


Abstract
Homologous recombination (HR) is a highly accurate mechanism of DNA repair that can be exploited for homology-directed gene targeting. Since in most cell types HR occurs very infrequently (∼10−6 to 10−8), its practical application has been largely restricted to specific experimental systems that allow selection of the few cells that become genetically modified. HR-mediated gene targeting has nonetheless revolutionized genetics by greatly facilitating the analysis of mammalian gene function. Recent studies showed that generation of double-strand DNA breaks at specific loci by designed endonucleases greatly increases the rate of homology-directed gene repair. These findings opened new perspectives for HR-based genome editing in higher eukaryotes. Here, we demonstrate by using donor DNA templates together with the adeno-associated virus (AAV) Rep78 and Rep68 proteins that sequence- and strand-specific cleavage at a native, predefined, human locus can also greatly enhance homology-directed gene targeting. Our findings argue for the development of other strategies besides direct induction of double-strand chromosomal breaks to achieve efficient and heritable targeted genetic modification of cells and organisms. Finally, harnessing the cellular HR pathway through Rep-mediated nicking expands the range of strategies that make use of AAV elements to bring about stable genetic modification of human cells.
INTRODUCTION
Homologous recombination (HR) ensures the high-fidelity repair of genomes by using homologous DNA sequences (e.g. sister chromatids) as templates for correction (1). Under normal conditions, HR is a rare event in most mammalian cell types. In HeLa and HT-1080 cells it occurs at frequencies of ∼10−7 to 10−8 (2,3) and 10−6 to 10−7 (3–5), respectively, whereas in human fibroblasts it has an incidence of ∼10−7 (6). Due to these low HR rates, homology-directed genome editing techniques have heavily depended on the use of stringent cell selection procedures that are not easily applicable beyond purely experimental systems. Even so, the exploitation of HR-mediated gene targeting has greatly impacted biological research by providing the principles to ‘knock in’ and ‘knock out’ genes (7). The observation that the induction of site-specific double-strand chromosomal breaks stimulates homology-directed gene repair (8,9) provided a rationale for the development of artificial zinc finger nucleases (ZFNs) (10–13). ZFNs consist of a modular assembly of zinc finger domains covalently linked to the nuclease motif of the Type IIS restriction endonuclease FokI. The former domains confer specificity to the double-strand DNA breaks generated by dimers of the latter. Indeed, ZFNs can cleave predefined sequences in the genomes of higher eukaryotes and thereby increase the frequency of HR between donor and recipient sequences by 3–4 orders of magnitude. These findings have greatly improved the prospects for the application of HR-based genome editing methods in clinical and industrial settings. For instance, efficient gene targeting at specific loci could be used to rescue genetic disease phenotypes while avoiding insertional oncogenesis as observed in clinical trials deploying γ-retrovirus vectors to treat X-linked severe combined immunodeficiency (14). Although ZFNs have great potential, the clinical application of these proteins awaits technical improvements such as the reduction of off-target chromosomal double-strand breaks and associated cytotoxicity as well as the control of their activity in target cells (15). An alternative HR-based gene editing strategy consists of exploiting the recombinogenic nature of adeno-associated virus (AAV) vector genomes (16). Several reports have demonstrated that AAV vectors can be tailored to introduce precise nucleotide alterations into the human genome at frequencies approaching 1% when very high multiplicities of infection are used (i.e. 105–106 genome copies per cell). In comparison with other methods, the AAV vector-mediated HR process seems to be less dependent on the extent of homology between donor and target templates. Currently, however, with this method, each targeted gene conversion event is accompanied by approximately 10 random DNA insertions (17).
Historically, single-strand and double-strand DNA breaks have both been invoked as the initiators of homology-directed DNA repair in HR models. However, experimental indications that single-strand DNA gaps or nicks may constitute, per se, triggers for HR have only recently been obtained (18). Instrumental to this conclusion was the deployment of mutant RAG proteins that preferentially nick instead of cleave their recombination signal sequences and a reporter gene expression rescue assay based on plasmids containing two non-functional but complementary cyan fluorescent protein gene segments (18).
