation rate of Ca from the myofilaments). Here, we discuss how stimulation of the Na/K-ATPase, mediated by phosphorylation of phospholemman (a small sarcolemmal protein that associates with and modulates Na/K-ATPase), is an additional important player in the sympathetic fight-or-flight response. Enhancement of Na/K-ATPase activity limits the rise in [Na]i caused by the higher level of Na influx and by doing so limits the rise in cellular and SR Ca load by favoring Ca extrusion via the Na/Ca exchanger. Thus, phospholemman-mediated activation of the Na/K-ATPase may prevent Ca overload and triggered arrhythmias during stress.
Sympathetic Stimulation of the Heart and β-Adrenergic Receptors
During activation of the sympathetic nervous system, cardiac performance is increased as part of the fight-or-flight stress response. The increase in contractility with sympathetic stimulation is an orchestrated combination of intrinsic inotropic, lusitropic and chronotropic effects, mediated mostly by activation of Gs-coupled β-adrenergic receptors (β-ARs). Indeed, sympathetic nerve endings and β-ARs are broadly distributed throughout the heart. Sympathetic neurotransmitters activate β-ARs to increase adenylyl cyclase activity, thus elevating intracellular cyclic AMP concentration, which in turn activates cyclic AMP-dependent protein kinase A (PKA) activity (Figure 1).
Figure 1.
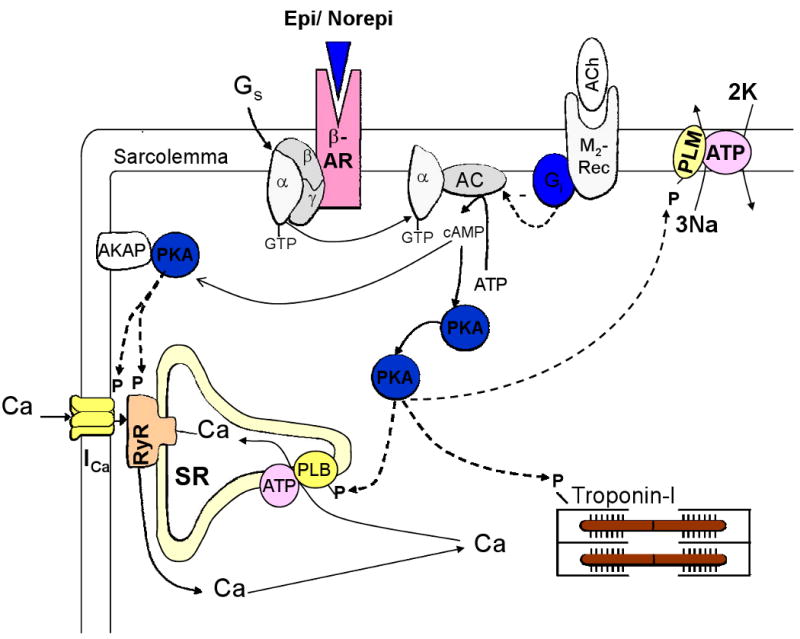
Pathway for activation of Ca cycling proteins in cardiac myocytes during β-adrenergic stimulation. β-AR = β adrenergic receptor; Epi = epinephrine; Norepi = norepinephrine, M2-rec = M2-muscarinic receptors, Ach = acetylcholine, AC = adenylyl cyclase, AKAP – PKA anchoring protein, PLB = phospholamban, PLM = phospholemman
The stimulatory effect of β-adrenergic agonists on adenylyl cyclase and cAMP production can be inhibited by another G-protein (Gi) which is activated by muscarinic M2 receptors (and some other receptors; Figure 1). Thus, parasympathetic release of acetylcholine can diminish the effect of sympathetic stimulation of β-ARs. While this clearly occurs in selected regions in the heart (e.g. sino-atrial node) and is important in the control of heart rate, there are fewer parasympathetic nerve endings in ventricular muscle. Thus, parasympathetic activation may have less anti-adrenergic effect on contractility than on heart rate. Nevertheless, there are muscarinic (and other) receptors in ventricular myocytes which can mediate Gi-dependent limitation of adenylyl cyclase activation.
Activation of β-ARs causes phosphorylation by PKA of several Ca cycling proteins in cardiac myocytes (Bers 2001; see Figure 1), including 1) sarcolemmal L-type Ca channels (increasing Ca current, ICa), 2) phospholamban (increasing SR Ca pump rate), 3) SR Ca release channels (modifying ryanodine receptor, RyR, gating) and 4) myofilament proteins (troponin I and myosin binding protein C, reducing myofilament Ca affinity and increasing crossbridge kinetics). This results in stronger and faster contractions and faster relaxation of the cardiac muscle (i.e., intrinsic inotropic and lusitropic effects). Moreover, β-AR stimulation also leads to phosphorylation of Ca cycling proteins by Ca/calmodulin-dependent protein kinase II (CaMKII) (Figure 2). This is partly secondary to PKA-dependent enhancement of Ca transients, but can also occur independent of cAMP and intracellular Ca elevation (Curran et al. 2007). Wang et al. (2004) showed that PKA activation mediates short-term (10 min) inotropic effect of β-AR stimulation, whereas CaMKII mediates the long-term (24 h) enhancement of contractility.
Figure 2.
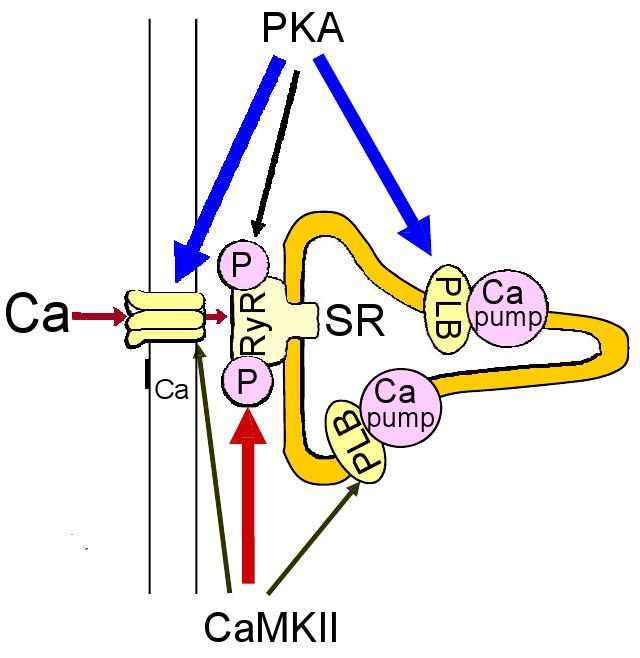
Both PKA and CaMKII phosphorylate L-type Ca channels, RyR and PLB during β-AR stimulation. PKA phosphorylation has significantly larger functional effects (vs. CaMKII phosphorylation) on L-type Ca channels and PLB. Conversely, CaMKII phosphorylation has stronger effects on RyR2 than those of PKA.
