

Abstract
The peroxiredoxins (Prxs) are conserved antioxidant proteins that utilize cysteine as the primary site of oxidation during the reduction of peroxides. Many organisms have more than one isoform of Prx. Deletion of TSA1, one of five Prxs in yeast Saccharomyces cerevisiae, results in accumulation of a broad spectrum of mutations including gross chromosomal rearrangements. Deletion of TSA1 is synthetically lethal with mutations in RAD6 and several key genes involved in DNA double-strand break repair. Here we have examined the function of human peroxiredoxins PrxI and PrxII, which share a high degree of sequence identity with Tsa1, by expressing them in S. cerevisiae cells under control of the native TSA1 promoter. We found that expression of PrxI, but not PrxII, was capable of complementing a tsa1Δ mutant for a variety of defects including genome instability, the synthetic lethality observed in rad6Δ tsa1Δ and rad51Δ tsa1Δ double mutants and mutagen sensitivity. Moreover, expression of either Tsa1 or PrxI prevented Bax-induced cell death. These data indicate that PrxI is an ortholog of Tsa1. PrxI and Tsa1 appear to act on the same substrates in vivo and share similar mechanisms of function. The observation that PrxI is involved in suppressing genome instability and protecting against cell death potentially provides a better understanding of the consequences of PrxI dysfunction in human cells. The S. cerevisiae system described here could provide a sensitive tool to uncover the mechanisms that underlie the function of human Prxs.
Keywords: peroxiredoxin, oxidative stress, genome instability, S. cerevisiae
Introduction
Reactive oxygen species (ROS), including the superoxide anion (O•), hydrogen peroxide (H2O2) and hydroxyl radical (OH•) are oxygen-derived reactive molecules. ROS are generated as byproducts of normal metabolism, particularly mitochondrial respiration, and as a result of exposure to various environmental agents (1). ROS are employed as intracellular signaling molecules by pathways involved in proliferation, stress response and apoptosis (2, 3). When ROS levels become too high, oxidative stress results. Failure to respond to oxidative stress can result in severe damage to lipids, proteins and DNA and ultimately lead to cell death. Pathologically increased ROS generation is linked to various human diseases (4, 5).
Peroxiredoxins (Prxs), previously termed the thioredoxin peroxidases, have received considerable attention in recent years as a new and expanding family of thiol-specific antioxidant proteins (6, 7). Prxs are abundant, ubiquitously distributed peroxidases that utilize cysteine (Cys) as the primary site of oxidation during the reduction of peroxides. Many organisms have multiple Prxs, with five Prxs having been identified in yeast Saccharomyces cerevisiae and six in human cells (6, 7). Prx isoforms are distributed differently within the cell. Based on structural and mechanistic data, Prxs can be divided into three subgroups: 2-Cys Prx proteins, atypical 2-Cys Prx proteins and 1-Cys Prx proteins. The 2-Cys Prxs, which contain both the N- and C-terminal-conserved Cys residues (peroxidatic and resolving Cys, respectively), and require both of them for catalytic function, constitute the largest class of Prxs. S. cerevisiae Tsa1 and Tsa2 and human PrxI, II, III and IV belong to the 2-Cys Prx subgroup.
The genetic consequences of deletion of S. cerevisiae genes encoding the 2-Cys Prxs Tsa1 and Tsa2 and three other peroxiredoxins (cTPxIII, nTPx and mTPx) have been investigated (8–11). Tsa1 is the most significant contributor to genome stability and prevents a broad spectrum of mutations including gross chromosomal rearrangements (GCRs) as well as base substitution and frameshift mutations (9). Tsa1 is also essential for cell survival in the absence of functional recombinational repair or the Rad6-mediated post-replication repair pathway (10). Recombinational repair involving Rad51 and Rad52 is a key mechanism for repair of double-strand DNA breaks while the Rad6-mediated post-replication repair supports both error-free and error-prone pathways that bypass replication-blocking DNA lesions (12, 13). The spontaneous genome instability and cell death associated with deletion of TSA1 appears to be predominantly due to endogenous ROS produced by oxygen metabolism (14). These dramatic phenotypes distinguish Tsa1 from other S. cerevisiae Prxs. Interestingly, mice lacking PrxI, which is highly homologous to Tsa1 in amino acid sequence, have increased erythrocyte ROS and increased risk of developing hemolytic anemia and several malignant cancers (15).
Given the importance of Tsa1 in maintaining genome stability and cell survival, and that human PrxI and PrxII are both cytoplasmic and share a high degree of sequence identity with Tsa1 (Fig. 1), we have explored the ability of human PrxI and PrxII to complement a variety of defects caused by a tsa1Δ mutation. Our results support the idea that PrxI, but not PrxII, is an ortholog of Tsa1 and is involved in maintaining genome stability and preventing cell death. Because PrxI and Tsa1 appear to act on the same substrates in vivo and share a similar mechanism of function, the S. cerevisiae system could provide a sensitive tool for the functional analysis of human Prxs.
Figure 1.
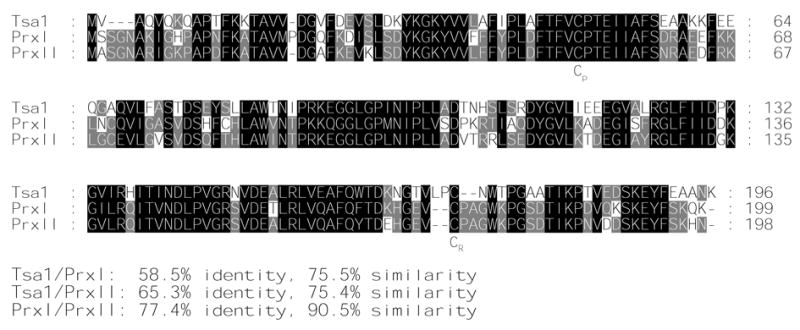
Alignment of the amino acid sequences of Tsa1, PrxI and PrxII using the program ClustalW. Residues identical in all three proteins are indicated by black boxes and those conserved in only two proteins by shaded boxes. The position of the conserved peroxidatic and resolving cysteines (Cp and CR) are indicated. The percentages of identity and similarity between sequences are shown at the bottom.
Materials and Methods
Gene and protein designations
The human peroxiredoxin I and II genes are referred to as PRXI and PRXII respectively, and the proteins are referred to as PrxI and PrxII. The homologous S. cerevisiae gene is TSA1, while the protein is Tsa1. The tsa1Δ is a complete deletion of the TSA1 gene. Strains in which TSA1 has been replaced by human PRXI or PRXII and expressed under control of the TSA1 promoter are designated tsa1Δ::PRXI or tsa1Δ::PRXII, respectively.