Here, we investigated whether a bona fide nicking endonuclease could stimulate HR at a predefined native human locus, and by doing so, could be used for the targeted chromosomal insertion of a functional transcription unit. For this purpose, we exploited the ability of the two largest AAV Rep proteins (i.e. Rep78 and Rep68) to introduce a single-strand DNA break in a locus on the long arm of human chromosome 19 designated AAVS1. Introduction into human cells of these sequence- and strand-specific endonucleases together with donor templates consisting of a 4.1-kb humanized Renilla reniformis green fluorescent protein (hrGFP) transcription unit flanked by sequences homologous to AAVS1 greatly enhanced homology-directed gene addition. These results demonstrate that a sequence- and strand-specific endonuclease can stimulate targeted insertion of new genetic information into a predefined human genomic region in its native chromosomal context.
MATERIALS AND METHODS
DNA constructions
The AAV rep78/68 expression plasmid pGAPDH.Rep78/68 has been described before (19). The annotated nucleotide sequences of the expression plasmids pGAPDH.Rep68 and pGAPDH.Rep68(Y156F) encoding endonuclease-proficient and -deficient versions of Rep68, respectively, as well as that of the targeting vector pA1.GFP.A2 can be retrieved through GenBank accession numbers, GQ380656, GQ380657 and GQ380658, respectively.
DNA transfections
Eighty thousand human cervical carcinoma (HeLa) cells (American Type Culture Collection) in wells of 24-well plates (Greiner Bio-One) were co-transfected with pA1.GFP.A2 and pGAPDH.Rep78/68 at a molar ratio of 2 : 1 or with pA1.GFP.A2 and an ‘empty plasmid’ using ExGen500 (Fermentas). The total amounts of transfection reagent and plasmid DNA were 2.4 µl and 0.4 µg per well, respectively. Twenty-four hours after transfection, the cells received fresh culture medium [Dulbecco's modified Eagle's medium (DMEM; Invitrogen) plus 10% fetal bovine serum (FBS; Invitrogen)]. Cultures exposed to the ‘empty’ plasmid and to the rep78/68 expression vector contained 69% ± 7.1 (n = 3) and 68.4% ± 2.1 (n = 3) hrGFP-positive cells, respectively, as determined by flow cytometry 48 h post-transfection (10 000 events measured per sample). The transfected cells as well as non-transfected control cells were sub-cultured every 3–4 days for 4 weeks in order to allow the identification, through direct fluorescence microscopy and flow cytometry, of stably transduced HeLa cells. Next, the cells were sorted to establish single-cell clones. Transfection experiments using constructs pGAPDH.Rep68 and pGAPDH.Rep68(Y156F) were performed as those described above using pGAPDH.Rep78/68.
Cell sorting and clonal expansion
hrGFP-based cell sorting was carried out 4 weeks post-transfection to permit loss of episomal pA1.GFP.A2 donor templates. Cells were collected in a 1 : 1 mixture of DMEM and FBS. The sorted hrGFP-positive cells were seeded in wells of 96-well plates (Greiner Bio-One) at a density of 0.3 cells per well in DMEM containing 10% FBS, 50 µM α-thioglycerol (Sigma-Aldrich) and 20 nM bathocuprione disulphonate (Sigma-Aldrich) to increase the cloning efficiency (20). Twenty-four randomly picked clones derived from the HeLa cells transfected with pA1.GFP.A2 and pGAPDH.Rep78/68 and 25 clones selected at random from the sorted HeLa cells that received pA1.GFP.A2 and the ‘empty vector’ were used for further analysis.
Flow cytometry
Quantification of hrGFP-positive cells was performed using a BD LSR II flow cytometer. Data were analyzed with the aid of BD FACSDiva software version 5.0.1 with non-transfected HeLa cells serving to set the background level of the assay at zero events. At least 10 000 events were acquired per sample.