Chronotropic Effect of Sympathetic Activation
Activation of β-ARs in pacemaker cells from the sino-atrial node is responsible for the chonotropic effect (acceleration of heart rate) of sympathetic stimulation. β-AR activation shifts the voltage-dependence of the inward pacemaker current If, which is activated by hyperpolarization, to more depolarized potentials. This results in a more rapid diastolic depolarization. Activation of β-ARs also accelerates the decline in potassium conductance, which may contribute to pacemaker activity (especially in cells with more positive diastolic potentials, such as sino-atrial nodal cells). Activation of SR Ca uptake by PLB phosphorylation also increases spontaneous SR Ca release in pacemaker cells, activating inward (depolarizing) Na/Ca exchange current, which accelerates diastolic depolarization. Thus, several effects contribute to the positive sympathetic chronotropic effect.
Increased heart rate by itself increases contractility (thus has an inotropic effect) and speeds relaxation (lusitropic effect). This is due to increased Na and Ca loading at higher frequency (and shorter diastolic interval) and faster SR Ca uptake (known as frequency-dependent acceleration of relaxation, FDAR; Bers 2001). FDAR also enhances SR Ca load. These PKA-independent effects of higher heart rate contribute to enhanced cardiac output during sympathetic stimulation.
Lusitropic Effect of Sympathetic Stimulation
The main mechanism by which sympathetic stimulation accelerates relaxation of the heart is the enhancement of SR Ca re-uptake. This is due to an increase in the SR Ca-ATPase (SERCA) activity brought about by PLB phosphorylation by PKA. PLB is an endogenous inhibitor of SERCA that decreases Ca transport and ATPase activity, especially at low [Ca], because it decreases SERCA affinity for Ca (Figure 3), without altering the maximum transport rate. In the mouse, SERCA is not saturated with PLB in situ, as indicated by studies in transgenic mice that overexpress PLB (Kadambi et al. 1996). However, PLM overexpression in rabbit myocytes did not alter SR Ca transport (Waggoner et al. 2009). The apparently smaller PLB:SERCA ratio may contribute to the larger contribution of SERCA (vs. Na/Ca exchanger) to Ca transient relaxation in mice vs. rabbits.
Figure 3.
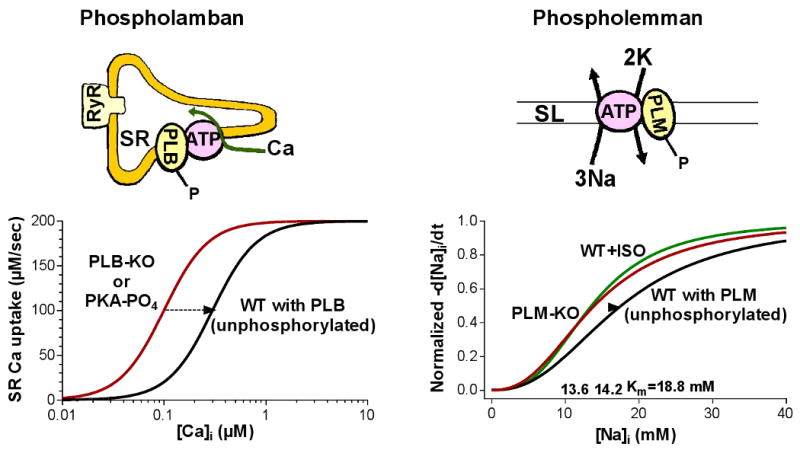
PLM regulates NKA the same way PLB modulates SERCA. Unphosphorylated PLB inhibits SERCA by reducing its Ca affinity and unphosphorylated PLM reduces the NKA apparent affinity for internal Na. Phosphorylation of PLB and PLM restores the ion affinity of the pumps and thus enhances the activity of SERCA and NKA, respectively.
PLB is phosphorylated by PKA at Ser16. This largely reverses the PLB-induced Ca affinity shift, increasing the Ca affinity of SERCA by 2-3 fold (Figure 3). Thus, for most physiologically relevant [Ca]i values (0.1-1 μM) there is a substantial increase in Ca-pump rate upon PLB phosphorylation. Application of a PLB antibody which interferes with the interaction between PLB and SERCA produces similar effects (Sham et al. 1991). Furthermore, when either the cardiac or skeletal muscle SR Ca-pump is expressed without PLB the Ca pumping and ATPase activity properties are like the endogenous PKA phosphorylated Ca-pump (e.g. Toyofuku et al. 1993). PLB is also phosphorylated by CaMKII (at Thr17) and this may synergize with PKA-dependent SERCA stimulation in the overall β-AR stimulation of the heart (Figure 2).