Media and strains
S. cerevisiae strains were grown in standard media including yeast extract peptone dextrose medium (YPD) or synthetic complete medium (SC) lacking appropriate amino acids as indicated (16). Cycloheximide resistant strains (CyhR) containing mutations in the CYH2 gene were selected on YPD agar plates containing 10 μg/mL cycloheximide. Canavanine resistant mutants (Canr) caused by inactivation of the CAN1 gene were selected on SC-arginine dropout plates containing 60 mg/L canavanine. Canavanine- and 5-fluoroorotic acid (5FOA)- resistant mutants (Canr-5FOAr) resulted from the loss of the region including CAN1 and URA3 on chromosome V were selected on SC-arginine and -uracil dropout plates containing 60 mg/L canavanine, 1 g/L 5FOA and 50 mg/L uracil (17).
The strains used for this study were all isogenic to the S288c based parental strain RDKY3615 MATa, ura3–52, leu2Δ1, trp1Δ63, his3Δ200, lys2ΔBgl, hom3–10, ade2Δ1, ade8, hxt13::URA3 (17). Gene replacements were made by standard PCR-based homology-directed methods (16). The construction of tsa1Δ::PRXI and tsa1Δ::PRXII strains in which the entire TSA1 ORF was replaced by PRXI or PRXII coding sequences under control of the native TSA1 promoter is illustrated schematically in Figure 2A. First, the RDKY3615 strain, auxotrophic for tryptophan and sensitive to cycloheximide (Trp− CyhS), was plated onto YPD plates containing 10 μg/mL cycloheximide to select spontaneous CyhR colonies. Genetic analysis confirmed that the CyhR phenotype was caused by a cyh2 mutation and this cyh2 mutant strain was designated MEHY1537 (Trp− CyhR). Second, a fragment containing the TRP1 and CYH2 genes flanked by sequences homologous to the upstream and downstream sequence of TSA1, obtained by PCR amplifying the TRP1-CYH2 genes present on plasmid p443 (18), was introduced into the MEHY1537 strain to replace the full TSA1 locus, resulting in strain MEHY1539 (Trp+ CyhS). Since wild-type CYH2 gene is dominant to the cyh2 mutation, cells containing wild-type CYH2 gene or both CYH2 and cyh2 exhibit a CyhS phenotype. Third, cDNA from the Epstein-Barr virus-transformed human lymphoblastoid B cell line D1 (19) was used as a template to amplify the entire human PRXI or PRXII coding sequence, which was then cloned between the BglII-KpnI restriction sites of the vector pLitmus28 (Ozyme, Saint Quentin Yvelines, France), resulting in pLitmus28-PRXI and pLitmus28-PRXII. Fragments containing PRXI or PRXII coding sequences flanked by sequences homologous to the upstream and downstream sequence of TSA1, generated by PCR using pLitmus28-PRXI or pLitmus28-PRXII as a template, were used to transform MEHY1539 (Trp+ CyhS) to replace TRP1-CYH2 at the TSA1 locus yielding the strains MEHY1384 (Trp− CyhR) and MEHY1389 (Trp− CyhR) in which the PRXI or PRXII coding sequence, respectively, is under the control of the native TSA1 promoter. Correct integration of PRXI or PRXII at TSA1 locus was confirmed by genomic PCR and by DNA sequencing.
Figure 2.
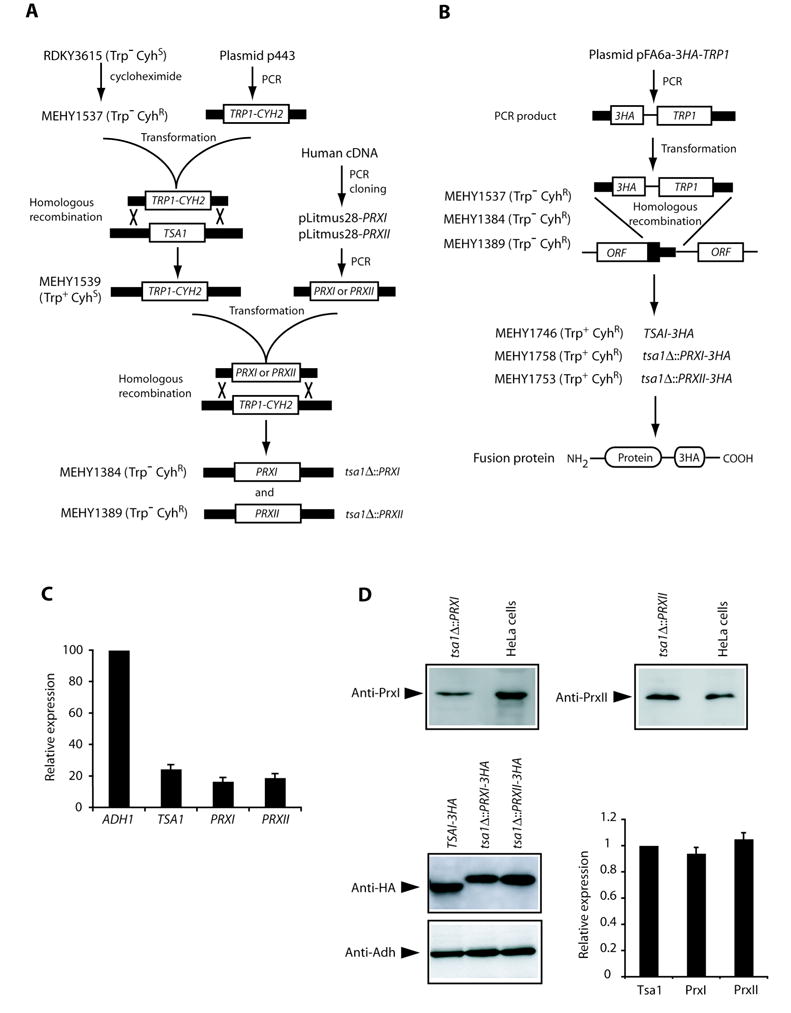
Construction of S. cerevisiae strains and analysis of TSA1, PRXI and PRXII expression. A, schematic illustration of construction of the S. cerevisiae strains expressing PRXI or PRXII coding sequence under control of the endogenous TSA1 promoter as described in Materials and Methods. Gene replacement cassettes containing nutritional or drug-selectable markers or desired coding sequences (PRXI or PRXII) were tailed by PCR with sequence homologous to regions flanking the chromosomal target ORF to allow targeted recombination. B, strategy for HA-tagging Tsa1, PrxI and PrxII proteins. PCR products containing the 3HA tag and a selectable marker gene were inserted at the C-terminus of each gene by homologous recombination. C, analysis of TSA1, PRXI and PRXII expression by qRT-PCR. Expression of ADH1 was used as an internal control. The ratio of TSA1, PRXI or PRXII mRNA levels to that of ADH1 was calculated to determine relative expression. Columns, mean of five determinations; bars, SD. D, analysis of protein expression by Western blot. PrxI and PrxII proteins were detected using specific anti-PrxI or anti-PrxII antibodies in lysates prepared from the indicated yeast strains and human HeLa cells for a positive control. Anti-PrxI antibody detected the same sized band between 20.6–28.9 kDa in extracts from both the tsa1Δ::PRXI strain and HeLa cells (top left panel). Wild-type and tsa1Δ strain extracts did not show any immunoreactivity (data not shown). Similarly, anti-PrxII antibody detected a specific band of expected size in extracts from both the tsa1Δ::PRXII strain and HeLa cells (top right panel). 3HA-tagged Tsa1, PrxI and PrxII were detected by anti-HA antibody and Adh (encoded by ADH1) served as a loading control (bottom left panel). For each experiment, the relative levels of Tsa1, PrxI or PrxII versus Adh were calculated and the ratio of Tsa1 to Adh was set as 1, with relative values of other ratios calculated accordingly (bottom right panel). Columns, mean of six independent experiments; bars, SD.