Detection of gene targeting events by Southern blot analysis
Chromosomal DNA was purified according to a published method (21). After overnight digestion with ApaLI (New England Biolabs), 10 µg DNA samples were resolved in a 0.8% agarose gel in 1× Tris–acetate–EDTA buffer. Next, the DNA was transferred by capillary action to an Amersham Hybond-XL membrane (GE Healthcare) using a standard Southern blot technique. The 393-bp AAVS1-specific probe was obtained by PCR amplification of human chromosomal DNA using 0.012 U/µl Phusion High-Fidelity DNA polymerase (Finnzymes), 200 µM deoxynucleoside triphosphates (dNTPs; Fermentas), 1× GC buffer (Finnzymes) plus 0.2 µM of primers #633 (5′-GGTCCCCAGCATGTCTTCCTA-3′) and #634 (5′-CTCCCGAACCTCAGATCTCC-3′). The resulting DNA fragment was purified after agarose gel electrophoresis with the aid of the QIAEX II gel extraction kit (Qiagen). The 738 bp hrGFP-specific probe was obtained by digestion of plasmid pU.CAG.hrGFP (22) with EcoRI and NotI (both from Fermentas) followed by preparative agarose gel electrophoresis. Both DNA probes were labeled with (α-32P)dATP (GE Healthcare) using the HexaLabel DNA labeling system (Fermentas). Prior to their application in hybridization experiments, the radiolabeled probes were separated from unincorporated dNTPs through size-exclusion chromatography using Sephadex-50 (GE Healthcare) columns. The PCR amplifications were carried out in a DNA Engine Tetrad 2 thermal cycler (Bio-Rad).
Detection of gene targeting events by PCR
PCR amplifications were performed on chromosomal DNA purified from hrGFP-positive cell clones derived from cell populations initially transfected with pA1.GFP.A2 and either the ‘empty plasmid’ or pGAPDH.Rep78/68. Cellular DNA extracted from parental non-transfected cells served as negative control. Samples containing 100 ng of genomic DNA were subjected to PCR with the AAVS1-specific primer #649 (5′-AGGCTTGCTCTGCACAACTT-3′), together with primer #651 (5′-TTCCTAACCCCAACACTTGC-3′) targeting the human elongation factor 1α (EF1α) promoter located in pA1.GFP.A2. Fifty microliters PCR mixtures containing 0.2 µM of each primer, 200 µM dNTPs, 0.6 U of Phusion High-Fidelity DNA polymerase and HF buffer (Finnzymes) at a 1× final concentration were placed in a DNA Engine Tetrad 2 thermal cycler and a touchdown PCR program was initiated by 1-min incubation at 98°C. This was followed by 36 cycles consisting of 20 s at 98°C, 30 s at 72°C with the temperature decreasing 0.5°C per cycle and 2 min at 72°C. When the PCR program reached the lower annealing temperature of 54°C, 16 additional cycles were carried out at this annealing temperature. The procedure was completed by 5-min incubation at 72°C. To control for the integrity of the genomic DNA, PCR amplifications with the hypoxanthine phosphoribosyltransferase 1 (HPRT1)-specific primers hHPRT.1 and hHPRT.2 (23) were carried out using the conditions and cycling parameters specified above. Semi-nested PCR amplifications were done by using primer #649 and the EF1α promoter-specific primer #186 (5′-CTATGTGGCCAACGCTAAG-3′) on 0.004% of the DNA synthesized with the aid of oligodeoxyribonucleotides #649 and #651. A touchdown PCR cycling program was initiated by 45-s incubation at 98°C. This was followed by 20 cycles consisting of 25 s at 98°C, 30 s at 72°C with the temperature decreasing 0.5°C per cycle and 2 min at 72°C. When the program reached the lower annealing temperature of 62°C, 24 additional cycles were carried out at this annealing temperature. The procedure was finished by 5-min incubation at 72°C.
Effect of Rep68 dose on HR-mediated gene targeting
To study the effect, in human cell cultures, of different Rep68 dosages on HR-mediated gene targeting, 8 × 104 HeLa cells were transfected essentially as specified above. Controls consisted of mock-transfected HeLa cells and of HeLa cells transfected with 0.2 μg of pA1.GFP.A2 and either 0.1 μg of ‘empty plasmid’ or 0.1 μg of pGAPDH.Rep68(Y156F). In parallel, HeLa cells were transfected with 0.2 μg of pA1.GFP.A2 mixed with increasing quantities of pGAPDH.Rep68 (i.e. 1, 3.3, 10, 33.3 and 100 ng). A total amount of 0.3 μg of recombinant DNA was transfected in each experimental condition by adding, whenever required, extra ‘empty plasmid’. At 3 days post-transfection live-cell light microscopy was performed to establish similar transfection efficiencies among the different experimental conditions. In this analysis, an Olympus IX51 inverse fluorescence microscope equipped with a ColorView II Peltier-cooled charge-coupled device camera was deployed. Images were archived by using analysis software (Soft Imaging Systems). At 4 days after transfection, genomic DNA was extracted (21) and 1 μg was incubated overnight at 37°C with DpnI to selectively digest possible remaining plasmid input. Next, the genomic DNA was analyzed by PCR and semi-nested PCR as described above except that for the PCR the cycling conditions of the semi-nested PCR were used. To control for the integrity of the genomic DNA, PCR amplifications with the HPRT1-specific primers hHPRT.1 and hHPRT.2 (23) were carried out using the same conditions and cycling parameters.