Cardiac TnI is also phosphorylated during sympathetic stimulation of the heart (at both Ser 23 and 24). This phosphorylation decreases myofilament Ca sensitivity in intact ventricular muscle and in skinned fibers, a result which is mimicked by cyclic AMP (Layland et al. 2005). The lower myofilament Ca sensitivity (and Ca-affinity) contributes to the more rapid relaxation during sympathetic stimulation, owing to faster Ca dissociation from troponin C. Mechanical factors, such as the rate of cross-bridge detachment and geometric considerations, might also be involved in the β-adrenergic induced acceleration of relaxation. Li et al. (2000) quantitatively compared the contributions of TnI and phospholamban phosphorylation in the lusitropic effect of isoproterenol. In mice lacking phospholamban (PLB-KO), isoproterenol had no effect at all on myocyte or unloaded muscle twitch relaxation, despite a substantial increase in TnI phosphorylation. However, when muscles from PLB-KO mice developed isometric force, the TnI-dependent lusitropic effect of isoproterenol was directly related to force development. This emphasizes the dynamic interplay between force, myofilament properties and Ca transients. Nevertheless, the lusitropic effect of isoproterenol in the PLB-KO mice was still very small, compared to that in wild-type mice. This indicates that the vast majority of the β-AR-dependent lusitropic effect (∼85%) is due to phospholamban phosphorylation vs. TnI phosphorylation.
Inotropic Effect of Sympathetic Stimulation
Sympathetic stimulation, mainly through PKA phosphorylation, causes a 2-4-fold increase in Ca current (ICa) through L-type Ca channels in ventricular myocytes and shifts the voltage-dependence of activation and inactivation to more negative membrane potentials (Em) (Kamp and Hell 2000). The shift in activation gating causes the ICa enhancement to be most prominent at negative Em and maximal ICa to be at more negative Em. At the single channel level PKA has no effect on unitary conductance, but there are fewer blank sweeps (without any openings) and an apparent increase in open times which may reflect a shift toward mode 2 gating (Yue et al. 1990).
PKA is probably anchored in the vicinity of the α1C subunit of the Ca channel via an A-kinase anchoring protein (AKAP15) (Hulme et al. 2006). The site of interaction of PKA and AKAP15 lies in the distal C-terminus of the α1C subunit, which appears to be cleaved from the remainder of the channel by in vivo proteolytic processing in myocytes. This distal C-terminal still interacts with the proximal C-terminal, serving as an autoinhibitory domain, and PKA phosphorylation may relieve this inhibition (Hulme et al. 2006). However, it is increasingly clear that the site once thought to be the PKA target (S1928) is not involved in ICa modulation (Lemke et al. 2008), but two PKA target sites on the β2A subunit (Ser-478 & 479) are also not required for the PKA-dependent increase of ICa (Ganesan et al. 2006). Thus, how PKA activates ICa is unresolved.
CaMKII activation downstream of β-AR also results in ICa facilitation, a moderate increase in ICa amplitude and slowing of inactivation that happens from one beat to the next. The molecular mechanism of ICa facilitation is not fully resolved, but CaMKII can associate directly with the carboxy-tail of the α1C subunit and β2a subunit, and phosphorylation of sites on both α1C itself and the β2 subunit have been implicated in mediating ICa facilitation (Hudmon et al. 2005; Gruter et al. 2006).
Owing to increased ICa, more Ca enters the cell at each excitation during sympathetic stimulation. Moreover, the SR accumulates a larger fraction of the cytosolic Ca pool during relaxation, due to SR Ca-pump stimulation via PLB phosphorylation (see above). Thus, the SR Ca load is significantly increased during β-AR stimulation. The increased ICa (and SR Ca) may also increase the fraction of SR Ca which is released. The result is a much larger peak [Ca]i. Na/Ca exchanger is unaltered by adrenergic agonists (Ginsburg and Bers 2005; Lin et al. 2006). Thus, the SR Ca-pump stimulation will also bias the competition between these two mechanisms in favor of the SR Ca-pump (even though the higher peak [Ca]i will also stimulate Ca extrusion via Na/Ca exchange). This bias toward the SR Ca-pump enhances SR Ca load and may shorten the time for the SR Ca release process to recover from inactivation.
SR Ca release channels (RyR2) also participate in the adrenergic fight-or-flight response. PKA-dependent RyR2 phosphorylation alters RyR gating in bilayers. Valdivia et al. (1995) found that PKA slightly decreased basal open probablility (Po) at 100 nM [Ca], but increased peak Po (to nearly 1.0) during a rapid photolytic increase of [Ca]. PKA also accelerated the subsequent decline in Po (attributed to adaptation). In contrast, Marx et al. (2000, 2001) found that PKA-dependent RyR phosphorylation at Ser-2809 enhanced steady state open probability of single RyRs in bilayers, and attributed this to displacement of FKBP12.6 from the RyR. This is controversial, as some groups find that FKBP12.6 still binds to RyR2 after PKA-dependent phosphorylation (Stange et al. 2002, Xiao et al. 2004). In more intact cellular systems, PKA-dependent RyR phosphorylation had no effect on resting Ca sparks in the absence of phospholamban phosphorylation (with unaltered SR Ca load) (Li et al. 2002). However, when phospholamban is phosphorylated, and SR Ca load is increased, Ca spark frequency is increased. Ginsburg et al. (2004) characterized the effect of PKA activation over a broad range of SR Ca loads and ICa trigger amplitudes. At a fixed SR Ca load and ICa amplitude, PKA activation had no effect on the amount of SR Ca-released, but it did increase the initial rate of release and timing of shut-off of release. Thus, PKA-dependent phosphorylation of the RyR speeds up the time course of release without affecting the amount.
CaMKII also phosphorylates RyR2 and alters RyR activity. In intact voltage clamped ventricular myocytes endogenous CaMKII increased the amount of SR Ca-release for a given SR Ca content and ICa trigger (Li et al. 1997). Overexpression of CaMKII-δC (the cytosolic isoform) (Maier et al. 2003) or direct application of pre-activated CaMKII to permeabilized myocytes (Guo et al. 2006) increased diastolic Ca spark frequency and fractional SR Ca-release during excitation-contraction coupling. Measures of SR Ca load are important in interpreting cellular results, because if CaMKII reduces SR Ca load, that could cause Ca transient depression (even if fractional release is enhanced; Maier et al. 2003).
Both PKA and CaMKII phosphorylate PLB, RyR and Ca channels at different target sites (Figure 2). Each has functional effects, but PKA has more profound quantitative effects on ICa and SR Ca uptake than does CaMKII. Conversely, CaMKII effects on RyR are stronger than those of PKA. The activating effects on ICa and SR Ca-ATPase can lead to increased contractility. However, RyR2 phosphorylation and the consequent increase in the fractional SR Ca release may not contribute appreciably to sustained inotropic effect of β-AR stimulation. This is because the acutely enhanced SR Ca release causes greater Ca extrusion via NCX, which in turn reduces SR Ca2+ content to the point where Ca transients return to normal.