To construct the S. cerevisiae strains expressing influenza virus hemagglutinin (HA) epitope-tagged Tsa1, PrxI or PrxII proteins, we designed three gene-specific forward oliogonucleotide primers and one common reverse primer (Fig. 2B). Each of the gene-specific forward primer has a shared 3′ end that allows for PCR amplification of a 3HA-TRP1 cassette in the plasmid pFA6a-3HA-TRP1 (20), as well as a gene-specific 5′ end that allow for precise integration of the amplified cassettes at the 3′ end of the genomic coding sequence through homologous recombination. The common reverse primer has 3′ end complementary to the 3HA-TRP1 cassette for PCR amplification and a 5′ end homologous to the downstream sequence of TSA1. These amplified cassettes were used to transform corresponding strains MEHY1537, MEHY1384 and MEHY1389 followed by selection on SC-tryptophan dropout plates. Insertion of the cassette and in-frame fusion of the 3HA tag at the C-terminal end of the coding region of each gene was confirmed by genomic PCR and sequencing. Sequences of primers used in strain constructions are available on request.
Quantitative real-time reverse transcription PCR (qRT-PCR)
Wild-type, tsa1Δ::PRXI and tsa1Δ::PRXII strains were grown to mid-exponential phase in YPD medium. Total RNA was extracted with acidic phenol (pH 5) according to an established procedure (21). Extracted RNA was treated with DNase and verified by conventional PCR to ensure the absence of trace DNA in samples. cDNA was generated using iScript cDNA synthesis Kit (Bio-Rad Laboratories, Hercules, CA). We designed gene-specific primers for TSA1, PRXI, PRXII, and ADH1 (encoding alcohol dehydrogenase and used as an internal control). Serial dilutions of cDNA from each strain were amplified using the appropriate primers and iQ SYBR Green Supermix (Bio-Rad). A single melt curve peak was observed for each sample used in data analysis, thus confirming the purity and specificity of all amplified products. The relative ratio of TSA1, PRXI or PRXII expression to ADH1 was calculated using the relative quantity (ΔCT) analysis formula in the Bio-Rad iQ5 Gene Expression Optical System Software. Sequences of gene-specific primers are available upon request.
Western blot analysis
Strains were grown to mid-logarithmic phase in YPD medium and whole cell extracts isolated by trichloroacetic acid (TCA) extraction (22). Protein concentrations were measured by the Bradford procedure (Bio-Rad) using BSA as a standard. Cell extracts were electrophoresed under reducing conditions on 12% SDS-PAGE minigels and electrophoretically transferred to nitrocellulose membrane that was probed with the following primary antibodies: goat anti-PrxI (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-PrxII (LabFrontier, Seoul, Korea), mouse anti-HA (Roche Diagnostics, Mannheim, Germany) or rabbit anti-yeast alcohol dehydrogenase (anti-Adh, Chemicon International, Hampshire, UK). Corresponding horseradish peroxidase-conjugated secondary antibody was detected with the Amersham ECL Western blotting analysis system (GE Healthcare, Buckinghamshire, UK). Human HeLa cell lysate was extracted and processed as described for S. cerevisiae cells and used as a positive control in Western blots. Band intensities were quantitated using ImageJ (http:/rsb.info.nih.gov/ij).
Measurement of Canr and GCR rates
The rate of accumulation of mutations was determined by fluctuation analysis as described previously (17, 23). For fluctuation analysis of Canr mutations, cells were diluted to approximately 100 cells/mL in 15 to 34 independent 2-mL YPD cultures per strain, grown to 1–2 ×108 cells/mL, harvested, washed and resuspended in sterile water. Appropriate dilutions were plated on SC-arginine dropout plates containing canavanine to identify mutations in CAN1, and on YPD for total cell counts. Colonies were counted after three or four days of growth at 30°C. The number of Canr colonies per culture was calculated and the median value for each strain was used to determine the mutation rate as described (24). GCR rates were determined in the same way except that cultures were 5 to 25 mL, depending on the strain, and that the washed cells were plated onto SC-arginine and -uracil dropout plates containing canavanine and 5FOA and added uracil. The 95% confidence intervals for a median rate were calculated based on order statistics with the formula available at http://www.math.unb.ca/~knight/utility/MedInt95.htm. Mutation rates were compared by the Mann-Whitney test using programs available at http:/faculty.vassar.edu/lowry/vshome.html. Canr mutation spectra were determined by PCR amplification and sequencing of the CAN1 gene from independent Canr isolates as previously described (23).
H2O2 and antimycin A sensitivity analysis
Sensitivity to H2O2 and antimycin A were assayed on solid media by spotting 10-fold serial dilutions of mid-logarithmic phase cells onto YPD plates or YPD plates containing H2O2 at indicated concentrations with 2.5 μg/mL antimycin A (Sigma-Aldrich, Steinheim, Germany). Plates were assessed after 2 days of growth at 30°C.
Bax-induced cell death analysis
The URA3 plasmid pCM189-Bax expressing human Bax under control of a Tet-Off promoter (repressed by the addition of doxycycline) was described previously (25). A fragment containing the promoter-Bax cassette was isolated from pCM189-Bax and inserted between the EcoRI-HindIII sites of the YCplac111 vector (26), generating YCplac111-Bax which contains a LEU2 selectable marker. For analysis of Bax-induced cell death, wild-type, tsa1Δ, tsa1Δ::PRXI and tsa1Δ::PRXII strains transformed with YCplac111-Bax were grown to exponential phase (~2 × 107 cells/mL) in SC-leucine supplemented with 10 μg/mL of doxycycline (Sigma-Aldrich, Steinheim, Germany). Then cells were harvested, washed and resuspended at a density of 106 cells/mL in SC-leucine medium without doxycycline. Samples were removed, diluted and plated onto SC-leucine plates supplemented with 10 μg/mL doxycycline. The remaining cultures were allowed to grow for an additional 6 hours before plating onto SC-leucine plates supplemented with doxycycline to repress Bax expression allowing surviving cells to recover and grow. The number of living cells in the cultures was estimated by counting the colonies that had grown after 48 hours. Cell survival percentage is the ratio of the number of colonies obtained in culture after induction of Bax expression for 6 hours to that from the aliquots removed prior to induction.