Molecular characterization of junctions between exogenous and endogenous DNA
The amplification of left (i.e. telomeric) transgene-host DNA junctions has been described above. Their centromeric (i.e. right) counterparts were amplified following essentially the same protocol except for the use of primers #635 (5′-GCACTTTGGGTGAATTGTAGG-3′) and #650 (5′-GGGAGGTGTGGGAGGTTTT-3′) and GC instead of HF buffer. Prior to nucleotide sequencing analysis using the 3730xl DNA analyzer and BigDye Terminator v3.1 cycle sequencing kit (both from Applied Biosystems), PCR products representing left- and right-hand AAVS1-transgene junctions were cloned into plasmids pCR-blunt-II TOPO (Invitrogen) and pJet1.2 (Fermentas), respectively, according to the manufacturers’ instructions.
Statistical analysis
Mean and SEM values were computed using GraphPad Prism software version 4.03. Student's t-test was used to compare data sets. P < 0.05 was considered significant.
RESULTS
Experimental model and strategy
The AAV rep gene encodes four overlapping nonstructural proteins (dubbed Rep78, Rep68, Rep52 and Rep40) from a single open reading frame (ORF) by a combination of alternative promoter usage and splicing. Biochemically, Rep78 and Rep68 are virtually indistinguishable (24). During AAV DNA replication, they catalyze the strand- and site-specific cleavage of viral replicative intermediates at the so-called terminal resolution site (trs) to generate a free 3′ hydroxyl group for DNA chain elongation. Nicking of AAV DNA at the trs is dependent on the binding of Rep78 and Rep68 to a nearby Rep-binding element (RBE). A trs and an adjacent, properly spaced RBE are also present in the human genome at 19q13.3-qter (AAVS1 locus). These elements are utilized for the Rep78/68-dependent insertion of AAV DNA at the AAVS1 locus in human chromosome 19 (25,26).
Here, we exploited the sequence- and strand-specific endonuclease activities of Rep78/68 to study whether a single-strand DNA break could serve as an initiator of HR between added foreign DNA and a predefined endogenous human locus. To serve as donor template for HR, we generated the targeting construct pA1.GFP.A2, which contains a human EF1α promoter-driven hrGFP expression unit flanked by DNA segments (‘arms’) homologous to sequences framing the AAV DNA pre-integration site on human chromosome 19 at 19q13.3-qter (Figure 1 A). The presence in pA1.GFP.A2 of a recombinant hrGFP gene allowed us to trace, accurately quantify and sort genetically modified cells independently of the mechanism by which genetic modification of the target cells was brought about. The latter aspect is important because it avoids the exclusive detection of homology-dependent gene targeting events to the detriment of non-targeted exogenous DNA insertions as occurs in commonly used assays based on HR-mediated rescue of reporter gene expression. Clearly, however, the occurrence of integration events involving either truncated reporter genes or reporter genes bearing function-impairing mutations can be scored by neither of the two assays.
Figure 1.