β-Adrenergic activation also decreases myofilament Ca sensitivity (owing to phosphorylation of TnI, see above), but the dramatic increase in Ca transients more than compensates for this, so contractile force is increased substantially (but less so than Ca transients). PKA also phosphorylates myosin binding protein C and increases crossbridge cycling (Tong et al. 2008); this may contribute to faster cardiac ejection.
PLB phosphorylation and enhanced SR Ca-ATPase is probably the most important single factor in PKA-induced inotropy. However, isoproterenol still induces significant inotropy in PLB-KO mice (presumably owing to ICa enhancement).
Na/K-ATPase Activity during Sympathetic Activation of the Heart; Role of Phospholemman
Sarcolemmal Na/K-ATPase is also affected by β-AR agonists. Early Na-selective microelectrode studies indicated that β-AR stimulation enhances NKA activity. Most myocyte studies of either pump current (IPump; voltage-clamp) or [Na]i measurements indicate that NKA function is increased by β-AR stimulation (Glitsch 2001). However, some IPump studies reported either no β-AR effects in rat myocytes or even an inhibition of guinea-pig IPump at intracellular [Ca] <150 nM (reviewed in Glitsch 2001). It has also been reported that β-AR stimulation of IPump only affects the α1 isoform of NKA (the major isoform in heart), but not α2 (Gao et al. 1999, Silverman et al. 2005), although we find that β-AR activates both α1 and α2 NKA function (Bossuyt et al. 2009).
The NKA α subunit can be phosphorylated by PKA at Ser936, the only serine in a consensus motif (RRNSF) for PKA. However, phosphorylation was observed only in the presence of detergents and not in living cells or tissues. Sweadner and Feschenko (2001) aligned the NKA sequence with the SR Ca-ATPase structure and predicted that Ser936 is located near the membrane and is unlikely to be accessible to the kinase in vivo. This prediction was recently confirmed by X-ray crystallography of the NKA (Morth et al. 2007).
Recently it was found that PKA-dependent phosphorylation of phospholemman (PLM) mediates β-AR-dependent activation of NKA activity in cardiac myocytes (Despa et al. 2005). PLM is a small (72 aminoacids), single membrane-spanning sarcolemmal protein, long known as a major PKA phosphorylation target in the heart, quantitatively comparable to that of troponin I and phospholamban (Presti et al. 1985, Palmer et al. 1991). Early work suggested that PLM forms taurine-selective channels in lipid bilayers (Moorman et al. 1995) and recent structural studies support the idea of PLM multimers (Beevers and Kukol, 2006, Bossuyt et al. 2006). Taurine-selective channels suggested that PLM might be involved in cell volume regulation. However, myocytes from WT mice and mice that lack PLM (PLM-KO mice) swell equally in response to a hypo-osmotic challenge (Bell et al. 2009), indicating that PLM is not essential in limiting water accumulation. A key insight into the physiological role of PLM was the identification that PLM belongs to a family of proteins that associate with and modulate Na/K ATPase in various tissues (Sweadner and Rael, 2000). The FXYD protein family, named for a conserved Pro-Phe-X-Tyr-Asp motif in the extracellular N-terminus domain, also includes the NKA γ-subunit (FXYD2) and the corticosteroid hormone-induced factor, CHIF (FXYD4), both found in the kidney, the mammary tumor marker Mat-8 (FXYD3) found in breast tumors, dysadherin (FXYD5), phosphohippolin (FXYD6) and FXYD7 (Geering, 2006). PLM (FXYD1) is the only FXYD protein that is highly expressed in the heart and is also unique within the family in that it has multiple phosphorylation sites at its cytosolic carboxy-terminus. PLM is phosphorylated at Ser68 by PKA and PKC, and at Ser63 by PKC.
PLM co-immunoprecipitates with the α-subunit of NKA both in native tissues and when co-expressed in heterologous systems (Crambert et al. 2002, Feschenko et al. 2003, Fuller et al. 2004, Bossuyt et al. 2005, Silverman et al. 2005), suggesting that these two proteins are physically associated in the membrane. There is some controversy regarding the isoform-specificity of the NKA α-subunit interaction with PLM. Bossuyt et al. (2005) found that NKA-α1, NKA-α2 and NKA-α3 isoforms all co-immunoprecipitate PLM in rabbit heart. This was also the case in heterologous expression systems (Crambert et al. 2002). In contrast, Silverman et al. (2005) reported that NKA-α1 but not NKA-α2 co-immunoprecipitate with PLM in guinea-pig ventricular myocytes. PLM phosphorylated by PKA or PKC remains associated with NKA (Fuller et al. 2004, Bossuyt et al. 2005, Bibert et al. 2008).
We have recently investigated (Bossuyt et al. 2006) the interaction between PLM and NKA α-subunit using fluorescence resonance energy transfer (FRET). FRET is a sensitive indicator of protein interaction and the physical distance and relative orientation of two fluorophores, a donor and an acceptor. We co-expressed NKA- α1-CFP (donor) and PLM-YFP (acceptor) in HEK293 cells and measured FRET as an increase in the fluorescence of the donor upon acceptor photobleaching or as the ratio between the fluorescence of the acceptor and that of the donor when exciting the donor. We found robust FRET between NKA and PLM that was largely prevented when over-expressing unlabeled PLM. Based on the FRET efficiency measured, we estimated that the NKA-PLM intermolecular distance is likely <5 nm (Bossuyt et al. 2006). Phosphorylation of PLM by either PKA or PKC reduced the FRET significantly (by 80-90%), suggesting that although PLM and NKA do not physically dissociate upon phosphorylation, as indicated by co-IP experiments, their interaction is altered.