Results
Expression of human PrxI and PrxII in a tsa1Δ yeast strain
We sought to develop S. cerevisiae strains that would express the full-length PRXI and PRXII genes under the control of the native TSA1 promoter to yield levels of the mature protein comparable to the physiological level of Tsa1. The resulting strains, designated tsa1Δ::PRXI and tsa1Δ::PRXII in which the complete TSA1 ORF (ATG-Stop) was replaced by the PRXI and PRXII coding sequences, respectively, were constructed as illustrated in Figure 2A. qRT-PCR assays confirmed that expression of PRXI and PRXII in yeast was approximately 68% and 77%, respectively, of that of TSA1 (Fig. 2C). The presence of S. cerevisiae-produced PrxI protein in tsa1Δ::PRXI cells and PrxII protein in tsa1Δ::PRXII cells was detected by Western blots using a specific anti-PrxI and anti-PrxII antibodies, respectively (Fig. 2D). To compare the protein levels of yeast-produced PrxI and PrxII and native Tsa1, we tagged the C-terminal ends of Tsa1, PrxI and PrxII with a 3HA epitope tag, enabling the immunodetection of these proteins with the same anti-HA antibody. All three fusion proteins were expressed at a similar level (Fig. 2D). Taken together, we conclude that the S. cerevisiae strains tsa1Δ::PRXI and tsa1Δ::PRXII produce corresponding mRNA and proteins that are comparable to the physiological level of TSA1 transcript and protein.
PrxI significantly decreases Canr and GCR mutation rates of a tsa1Δ mutant
The absence of Tsa1 causes the accumulation of a broad spectrum of mutations as well as GCRs, indicating Tsa1 is a significant contributor to genome stability (9). We first examined the ability of PrxI and PrxII to complement the mutator phenotypes of a tsa1Δ strain using Canr and GCR assays. The Canr assay detects inactivating mutations can occur in the 1.8-kb CAN1 gene, including base substitution and frameshift mutations (9, 23). The GCR assay detects a broad spectrum of genome rearrangements such as translocations, chromosome fusions, large interstitial deletions and terminal deletions with associated de novo telomere additions (17). As previously observed (9), deletion of TSA1 increased the Canr rate and GCR rate compared to a wild-type strain (Table 1). However, the Canr rate and the GCR rate of the tsa1Δ::PRXI strain were significantly reduced compared to the tsa1 mutant (p < 0.01 in both cases; Mann-Whitney test), although the Canr rate was still higher than that of the wild-type strain (p < 0.01, Mann-Whitney test). In order to determine whether PrxI preferentially suppresses some types of mutation produced in the tsa1Δ strain over others, 40 independent Canr mutations arising in the tsa1Δ::PRXI strain were sequenced. We found that 70% of Canr mutations were single-base substitutions, 2.5% double-base substitutions, 20% single base insertion/deletion mutations (primarily deletions), 2.5% large deletion mutations and 5% complex events. Thus, frequencies of the different classes of mutations for the tsa1Δ::PRXI strain were similar to those observed in the Canr mutation spectra of the tsa1Δ strain that was determined previously (9). The frequency of each type of single-base substitution mutation was also determined (data not shown) and was found to be similar to frequencies of the different types of base substitution mutations in the Canr mutation spectra of the tsa1Δ strain (9). Deletion of TSA1 results in a general increase of various types of mutations as the rate of each type of mutation, obtained by multiplying the frequency of each mutation by the total mutation rate, is generally higher than that in wild-type (9). By then calculating the rates of occurrence of each of the different classes of mutations and the different types of base substitution mutations observed in the tsa1Δ::PRXI strain and comparing them to the corresponding rates for the tsa1Δ strain (9), we determined that each type of mutation was suppressed from 2.7- to 4.5-fold by the tsa1Δ::PRXI allele, indicative of a general suppression of various types of mutations resulting due to the tsa1Δ mutation. In contrast to the tsa1Δ::PRXI cells, the Canr mutation rate and the GCR rate of the tsa1Δ::PRXII cells were not significantly reduced compared to that of the tsa1Δ mutant (p > 0.05 in both cases; Mann-Whitney test), although it cannot be excluded that PrxII has some minor effect (Table 1).
Table 1.
Prx I suppresses tsa1-associated Canr and GCR mutation rates
| Strain | Relevant Genotype | Canr rate (× 10−7) | GCR rate (× 10−10) |
|---|---|---|---|
| RDKY3615 | Wild-type | 2.7 (2.1–5.9) | 4.0 (2.6–7.0) |
| RDKY5502 | tsa1Δ | 35.6 (29.1–38.0) | 80.5 (51.7–129.1) |
| MEHY1384 | tsa1Δ::PRXI | 10.3 (9.2–12.2) | 7.3 (3.9–11.3) |
| MEHY1389 | tsa1Δ::PRXII | 26.9 (25.1–35.7) | 51.4 (30.4–83.7) |
| RDKY5504 | ogg1Δ | ND | 97.4 (72.5–145.6) |
| RDKY5505 | ogg1Δ tsa1Δ | ND | 1410.6 (1102.3–2221.2) |
| MEHY1631 | ogg1Δ tsa1Δ::PRXI | ND | 227.9 (167.6–279.9) |
| MEHY1635 | ogg1Δ tsa1Δ::PRXII | ND | 912.8 (886.0–1170,5) |
The numbers in parentheses indicate the low and high values for the 95% confidence interval for each rate obtained by using the confidence interval for the median test.
ND: not determined.
To confirm the observed effects of PrxI and PrxII on GCR formation, we incorporated an ogg1 deletion into each strain to enhance the basal GCR rate and increase the sensitivity of this assay (10). OGG1 encodes a glycosylase that acts in the base excision repair pathway by removing 8-oxo-dG from 8-oxo-dG:C base pairs (27). As previously observed (10), deletion of OGG1 in a tsa1Δ strain causes a synergistic increase in the GCR rate compared to the single mutants (Table 1). Expression of PrxI largely suppressed the GCR rate of the tsa1Δ ogg1Δ strain (p < 0.01, Mann-Whitney test) (Table 1). However, the observed rate was still higher than that of the ogg1Δ single mutant (p < 0.01, Mann-Whitney test). Again, the expression of PrxII did not significantly reduce the GCR rate of tsa1Δ ogg1Δ double mutant (p > 0.05; Mann-Whitney test), although the rate of the tsa1Δ::PRXII strain appeared to be slightly lower than that of the tsa1Δ ogg1Δ double mutant. Taken together, these observations indicate that PrxI is capable of largely replacing the function of Tsa1 in regard to suppression of genome instability, while the effect of PrxII, if any, is not significant.