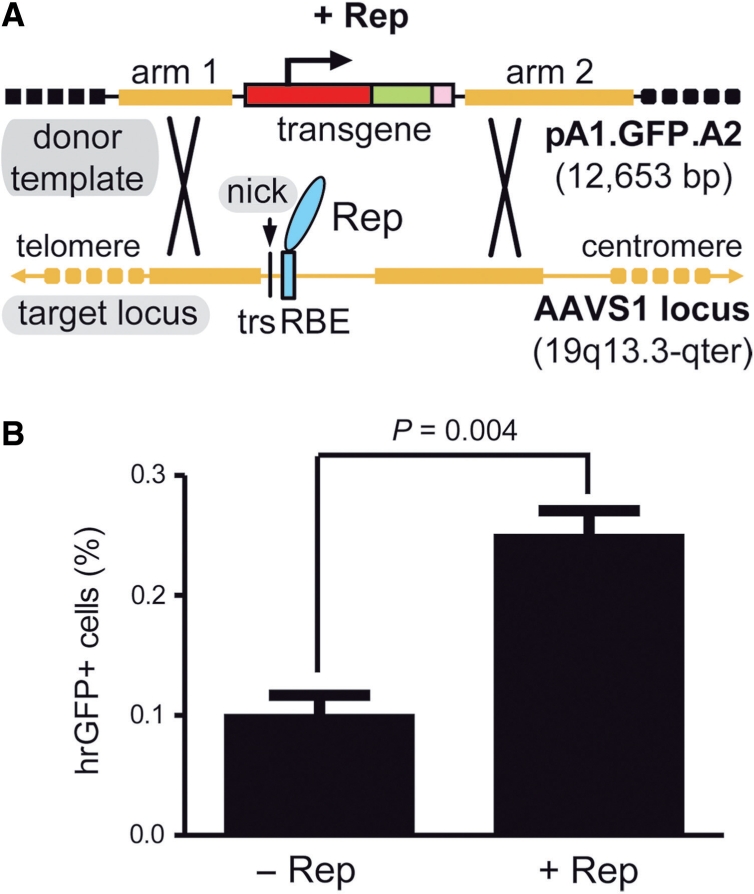
Stable genetic modification of human cells with a targeting vector containing sequences homologous to the genomic region flanking the RBE and trs of AAVS1. (A) Schematic representation of the experimental setup used to test the capacity of a nicking endonuclease to induce homology-directed gene targeting at a native human locus. The episomal donor template pA1.GFP.A2 (12.7 kb) contains a 4.1-kb transcription unit consisting of the EF1α promoter (red box), the hrGFP ORF (green box) and the simian virus 40 polyadenylation signal (pink box). This transcription unit is flanked by sequences homologous to those framing the trs (vertical thin black line) and RBE (cyan box) at the chromosomal acceptor site (i.e. the AAVS1 locus on human chromosome 19 at 19q13.3-qter; thick horizontal yellow lines). Homology arms 1 and 2 are 2063 and 4381 bp in length, respectively. Black crosses represent crossing-over events between homologous DNA segments that lead to targeted gene addition. (B) Flow cytometric quantification of stably transduced human cells. HeLa cells were co-transfected with pA1.GFP.A2 and an ‘empty’ control vector (−Rep) or with pA1.GFP.A2 and the AAV serotype 2 rep78/68-expression construct pGAPDH.Rep78/68 (+Rep). The overall frequency of genetically modified cells (i.e. irrespective of whether they were stably transduced via random or via AAVS1-targeted exogenous DNA insertion events) was determined at 44 days post-transfection through hrGFP-based flow cytometric analysis of 100 000 events per sample. Bars represent mean ± SEM of three independent experiments.
The AAV Rep78/68 proteins stimulate homology-directed gene targeting of an entire transcription unit into AAVS1
HeLa cells were co-transfected with pA1.GFP.A2 and either the AAV rep78/68 expression plasmid pGAPDH.Rep78/68 or an ‘empty’ control vector. Stably transduced cells present in both target cell populations were identified following extensive subculturing by virtue of their hrGFP-specific fluorescence. Forty-four days after transfection, the frequency of hrGFP-positive cells had stabilized in both types of cell populations. However, the cultures derived from the pGAPDH.Rep78/68-transfected HeLa cells contained more stably transduced cells than those of HeLa cells not exposed to the rep78/68 expression plasmid (Figure 1B).