Studies in heterologous expression systems show that PLM significantly reduces the Na affinity and, to a lesser extent, the K affinity of NKA and has no effect on the NKA maximal transport rate, Vmax (Crambert et al. 2002, Lifshitz et al. 2006). We measured the rate of NKA-mediated Na extrusion as a function of [Na]i in intact cardiac myocytes isolated from WT and PLM-KO mice (Despa et al. 2005). We found that the apparent Km for internal Na was 4-5 mM higher in myocytes from WT than PLM-KO mice (Figure 3) while Vmax was not significantly different. A 4-5 mM difference in the Km may seem small, but because the Km is in the physiological [Na+]i range in cardiac myocytes, Na-pump function can be increased nearly 2-fold. This is enough to significantly affect the activity of the Na/Ca exchanger and therefore intracellular Ca and contractility (Bers 2001). Thus, endogenous PLM also significantly reduces the NKA affinity for intracellular Na and therefore inhibits NKA in the heart. A possible effect on the Vmax is still controversial.
We also determined the effect of β-AR stimulation on Na-pump function in ventricular myocyte from WT and PLM knockout (KO) mice (Despa et al. 2005). In WT mice, ISO caused phosphorylation of PLM at Ser-68 and stimulated NKA by inducing a leftward shift in the Na-activation curve. With ISO, the apparent Km for internal Na in myocytes from WT mice became comparable to that found in PLM-KO mice (Figure 3). In contrast, ISO had no significant effect on Na/K pump function in myocytes from PLM-KO mice. Thus, PLM phosphorylation is necessary for β-adrenergic activation to stimulate NKA. A recent study on the NKA expressed in Xenopus oocytes (Bibert et al. 2008) confirmed this conclusion. β-AR activation stimulated NKA only when PLM was co-expressed. No effect was observed when PLM mutants that cannot be phosphorylated by PKA (S68A and S63D/Ser68D) were co-expressed with NKA. Thus, unphosphorylated PLM inhibits NKA, mostly by reducing its affinity for internal Na, while PLM phosphorylation enhances NKA activity by relieving this inhibition. This mechanism is similar to the way phospholamban modulates SERCA, a P-type pump closely related to NKA (see Figure 3).
PLM-mediated Na/K-ATPase Activation limits the Sympathetic Inotropy and Arrhythmias
During β-AR stimulation in the sympathetic fight-or-flight response, Na influx into cardiac myocytes is greatly increased. This is due to more frequent Na current (which may also be larger owing to PKA phosphorylation) and Ca transient-driven Na entry via NCX. The larger Ca transients during β-AR stimulation drive greater Ca extrusion, and thus greater Na influx, via NCX at each beat (even without any direct β-AR effect on NCX). Looked at another way, greater Ca current enhances Ca influx upon β-AR stimulation, and Ca extrusion (coupled to Na influx via NCX) must also be increased to attain Ca flux balance. This increase in Na influx could increase [Na]i. The elevated [Na]i may add another component to the inotropic effect of β-AR stimulation by favoring more Ca influx and limiting Ca efflux through the Na/Ca exchanger, mechanistically analogous to cardiac glycosides-induced inotropy. Phosphorylation of PLM and the consequent enhancement of Na/K pump activity may limit the rise in [Na]i and thus may limit the inotropic state.
To investigate this, we recently measured the effect of β-AR activation on [Na]i and [Ca]i in isolated myocytes from WT and PLM-KO mice (Despa et al. 2008). [Na]i is determined by a balance of Na efflux via NKA and Na influx via several mechanisms (NCX, Na channels, Na/H exchanger). Resting [Na]i was comparable in myocytes from the WT and PLM-KO mice under basal conditions (time 0 in Figure 4). This might be in part because the increased Na affinity of NKA in PLM-KO mice is offset by a lower number of NKA molecules (Despa et al. 2005), resulting in relatively normal [Na]i and NKA rate at that [Na]i. An increased Na influx in PLM-KO mice might also balance the effect of enhanced Na affinity of NKA. The mean resting Na influx, calculated as the initial rate of [Na]i rise upon NKA blockade, was slightly but not significantly higher in PLM-KO vs. WT myocytes (Despa et al. 2005).
Figure 4.
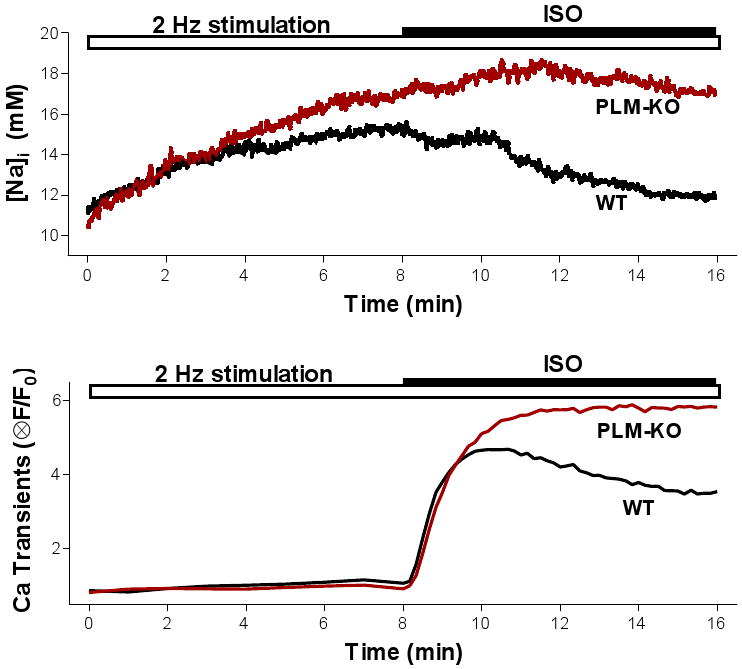
Time course of changes in [Na]i (top panel) and Ca transient amplitude (bottom panel) induced by field stimulation (2 Hz) and 1 μM isoproterenol (ISO) in ventricular myocytes from WT and PLM-KO mice. Replotted from Despa et al. (2008).