PrxI restores cellular viability of rad6Δ tsa1Δ and rad51Δ tsa1Δ double mutants
We have previously shown that combining a tsa1Δ mutation with rad6Δ or rad51Δ mutations, among others, results in cell death (10). The synthetic lethality of these double mutants is likely due to excessive ROS-related DNA lesions that occur in the absence of TSA1, in combination with the absence of appropriate repair, as anaerobic growth conditions restore viability of these mutants (14). To further assess the ability of PrxI and PrxII to replace the functions of Tsa1, we tested their ability to complement the synthetic lethality of rad6Δ tsa1Δ and rad51Δ tsa1Δ mutants. Four diploid strains (rad6Δ/RAD6 tsa1Δ/TSA1; rad51Δ/RAD51 tsa1Δ/TSA1; rad6Δ/RAD6 tsa1Δ::PRXI/TSA1; and rad51Δ/RAD51 tsa1Δ::PRXI/TSA1) were sporulated and dissected, and the spore dissection plates were incubated under aerobic conditions for 4 days (Fig. 3A, B). As previously reported (10), a rad6Δ mutation was found to be lethal in combination with tsa1Δ. In contrast, spore products from diploid strains carrying tsa1Δ::PRXI showed an increased frequency of viable spores, indicating complementation. Genotyping of spore colonies confirmed that rad6Δ tsa1Δ::PRXI mutants were viable. A tsa1Δ mutation in combination with a rad51Δ mutation also resulted in lethality as previously reported, and this lethality was entirely restored by expression of PrxI (Fig. 3A, B). Identical experiments were performed with diploid strains containing the tsa1Δ::PRXII allele (rad6Δ/RAD6 tsa1Δ::PRXII/TSA1; rad51Δ/RAD51tsa1Δ::PRXII/TSA1) to analyze the effect of PrxII expression. PrxII was not able to restore the viability of rad6Δ tsa1Δ or rad51Δ tsa1Δ double mutants (Fig. 3C). Therefore, expression of human PrxI but not PrxII complements the synthetic lethality of rad6Δ tsa1Δ and rad51Δ tsa1Δ double mutants.
Figure 3.

Human PrxI complements the synthetic lethality of rad6Δ tsa1Δ and rad51Δ tsa1Δ double mutant strains. Tetrads from heterozygous diploids were dissected and grown on YPD at 30°C for 4 days. A, tetrad dissection plates of heterozygous diploids rad6Δ/RAD6 tsa1Δ/TSA1 and rad51Δ/RAD51 tsa1Δ/TSA1. B, tetrad dissection plates of heterozygous diploids rad6Δ/RAD6 tsa1Δ::PRXI/TSA1 and rad51Δ/RAD51 tsa1Δ::PRXI/TSA1. C, tetrad dissection plates of heterozygous diploids rad6Δ/RAD6 tsa1Δ::PRXII/TSA1 and rad51Δ/RAD51 tsa1Δ::PRXII/TSA1. Circles indicate either the inferred or determined mutants rad6Δ tsa1Δ or rad51Δ tsa1Δ (A)rad6Δ tsa1Δ::PRXI or rad51Δ tsa1Δ::PRXI (B), and rad6Δ tsa1Δ::PRXII or rad51Δ tsa1Δ::PRXII (C).
PrxI complements the sensitivity of tsa1Δ cells to exogenous oxidant insult
The role of Tsa1 in the protection of cells against oxidant-induced killing has been investigated and in most cases, the tsa1Δ mutant shows moderately increased sensitivity to H2O2 (8, 11, 28–30). Antimycin A, a drug that inhibits respiratory complex III and when used alone does not affect cell growth, enhances sensitivity of the tsa1Δ mutant to H2O2 (30). We therefore compared the sensitivities of wild-type, tsa1Δ, tsa1Δ::PRXI and tsa1Δ::PRXII strains to H2O2 in the presence of antimycin A using spot assays. The tsa1Δ strain was more sensitive to H2O2 plus antimycin A than wild-type under the conditions tested (Fig. 4), consistent with previously published results (30). The tsa1Δ::PRXI cells were more resistant than the tsa1Δ mutant to treatment with H2O2 plus antimycin. Growth of the tsa1Δ::PRXI strain appeared to be very similar to wild-type at H2O2 concentrations of up to 1 mM. At levels of 1.5 mM H2O2 and higher, the tsa1Δ::PRXI strain appeared to be more sensitive than the wild-type strain, although still considerably less sensitive than the tsa1Δ strain. In contrast, tsa1Δ::PRXII and tsa1Δ strains showed similar sensitivity to H2O2 plus antimycin A under the conditions tested, indicating a lack of complementation by PRXII (Fig. 4).
Figure 4.

Human PrxI complements the H2O2 plus antimycin sensitivity of a tsa1Δ mutant. Ten-fold serial dilutions of exponentially growing cultures of indicated strains were spotted on YPD media and YPD containing indicated concentrations of H2O2 combined with 2.5 μg/mL antimycin A and grown for 2 days at 30°C.
PrxI and Tsa1 protect S. cerevisiae cells from Bax-induced cell death
Human PrxI and PrxII are thought to have functions as inhibitors of cell death (31, 32). Elevated expression of PrxI has been observed in various human cancers (33). PrxI suppresses radiation-induced c-Jun-NH2-kinase signaling and apoptosis in lung cancer cells (31). Therefore, we investigated whether Tsa1, PrxI and PrxII might protect against mammalian pro-apoptotic protein Bax-induced yeast cell death (34, 35). Expression of Bax was under the control of a Tet-Off promoter regulated by the presence or the absence of doxycycline. The wild-type, tsa1Δ, tsa1Δ::PRXI and tsa1Δ::PRXII strains with empty vector showed normal survival in the absence of doxycycline (data not shown). In contrast, induction of Bax expression for 6 hours in these strains reduced viability (Fig. 5). Bax expression caused a greater reduction in cell survival in a tsa1Δ strain. This increased cell death was largely prevented by the expression of PrxI but not PrxII (Fig. 5). These results indicate that yeast Tsa1 as well as human PrxI can prevent induction of apoptosis-like cell death by Bax expression. This correlates with the inferred role of Prx enzymes during cellular response to oxidative stress. Of note, accumulating evidence points to ROS as key regulators of apoptosis, including Bax-induced yeast cell death (36–38).
Figure 5.