We next asked whether stable genetic modification of cells in each of these experimental groups was brought about by targeted or by random DNA insertion. To this end, hrGFP-positive cells from the two different setups were independently sorted and clonally expanded. Subsequently, genomic DNA extracted from randomly selected cell clones (n = 49) was subjected to PCR analysis using primers #649 and #651 specific for the AAVS1 locus and the EF1α promoter, respectively (Figure 2 A). Parental, non-transfected HeLa cells served as negative control. Homology-directed gene targeting at AAVS1 following transfection of pA1.GFP.A2 should originate a 2652 bp PCR fragment (Figure 2A). Products with a size consistent with this process could readily be detected in 71% (n = 24) of PCR samples corresponding to DNA of cell clones derived from cultures exposed to pA1.GFP.A2 and pGAPDH.Rep78/68 (Figure 2B, upper GT panel). Conversely, consistent with the well-established very low frequency of HR in HeLa cells (i.e. 10−7 to 10−8), the PCR assay performed on DNA of cell clones established from pA1.GFP.A2-positive cells that did not receive the AAV rep78/68 expression plasmid yielded non-specific amplification products (n = 25) (Figure 2B, lower GT panel). The identity of the PCR products corresponding to three clones whose founder cells had been co-transfected with pA1.GFP.A2 and pGAPDH.Rep78/68 was further investigated by semi-nested PCR using the primer pairs #649 and #186 (Figure 2A). Unambiguous detection of specific 2.3-kb semi-nested PCR products displaying diagnostic restriction patterns after digestion with ApaLI, PauI or SmaI confirmed that these DNA molecules indeed represented AAVS1-transgene junctions (Figure 2C).
Figure 2.

PCR assays to identify cells genetically modified through homology-directed gene targeting. (A) Overview of the PCR assays carried out on genomic DNA of stably transduced HeLa cells clonally expanded from cultures exposed (+Rep) or not exposed (−Rep) to pGAPDH.Rep78/68. The AAVS1 locus is depicted before and after HR with the donor template pA1.GFP.A2. HR-mediated transgene insertion should yield 2.7-kb PCR products using primers #649 and #651 (horizontal dark blue bar). The specificity of these amplicons can be confirmed by semi-nested PCR using primers #649 and #186 combined with restriction fragment length analyses of the resulting 2.3-kb DNA species (horizontal brown bars). The numbers above the brown bars refer to restriction fragment sizes. Open circle, prokaryotic origin of replication. For an explanation of the other symbols see the legend of Figure 1. (B) PCR analysis carried out on chromosomal DNA of parental HeLa cells (HeLa) and of clones derived from HeLa cells co-transfected with pA1.GFP.A1 and pGAPDH.Rep78/68 (+Rep) or with pA1.GFP.A2 and ‘empty plasmid’ (−Rep). The panels labeled GT display the results of amplification reactions performed with primers #649 and #651, which are specific for the AAVS1 locus and the EF1α promoter, respectively. PCR amplifications targeting a 1.9-kb segment of the HPRT1 were carried out in parallel to ascertain the integrity of the genomic DNA corresponding to individual HeLa cell clones (see panels marked HPRT1). PCR mixtures containing water instead of DNA served as a negative control (H2O). The positions and sizes (in kilo base pairs) of specific PCR products are indicated at the right. Marker, Gene Ruler DNA Ladder Mix molecular weight marker (Fermentas). (C) Characterization of the 2.7-kb amplification products obtained with primers #649 and #651 by semi-nested PCR and restriction enzyme fragment length analyses. A semi-nested PCR using primers #649 and #186 was performed on the 2.7-kb amplicons corresponding to clones 2, 4 and 5. The resulting 2.3-kb PCR products were subjected to agarose gel electrophoresis after digestion with ApaLI, PauI or SmaI. Und., undigested PCR fragments.