Myocytes were paced at 2 Hz, to simulate the pure chronotropic effect of β-AR stimulation. As expected, [Na]i increased similarly upon pacing in both WT and PLM-KO mice (from ∼11 to 15 mM; see Figure 4). When 1 μM isoproterenol was applied in WT mice, [Na]i started to decrease within 2 min (Figure 4). This caused steady-state [Na]i in the presence of isoproterenol to return near the resting level. In contrast, in PLM-KO mice isoproterenol did not change [Na+]i significantly. Thus, PLM-dependent enhancement of NKA activity limits [Na]i rise during β-AR activation in ventricular myocytes.
Isoproterenol also increased Ca transient amplitude in both WT and PLM-KO mice (Figure 4B; Despa et al. 2008). However, the Ca transient increase in PLM-KO was monotonic, whereas in WT mice there was a secondary decline in Ca transient amplitude toward a new steady state, and that paralleled the decline in [Na]i seen in WT. We infer that the decline in Ca transient amplitude that follows the initial increase in WT mice is due to the decline in [Na]i and the effect that this has on the Na/Ca exchanger and SR Ca content. The 2-min delay in [Na]i decline vs. activation of Ca cycling (Figure 4) indicates that β-AR dependent activation of Na/K pump (and the resultant decline in [Na]i lags behind activation of Ca cycling. This agrees with data indicating a slower time course for PKA phosphorylation of PLM at Ser68 (starts in 30 sec, reaching maximum in ∼15 min; Despa et al. 2005) compared to key proteins involved in Ca cycling such as phospholamban and L-type Ca channels.
At steady-state, Ca transients were 5.2±0.4 (mean±SE) times larger in the presence of ISO than under control conditions in WT mice, and increased by more (7.1±0.5 fold) in PLM-KO mice (Despa et al. 2008). Interestingly, Ca transient amplitude was similar in myocytes from WT and PLM-KO mice at the time of the peak in myocytes from WT mice (2 min after ISO addition). This suggests similar β-AR stimulation effects on SERCA (via PLB phosphorylation), L-type Ca channels and ryanodine receptors between WT and PLM-KO mice. The higher Ca transient during β-AR in PLM-KO mouse was also associated with higher SR Ca content (assessed by caffeine-induced Ca transients) and an increased propensity for spontaneous Ca transients and contractions compared to WT mice (Despa et al. 2008). We conclude that the higher [Na]i in the PLM-KO with isoproterenol causes the higher SR Ca content (via NCX), Ca transient amplitude, and arrhythmogenic spontaneous activity.
These data indicate that the PLM-dependent activation of the NKA limits the rise in [Na]i and Ca transient amplitude during β-AR stimulation. This enhancement of NKA activity may thus be a heretofore underappreciated integral part of the sympathetic fight-or-flight response of the heart. In particular, enhancing Na extrusion may better keep up with the higher level of Na influx (caused by the combined inotropic and chronotropic effects on the heart). While this may limit the maximal inotropic effect of β-AR activation physiologically, it would also be protective with respect to triggered arrhythmias. Indeed, the inability of NKA to be activated by β-AR stimulation in the PLM-KO mouse leads to excessive [Na]i elevation during sympathetic activation, which has both the benefits and the risks associated with NKA inhibition by cardiac glycosides (inotropy, but enhanced arrhythmogenesis). Thus, the physiological role of PLM may be to prevent Ca overload and triggered arrhythmias by limiting the rise of [Na]i during normal sympathetic stimulation of the heart. Indeed, PLM creates a rapidly recruitable NKA functional reserve during sympathetic activation. PKC also phosphorylates PLM at both S68 and S63, both of which stimulate NKA activity in myocytes (Han et al. 2007). So, co-activation of myocyte α-ARs may also contribute to the overall sympathetic response of cardiac myocytes described here for β-AR.
PLM has also been reported to interact with NCX and inhibit NCX function, especially when PLM is phosphorylated by PKA (Zhang et al. 2006a). We have not detected robust PLM-NCX interaction in our own immunoprecipitation or FRET studies, and NCX inhibition would be expected to enhance rather than limit the β-AR-induced inotropy described in Figure 4. While we cannot rule out PLM effects on NCX, our current view is that any such effects must be of smaller significance compared to the effects on NKA discussed here.
PLM expression and phosphorylation level is altered in heart disease. Total PLM is lower and a larger fraction of it is phosphorylated at Ser68 in both human and rabbit chronic failing hearts (Bossuyt et al. 2005). Both the lower PLM amount and increased fraction of PLM phosphorylated would diminish the inhibitory effect of PLM on NKA function. NKA protein expression is also reduced in HF, but to a lesser extent than PLM. This might explain why we found that basal NKA-mediated Na extrusion was not appreciably different in myocytes from failing vs. control rabbit hearts (i.e. fewer NKA molecules, but where the average NKA molecule is functioning closer to Vmax). This scenario would tend to limit the NKA reserve activity in the failing heart, along with the fact that [Na]i is already elevated in heart failure (Despa et al. 2002). The smaller NKA reserve in heart failure could result in greater [Na]i elevation during sympathetic activity, which might contribute to an increased propensity for β-AR-induced triggered arrhythmias. Not all pathophysiological states may be the same. For example, postmyocardial infarction rat hearts (in the short term) show increased PLM expression (Zhang et al. 2006b).
Conclusion
The increase in cardiac performance with sympathetic stimulation is an orchestrated combination of intrinsic inotropic, lusitropic and chronotropic effects, mediated in part by activation of β-adrenergic receptors and protein kinase A. Stimulation of the Na/K-ATPase (mediated by phosphorylation of phospholemman) limits the rise in [Na]i and Ca transient amplitude and may reduce the propensity for triggered arrhythmias during stress. In this way, β-adrenergic activation of Na/K-ATPase is an integral part of the sympathetic fight-or-flight response in the heart.