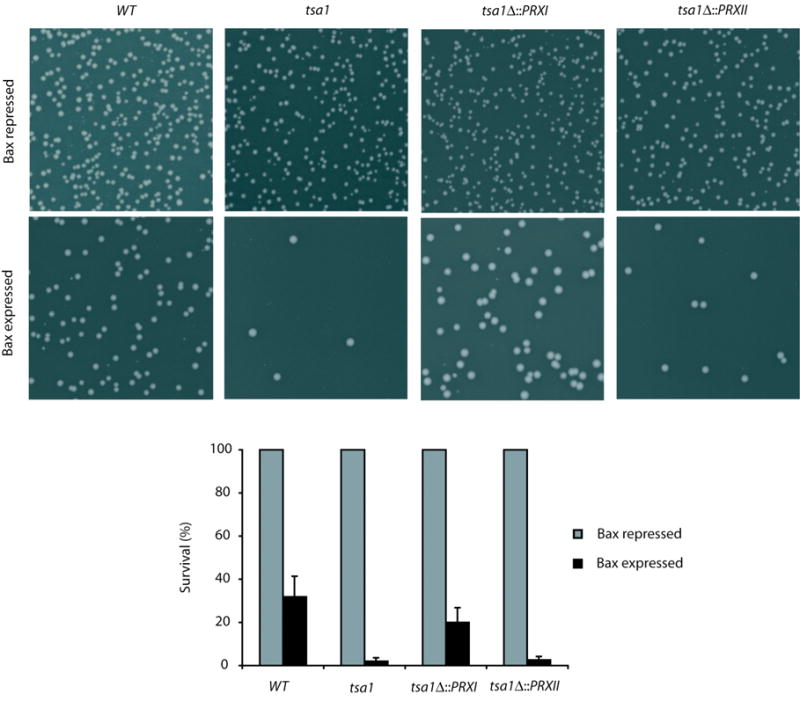
Human PrxI and yeast Tsa1 protect against Bax-induced cell death. Appropriate dilutions of cells before and after Bax expression were plated on SC-leucine plates supplemented with 10 μg/mL doxycycline (Bax repressed). A, representative images show significant reduction in number of colonies after induction of Bax expression for 6 hours. The tsa1Δ cells displayed a more drastic reduction than wild-type cells following the same treatment. PrxI but not PrxII largely suppressed this reduction. B, for each strain, number of colonies formed from the culture prior to the Bax expression was set to 100%. The mean survival percentage from the corresponding culture after a 6-hour induction of Bax expression was calculated. Averages and standard deviations of three independent experiments are reported.
Discussion
Human PrxI and PrxII share a number of features with S. cerevisiae Tsa1. First, all three are typical 2-Cys Prxs and their peroxidase activity relies on both the N- and C-terminal conserved Cys residues (6). Second, they are fairly abundant proteins localized in the cytosol (6). And third, they share high degree of amino acid identity including the critical catalytic Cys residues (Fig. 1) (6, 39). Therefore, it was anticipated that the cellular role of human PrxI or PrxII could be studied in S. cerevisiae, where it would be possible for the first time to examine whether PrxI or PrxII might function in the maintenance of genome stability and interact with other aspects of DNA metabolism. The results presented here indicate that the expression of PrxI largely suppressed the increased rates of accumulation of mutations and GCRs in tsa1Δ mutants, suppressed the synthetic lethality interactions seen in rad6Δ tsa1Δ and rad51Δ tsa1Δ double mutants and suppressed sensitivity to exogenous treatment with H2O2 plus antimycin. In addition, expression of Tsa1 or PrxI prevented Bax-induced cell death. Expression of PrxII did not complement tsa1Δ in any of these assays. These results indicate that PrxI is the human ortholog of Tsa1 and that it is functional in S. cerevisiae cells.
PrxI and Tsa1 are functionally almost interchangeable with regard to maintenance of genome stability in S. cerevisiae, as expression of PrxI decreased the high rates of Canr mutations and GCRs in tsa1Δ mutants. The mechanism of accumulation of tsa1Δ-associated Canr mutations, which are primarily base substitutions (9), seems likely to involve increased oxidation of DNA in the absence of the antioxidant activity of Tsa1, resulting in increased misincorporations during DNA replication. The mechanism underlying the formation of tsa1Δ-associated GCRs, which are predominantly broken chromosomes healed by de novo telomere addition (9), is less clear. It seems likely that excessive oxidative damage to DNA in tsa1Δ mutants leads to the accumulation of damaged replication intermediates ultimately resulting in broken DNA molecules that are substrates for some error-prone DNA repair pathways such as de novo telomere addition (14). The synthetic lethality resulting from combination of a tsa1Δ mutation with defects in postreplication repair or recombination is consistent with such a mechanism underlying the increased genome instability in tsa1Δ mutants. PrxI was initially proposed to be a tumor suppressor based on its ability to interact with, and modulate the activities of, both c-Myc and c-Abl oncoproteins (40, 41). However, the ability of PrxI to suppress the mutator phenotype of yeast tsa1Δ mutants, which lack equivalents of c-Abl and c-Myc, suggests that the suppression of mutations and genome rearrangements may be partially responsible for the role of PrxI as a tumor suppressor. Increased genome instability could be the driving force of the increased carcinogenesis seen in PrxI-deficient mice (15).
Human PrxI and PrxII are thought to have functions as inhibitors of cell death. An anti-apoptotic role for PrxI in irradiated lung cancer cells is thought to be mediated through interaction with the c-Jun NH2-terminal kinase (JNK)-glutathione S-transferase pi (GSTpi) complex, reducing JNK release/activation (31). JNK is a critical regulator of cell proliferation, cell survival, cell death, DNA repair and metabolism (42). Suppression of PrxII expression by antisense cDNA causes cancer cells to be more susceptible to radiation-induced apoptosis (32); this is consistent with the view that radiation-induced cytotoxicity is mediated primarily by the generation of ROS and ROS-driven oxidative stress (43, 44). In the present study, we found that the absence of Tsa1 enhanced Bax-induced cell death and PrxI suppressed this phenotype, suggesting that Tsa1 and PrxI are critical regulators of Bax-induced cell death. In contrast, expression of PrxII did not have such an effect in our system. The ability of Tsa1 and PrxI to suppress Bax-induced cell death might be due to the ability of their antioxidant activity suppressing ROS-mediated DNA damage and genome instability. Another possibility that could explain the protective effect of Tsa1 and PrxI is that they interfere with ROS-mediated signaling in the induction of apoptosis. In fact, although S. cerevisiae does not contain endogenous Bcl-2 family members, the initial events underlying Bax activity in S. cerevisiae and mammalian cells are similar, including translocation of the protein to mitochondria, release of cytochrome c, and alterations in mitochondrial function (36). The downstream effectors of Bax-induced cell death in S. cerevisiae are less well established, but appear to include ROS generated in the mitochondria. Regardless of the mechanism, our results suggest that the increased PrxI expression often observed in various human cancers may inhibit apoptosis in cancer cells and promote resistance to radiotherapy and chemotherapy. In agreement with this view, the thioredoxin system, which is a major intracellular antioxidant activity and an electron donor for peroxiredoxins, has been linked to evasion of apoptosis and to drug and radiation resistance (45).
Although the 2-Cys peroxiredoxins PrxI and PrxII share a high degree of sequence identity and the same catalytic mechanism, our results suggest differences in their functionality. In contrast to PrxI, PrxII was not able to functionally substitute for Tsa1. It is possible that the PrxII expressed in S. cerevisiae was simply not active, although this seems unlikely given the high degree of sequence identity with PrxI (77.4% amino acid identity and 90.5% similarity) and the fact that levels of S. cerevisiae-produced PrxI and PrxII proteins were quite similar (Fig. 2D). Assuming that PrxII is active in yeast, it is possible that PrxI is able to interact with a critical endogenous pathway in S. cerevisiae but that PrxII cannot. Another possibility is that PrxI and PrxII play distinct cellular roles and possess unique regulatory mechanisms (46). Indeed, the biological consequences of the absence of PrxI and PrxII in higher eukaryotes are different. Notably, PrxI-deficient mice develop malignant tumors in various tissues (15), whereas PrxII-deficient mice tend to develop only red blood cell abnormalities (47). In this regard, it is interesting to note that S. cerevisiae Tsa1 and Tsa2 share 86% identity in amino acid sequence but their roles in response to oxidative or nitrosative stress and in suppressing genome instability are distinct (9, 11, 28, 48). All these data highlight the diverse roles of 2-Cys Prxs in intracellular H2O2 signaling and oxidative stress. Different Prxs may direct selective and specific regulation of endogenous H2O2 and other free radicals depending on the sources and targets in various physiological and pathophysiological conditions. The S. cerevisiae system described here could provide a sensitive tool to help define the mechanisms that underlie the function of human Prxs.
Acknowledgments
Grant support: Action thématique et incitative sur programme (ATIP) of Centre National de la Recherche Scientifique, Institut Curie, and Fondation Recherche Medicale (M-E. Huang); and NIH Grant GM26017 (R.D. Kolodner).
We thank G. Baldacci and S. Shell for comments on the manuscript, and members of the Kolodner and Baldacci laboratories for helpful discussions. We thank S. Manon and L. Vernis for generously supplying plasmid pCM189-Bax, Rosine Onclercq-Delic for the cDNA from the Epstein-Barr virus-transformed human lymphoblastoid B cell line D1, and G. Kienda for technical assistance.
References
- 1.Newcomb TG, Loeb LA. Oxidative DNA damage and mutagenesis. In: Nickoloff JA, Hoekstra MF, editors. DNA repair in prokaryotes and lower eukaryotes. Totawa: Humana Press; 1998. pp. 65–84. [Google Scholar]
- 2.Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–54. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 3.D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–24. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 4.Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3:276–85. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 5.Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. 2004;10 (Suppl):S18–25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- 6.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–52. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 7.Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 8.Park SG, Cha MK, Jeong W, Kim IH. Distinct physiological functions of thiol peroxidase isoenzymes in Saccharomyces cerevisiae. J Biol Chem. 2000;275:5723–32. doi: 10.1074/jbc.275.8.5723. [DOI] [PubMed] [Google Scholar]
- 9.Huang ME, Rio AG, Nicolas A, Kolodner RD. A genomewide screen in Saccharomyces cerevisiae for genes that suppress the accumulation of mutations. Proc Natl Acad Sci U S A. 2003;100:11529–34. doi: 10.1073/pnas.2035018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang ME, Kolodner RD. A biological network in Saccharomyces cerevisiae prevents the deleterious effects of endogenous oxidative DNA damage. Mol Cell. 2005;17:709–20. doi: 10.1016/j.molcel.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 11.Wong CM, Siu KL, Jin DY. Peroxiredoxin-null yeast cells are hypersensitive to oxidative stress and are genomically unstable. J Biol Chem. 2004;279:23207–13. doi: 10.1074/jbc.M402095200. [DOI] [PubMed] [Google Scholar]
- 12.Krogh BO, Symington LS. Recombination proteins in yeast. Annu Rev Genet. 2004;38:233–71. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- 13.Barbour L, Xiao W. Regulation of alternative replication bypass pathways at stalled replication forks and its effects on genome stability: a yeast model. Mutat Res. 2003;532:137–55. doi: 10.1016/j.mrfmmm.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 14.Ragu S, Faye G, Iraqui I, Masurel-Heneman A, Kolodner RD, Huang ME. Oxygen metabolism and reactive oxygen species cause chromosomal rearrangements and cell death. Proc Natl Acad Sci U S A. 2007;104:9747–52. doi: 10.1073/pnas.0703192104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neumann CA, Krause DS, Carman CV, et al. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424:561–5. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- 16.Burke D, Dawson D, Stearns T. Methods in yeast genetics. Cold spring harbor: Cold spring harbor laboratory press; 2000. [Google Scholar]
- 17.Schmidt KH, Pennaneach V, Putnam CD, Kolodner RD. Analysis of gross-chromosomal rearrangements in Saccharomyces cerevisiae. Methods Enzymol. 2006;409:462–76. doi: 10.1016/S0076-6879(05)09027-0. [DOI] [PubMed] [Google Scholar]
- 18.Reynaud A, Facca C, Sor F, Faye G. Disruption and functional analysis of six ORFs of chromosome IV: YDL103c (QRI1), YDL105w (QRI2), YDL112w (TRM3), YDL113c, YDL116w (NUP84) and YDL167c (NRP1) Yeast. 2001;18:273–82. doi: 10.1002/1097-0061(200102)18:3<273::AID-YEA665>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 19.Ababou M, Dutertre S, Lecluse Y, Onclercq R, Chatton B, Amor-Gueret M. ATM-dependent phosphorylation and accumulation of endogenous BLM protein in response to ionizing radiation. Oncogene. 2000;19:5955–63. doi: 10.1038/sj.onc.1204003. [DOI] [PubMed] [Google Scholar]
- 20.Longtine MS, McKenzie A, 3rd, Demarini DJ, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–61. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 21.Ausubel FM, Brent R, Kingston RE, et al. Current protocols in molecular biology. New York: John Wiley & Sons, Inc; 2006. [Google Scholar]