Acknowledgments
Supported by grants from the National Institutes of Health HL-81526 and HL-64724 (DMB) and American Heart Association (0735084N to SD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beevers AJ, Kukol A. Secondary structure, orientation and oligomerization of phospholemman, a cardiac transmembrane protein. Protein Sci. 2006;15:1127–1132. doi: 10.1110/ps.051899406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JR, Lloyd D, Curl CL, et al. Cell volume control in phospholemman (PLM) knockout mice: Do cardiac myocytes demonstrate a regulatory volume decrease and is this influenced by deletion of PLM? Exp Physiol. 2009;94:330–343. doi: 10.1113/expphysiol.2008.045823. [DOI] [PubMed] [Google Scholar]
- Bers DM. Excitation-contraction coupling and cardiac contractile force. 2nd. Dordrecht, The Netherlands: Kluwer Academic Press; 2001. p. 427. [Google Scholar]
- Bibert S, Roy S, Schaer D, et al. Phosphorylation of phospholemman (FXYD1) by protein kinases A and C modulates distinct Na,K-ATPase isozymes. J Biol Chem. 2008;283:476–486. doi: 10.1074/jbc.M705830200. [DOI] [PubMed] [Google Scholar]
- Bossuyt J, Ai X, Moorman RJ, et al. Expression and phosphorylation the Na+-pump regulatory subunit phospholemman in heart failure. Circ Res. 2005;97:558–565. doi: 10.1161/01.RES.0000181172.27931.c3. [DOI] [PubMed] [Google Scholar]
- Bossuyt J, Despa S, Martin JL, et al. Phospholemman phosphorylation alters its association with the Na-pump as assessed by FRET. J Biol Chem. 2006;281:32765–32773. doi: 10.1074/jbc.M606254200. [DOI] [PubMed] [Google Scholar]
- Bossuyt J, Despa S, Han F, et al. Isoform-specificity of the Na/K-ATPase association and regulation by phospholemman. J Biol Chem. 2009 doi: 10.1074/jbc.M109.047357. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crambert G, Fuzesi M, Garty H, et al. Phospholemman (FXYD1) associates with Na+,K+-ATPase and regulates its transport properties. Proc Natl Acad Sci USA. 2002;99:11476–11481. doi: 10.1073/pnas.182267299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran J, Hinton MJ, Rios E, et al. β-adrenergic enhancement of sarcoplasmic reticulum Ca leak in cardiac myocytes is mediated by calcium/calmodulin – dependent protein kinase. Circ Res. 2007;100:391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- Despa S, Bossuyt J, Han F, et al. Phospholemman -phosphorylation mediates the β-adrenergic effects on Na/K pump function in cardiac myocytes. Circ Res. 2005;97:252–259. doi: 10.1161/01.RES.0000176532.97731.e5. [DOI] [PubMed] [Google Scholar]
- Despa S, Islam MA, Weber CR, et al. Intracellular Na+ concentration is elevated in heart failure, but Na/K-Pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- Despa S, Tucker AL, Bers DM. PLM-mediated activation of Na/K – ATPase limits [Na]i and inotropic state during β-adrenergic stimulation in mouse ventricular myocytes. Circulation. 2008;117:1849–1855. doi: 10.1161/CIRCULATIONAHA.107.754051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller W, Eaton P, Bell JR, et al. Ischemia-induced phosphorylation of phospholemman directly activates rat cardiac Na+/K+-ATPase. FASEB J. 2004;18:197–199. doi: 10.1096/fj.03-0213fje. [DOI] [PubMed] [Google Scholar]
- Ganesan AN, Maack C, Johns DC, et al. β-adrenergic stimulation of L-type Ca2+ channels in cardiac myocytes requires the distal carboxyl terminus of α1C but not serine 1928. Circ Res. 2006;98:e11–18. doi: 10.1161/01.RES.0000202692.23001.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Wymore R, Wymore RT, et al. Isoform-specific regulation of the sodium pump by α- and β-adrenergic agonists in the guinea-pig ventricle. J Physiol. 1999;516:377–383. doi: 10.1111/j.1469-7793.1999.0377v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geering K. FXYD proteins: new regulators of Na-K-ATPase. Am J Physiol Renal Physiol. 2006;290:F241–250. doi: 10.1152/ajprenal.00126.2005. [DOI] [PubMed] [Google Scholar]
- Ginsburg KS, Bers DM. Isoproterenol does not enhance Ca-dependent Na/Ca exchange current in intact rabbit ventricular myocytes. J Mol Cell Cardiol. 2005;39:972–981. doi: 10.1016/j.yjmcc.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Ginsburg KS, Bers DM. Modulation of excitation–contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca load and ICa trigger. J Physiol. 2004;556:463–480. doi: 10.1113/jphysiol.2003.055384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glitsch HG. Electrophysiology of the sodium-potassium-ATPase in cardiac cells. Physiol Rev. 2001;81:1791–1826. doi: 10.1152/physrev.2001.81.4.1791. [DOI] [PubMed] [Google Scholar]
- Guo T, Zhang T, Mestril R, et al. Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Schulman H, Kim J, et al. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005;171:537–47. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme JT, Yarov-Yarovoy V, Lin TW, et al. Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. J Physiol. 2006;576:87–102. doi: 10.1113/jphysiol.2006.111799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadambi VJ, Ponniah S, Harrer JM, et al. Cardiac-specific overexpression of phospholamban alters calcium kinetics and resultant cardiomyocyte mechanics in transgenic mice. J Clin Invest. 1996;97:533–539. doi: 10.1172/JCI118446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp TJ, Hell JW. Regulation of cardiac L-Type calcium channels by protein kinase A and protein kinase C. Circ Res. 2000;87:1095–1102. doi: 10.1161/01.res.87.12.1095. [DOI] [PubMed] [Google Scholar]
- Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res. 2005;66:12–21. doi: 10.1016/j.cardiores.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Lemke T, Welling A, Christel CJ, et al. Unchanged beta-adrenergic stimulation of cardiac L-type calcium channels in Cav1.2 phosphorylation site S1928A mutant mice. J Biol Chem. 2008;283:34738–34744. doi: 10.1074/jbc.M804981200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Desantiago J, Chu G, et al. Phosphorylation of phospholamban and troponin I in β-adrenergic-induced acceleration of cardiac relaxation. Am J Physiol. 2000;278:H769–H779. doi: 10.1152/ajpheart.2000.278.3.H769. [DOI] [PubMed] [Google Scholar]