- 22.Foiani M, Marini F, Gamba D, Lucchini G, Plevani P. The B subunit of the DNA polymerase alpha-primase complex in Saccharomyces cerevisiae executes an essential function at the initial stage of DNA replication. Mol Cell Biol. 1994;14:923–33. doi: 10.1128/mcb.14.2.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang ME, Rio AG, Galibert MD, Galibert F. Pol32, a subunit of Saccharomyces cerevisiae DNA polymerase delta, suppresses genomic deletions and is involved in the mutagenic bypass pathway. Genetics. 2002;160:1409–22. doi: 10.1093/genetics/160.4.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lea DE, Coulson CA. The distribution of the numbers of mutants in bacterial populations. J Genet. 1948;49:264–85. doi: 10.1007/BF02986080. [DOI] [PubMed] [Google Scholar]
- 25.Priault M, Camougrand N, Chaudhuri B, Schaeffer J, Manon S. Comparison of the effects of bax-expression in yeast under fermentative and respiratory conditions: investigation of the role of adenine nucleotides carrier and cytochrome c. FEBS Lett. 1999;456:232–8. doi: 10.1016/s0014-5793(99)00957-6. [DOI] [PubMed] [Google Scholar]
- 26.Gietz RD, Sugino A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988;74:527–34. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- 27.Boiteux S, Gellon L, Guibourt N. Repair of 8-oxoguanine in Saccharomyces cerevisiae: interplay of DNA repair and replication mechanisms. Free Radic Biol Med. 2002;32:1244–53. doi: 10.1016/s0891-5849(02)00822-5. [DOI] [PubMed] [Google Scholar]
- 28.Wong CM, Zhou Y, Ng RW, Kung Hf HF, Jin DY. Cooperation of yeast peroxiredoxins Tsa1p and Tsa2p in the cellular defense against oxidative and nitrosative stress. J Biol Chem. 2002;277:5385–94. doi: 10.1074/jbc.M106846200. [DOI] [PubMed] [Google Scholar]
- 29.Demasi AP, Pereira GA, Netto LE. Cytosolic thioredoxin peroxidase I is essential for the antioxidant defense of yeast with dysfunctional mitochondria. FEBS Lett. 2001;509:430–4. doi: 10.1016/s0014-5793(01)03215-x. [DOI] [PubMed] [Google Scholar]
- 30.Demasi AP, Pereira GA, Netto LE. Yeast oxidative stress response. Influences of cytosolic thioredoxin peroxidase I and of the mitochondrial functional state. FEBS J. 2006;273:805–16. doi: 10.1111/j.1742-4658.2006.05116.x. [DOI] [PubMed] [Google Scholar]
- 31.Kim YJ, Lee WS, Ip C, Chae HZ, Park EM, Park YM. Prx1 suppresses radiation-induced c-Jun NH2-terminal kinase signaling in lung cancer cells through interaction with the glutathione S-transferase Pi/c-Jun NH2-terminal kinase complex. Cancer Res. 2006;66:7136–42. doi: 10.1158/0008-5472.CAN-05-4446. [DOI] [PubMed] [Google Scholar]
- 32.Park SH, Chung YM, Lee YS, et al. Antisense of human peroxiredoxin II enhances radiation-induced cell death. Clin Cancer Res. 2000;6:4915–20. [PubMed] [Google Scholar]
- 33.Kang SW, Rhee SG, Chang TS, Jeong W, Choi MH. 2-Cys peroxiredoxin function in intracellular signal transduction: therapeutic implications. Trends Mol Med. 2005;11:571–8. doi: 10.1016/j.molmed.2005.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Priault M, Camougrand N, Kinnally KW, Vallette FM, Manon S. Yeast as a tool to study Bax/mitochondrial interactions in cell death. FEMS Yeast Res. 2003;4:15–27. doi: 10.1016/S1567-1356(03)00143-0. [DOI] [PubMed] [Google Scholar]
- 35.Ligr M, Madeo F, Frohlich E, Hilt W, Frohlich KU, Wolf DH. Mammalian Bax triggers apoptotic changes in yeast. FEBS Lett. 1998;438:61–5. doi: 10.1016/s0014-5793(98)01227-7. [DOI] [PubMed] [Google Scholar]
- 36.Jin C, Reed JC. Yeast and apoptosis. Nat Rev Mol Cell Biol. 2002;3:453–9. doi: 10.1038/nrm832. [DOI] [PubMed] [Google Scholar]
- 37.Madeo F, Frohlich E, Ligr M, et al. Oxygen stress: a regulator of apoptosis in yeast. J Cell Biol. 1999;145:757–67. doi: 10.1083/jcb.145.4.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osiewacz HD, Scheckhuber CQ. Impact of ROS on ageing of two fungal model systems: Saccharomyces cerevisiae and Podospora anserina. Free Radic Res. 2006;40:1350–8. doi: 10.1080/10715760600921153. [DOI] [PubMed] [Google Scholar]
- 39.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–3. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 40.Mu ZM, Yin XY, Prochownik EV. Pag, a putative tumor suppressor, interacts with the Myc Box II domain of c-Myc and selectively alters its biological function and target gene expression. J Biol Chem. 2002;277:43175–84. doi: 10.1074/jbc.M206066200. [DOI] [PubMed] [Google Scholar]
- 41.Wen ST, Van Etten RA. The PAG gene product, a stress-induced protein with antioxidant properties, is an Abl SH3-binding protein and a physiological inhibitor of c-Abl tyrosine kinase activity. Genes Dev. 1997;11:2456–67. doi: 10.1101/gad.11.19.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karin M, Gallagher E. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life. 2005;57:283–95. doi: 10.1080/15216540500097111. [DOI] [PubMed] [Google Scholar]
- 43.Wallace SS. Enzymatic processing of radiation-induced free radical damage in DNA. Radiat Res. 1998;150:S60–79. [PubMed] [Google Scholar]
- 44.Riley PA. Free radicals in biology: oxidative stress and the effects of ionizing radiation. Int J Radiat Biol. 1994;65:27–33. doi: 10.1080/09553009414550041. [DOI] [PubMed] [Google Scholar]
- 45.Arner ES, Holmgren A. The thioredoxin system in cancer. Semin Cancer Biol. 2006;16:420–6. doi: 10.1016/j.semcancer.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 46.Lee W, Choi KS, Riddell J, et al. Human peroxiredoxin 1 and 2 are not duplicate proteins: the unique presence of CYS83 in Prx1 underscores the structural and functional differences between Prx1 and Prx2. J Biol Chem. 2007;282:22011–22. doi: 10.1074/jbc.M610330200. [DOI] [PubMed] [Google Scholar]
- 47.Lee TH, Kim SU, Yu SL, et al. Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood. 2003;101:5033–8. doi: 10.1182/blood-2002-08-2548. [DOI] [PubMed] [Google Scholar]
- 48.Munhoz DC, Netto LE. Cytosolic thioredoxin peroxidase I and II are important defenses of yeast against organic hydroperoxide insult: catalases and peroxiredoxins cooperate in the decomposition of H2O2 by yeast. J Biol Chem. 2004;279:35219–27. doi: 10.1074/jbc.M313773200. [DOI] [PubMed] [Google Scholar]