- Li L, Satoh H, Ginsburg KS, et al. The effect of Ca–calmodulin-dependent protein kinase II on cardiac excitation–contraction coupling in ferret ventricular myocytes. J Physiol. 1997;501:17–32. doi: 10.1111/j.1469-7793.1997.017bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifshitz Y, Lindzen M, Garty H, et al. Functional interactions of phospholemman (PLM) (FXYD1) with Na+,K+-ATPase. Purification of α1/β1/PLM complexes expressed in Pichia pastoris. J Biol Chem. 2006;281:15790–15799. doi: 10.1074/jbc.M601993200. [DOI] [PubMed] [Google Scholar]
- Lin X, Jo H, Sakakibara Y, et al. Beta-adrenergic stimulation does not activate Na+/Ca2+ exchange current in guinea pig, mouse, and rat ventricular myocytes. Am J Physiol Cell Physiol. 2006;290:C601–C608. doi: 10.1152/ajpcell.00452.2005. [DOI] [PubMed] [Google Scholar]
- Maier LS, Zhang T, Chen L, et al. Transgenic CaMKII-δC overexpression uniquely alters cardiac myocyte Ca handling: reduced SR Ca load, but increased fractional release. Circ Res. 2003;92:904–911. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, et al. Phosphorylation-dependent regulation of ryanodine receptors: a novel role for leucine/isoleucine zippers. J Cell Biol. 2001;153:699–708. doi: 10.1083/jcb.153.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Moorman JR, Ackerman SJ, Kowdley GC, et al. Unitary anion currents through phospholemman channel molecules. Nature. 1995;377:737–740. doi: 10.1038/377737a0. [DOI] [PubMed] [Google Scholar]
- Morth JP, Pedersen BP, Toustrup-Jensen MS, et al. Crystal structure of the sodium – potassium pump. Nature. 2007;450:1043–1049. doi: 10.1038/nature06419. [DOI] [PubMed] [Google Scholar]
- Palmer CJ, Scott BT, Jones LR. Purification and complete sequence determination of the major plasma membrane substrate for cAMP-dependent protein kinase and protein kinase C in myocardium. J Biol Chem. 1991;266:11126–11130. [PubMed] [Google Scholar]
- Presti CF, Jones LR, Lindemann JP. Isoproterenol-induced phosphorylation of a 15-kilodalton sarcolemmal protein in intact myocardium. J Biol Chem. 1985;260:3860–3867. [PubMed] [Google Scholar]
- Sham JSK, Jones LR, Morad M. Phospholambam mediates the β-adrenergic-enhanced Ca2+ uptake in mammalian ventricular myocytes. Am J Physiol. 1991;261:H1344–H1349. doi: 10.1152/ajpheart.1991.261.4.H1344. [DOI] [PubMed] [Google Scholar]
- Silverman BD, Fuller W, Eaton P, et al. Serine 68 phosphorylation of phospholemman: acute isoform-specific activation of cardiac Na/K ATPase. Cardiovasc Res. 2005;65:93–103. doi: 10.1016/j.cardiores.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Stange M, Xu L, Balshaw D, et al. Characterization of recombinant skeletal muscle (Ser-2843) and cardiac muscle (Ser-2809) ryanodine receptor phosphorylation mutants. J Biol Chem. 2003;278:51693–51702. doi: 10.1074/jbc.M310406200. [DOI] [PubMed] [Google Scholar]
- Sweadner KJ, Feschenko MS. Predicted location and limited accessibility of protein kinase A phosphorylation site on Na-K-ATPase. Am J Physiol. 2001;280:C1017–C1026. doi: 10.1152/ajpcell.2001.280.4.C1017. [DOI] [PubMed] [Google Scholar]
- Sweadner KJ, Rael E. The FXYD gene family of small ion transport regulators or channels: cDNA sequence, protein signature sequence, and expression. Genomics. 2000;68:41–56. doi: 10.1006/geno.2000.6274. [DOI] [PubMed] [Google Scholar]
- Tong CW, Stelzer JE, Greaser ML, et al. Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ Res. 2008;103:974–982. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku T, Kurzydlowski K, Tada M, et al. Identification of regions in the Ca2+-ATPase of the sarcoplasmic reticulum that affect functional association with phospholamban. J Biol Chem. 1993;268:2809–2815. [PubMed] [Google Scholar]
- Valdivia HH, Kaplan JH, Ellis-Davies GCR, et al. Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science. 1995;267:1997–2000. doi: 10.1126/science.7701323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner JR, Ginsburg KS, Mitton BA, et al. Phospholamban overexpression in rabbit ventricular myocytes does not alter sarcoplasmic reticulum calcium transport. Am J Physiol – Heart Circ Physiol. 2009;296:H698–H703. doi: 10.1152/ajpheart.00272.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Zhu W, Wang S, et al. Sustained β1-adrenergic stimulation modulates cardiac contractility by Ca2+/calmodulin kinase signaling pathway. Circ Res. 2004;95:798–806. doi: 10.1161/01.RES.0000145361.50017.aa. [DOI] [PubMed] [Google Scholar]
- Xiao B, Sutherland C, Walsh MP, et al. Protein kinase A phosphorylation at Serine-2808 of the cardiac Ca2+-release channel (ryanodine receptor) does not dissociate 12.6-kDa FK506-binding protein (FKBP12.6) Circ Res. 2004;94:487–495. doi: 10.1161/01.RES.0000115945.89741.22. [DOI] [PubMed] [Google Scholar]
- Yue DT, Herzig S, Marban E. B-Adrenergic stimulation of calcium channels occurs by potentiation of high-activity gating modes. Proc Natl Acad Sci USA. 1990;87:753–757. doi: 10.1073/pnas.87.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XQ, Ahlers BA, Tucker AL, et al. Phospholemman inhibition of the cardiac Na+/Ca2+ exchanger; role of phosphorylation. J Biol Chem. 2006a;281:7784–7792. doi: 10.1074/jbc.M512092200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XQ, Moorman JR, Ahlers BA, et al. Phospholemman overexpression inhibits Na+-K+-ATPase in adult rat cardiac myocytes: relevance to decreased Na+ pump activity in post-infarction myocytes. J Appl Physiol. 2006b;100:212–220. doi: 10.1152/japplphysiol.00757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]