To further substantiate these results through an independent method, we deployed Southern blot analyses of ApaLI-digested genomic DNA extracted from randomly selected clones originating from pA1.GFP.A2-transfected HeLa cells that had or had not been exposed to pGAPDH.Rep78/68. Gene conversion resulting from HR between pA1.GFP.A2 and endogenous AAVS1 sequences should yield 9.9-kb DNA species while undisrupted/non-targeted alleles should give rise to 7.1-kb DNA molecules (Figure 3 A). Autoradiograms of Southern blots incubated with the AAVS1-specific probe showed the presence of 9.9-kb DNA fragments consistent with HR-mediated transgene insertion at AAVS1 (Figure 3B, upper panel, open arrowheads). Importantly, these DNA species occurred exclusively in chromosomal DNA of cell clones derived from cultures transfected with pGAPDH.Rep78/68. In genomic DNA of cell clones isolated from cultures that received the ‘empty’ control vector, the 7.1-kb ApaLI fragments (resulting from unmodified AAVS1 alleles) were the only DNA species detected (Figure 3B, upper panel, solid arrowhead). These results are in agreement with those obtained by PCR analysis (Figure 2). To strengthen the assertion that the 9.9-kb molecules represent HR-dependent gene targeting events at AAVS1, the human chromosome 19-specific probe was removed and the stripped membrane was incubated with a transgene-specific probe. The hybridization of the 9.9-kb DNA fragments to the new probe indicates that they are specific and result from HR-mediated transgene insertion at AAVS1 (Figure 3B, lower panel, open arrowheads). Of note is also the co-labeling by both probes of DNA species with higher molecular weights in lanes containing genomic DNA of cells expanded from pGAPDH.Rep78/68-transfected cultures (Figure 3B, open arrows). Possibly, these DNA fragments represent exogenous DNA insertion events at AAVS1 loci that went through local Rep78/68-induced DNA amplification/rearrangement [see e.g. ref. (27)] prior to or concomitant with exogenous gene addition. These events might have occurred via a homology-independent pathway such as that at play during locus-specific wild-type AAV DNA integration (25–27) or non-homologous end joining or, alternatively, through homology-directed DNA targeting as found in the other cells (Figure 3B, open arrowheads). Detailed molecular analysis of the structural organization of the integrated exogenous DNA and its flanking sequences will provide clues as to which mechanism was involved.
Figure 3.

Investigation of targeted versus random chromosomal insertion of exogenous DNA through restriction mapping and Southern blot analyses. (A) Diagram of the Southern blot assay performed on ApaLI-digested chromosomal DNA extracted from randomly selected clones of pA1.GFP.A2-transfected HeLa cells. The ApaLI restriction map of the AAVS1 locus is depicted before and after HR with the donor template pA1.GFP.A2. Non-targeted and targeted human chromosome 19 alleles are expected to give rise to 7.1- and 9.9-kb ApaLI DNA fragments, respectively, using the AAVS1-specific probe (orange horizontal bar). The 9.9-kb DNA species should also be specifically recognized by the transgene-specific probe, which spans the entire hrGFP ORF (green horizontal bar). Both DNA probes are drawn in relation to their target sequences. For an explanation of the other symbols see the legend of Figure 1. (B) Southern blots of ApaLI-digested chromosomal DNA of parental HeLa cells (HeLa) and of clones derived from HeLa cells co-transfected with pA1.GFP.A1 and pGAPDH.Rep78/68 (+Rep) or with pA1.GFP.A2 and ‘empty plasmid’ (−Rep). The open arrowheads point at the 9.9-kb DNA fragments expected to emerge following homology-directed gene targeting at AAVS1. The open arrows mark higher molecular weight DNA species that are also recognized by both probes indicating insertion of exogenous DNA at AAVS1 loci that underwent local Rep78/68-induced DNA amplification/rearrangement. The 12.7-kb ApaLI-linearized pA1.GFP.A2 DNA (solid arrowhead in lower panel) served as an internal control for probe binding specificity and removal. The solid arrowhead in the upper panel points at the 7.1-kb ApaLI fragments derived from unmodified AAVS1 alleles. Marker, Gene Ruler DNA Ladder Mix molecular weight ladder (Fermentas).
The observation that chromosomal DNA extracted from clones 26, 30 and 32 not only gave rise to high-molecular-weight ApaLI fragments (>12.7 kb) recognized by both Southern blot probes but also yielded the 2652 bp PCR species, suggests the involvement of HR at some stage during their genetic modification. Furthermore, in agreement with the aforementioned lack of AAVS1 disruptions, genomic DNA of cell clones derived from Rep78/68-negative cells gave rise to transgene-specific fragments with different sizes (Figure 3B, lower panel). Importantly, none of these fragments was recognized by the AAVS1-specific probe confirming that they originate exclusively from random chromosomal insertion of exogenous DNA.
The Rep68 protein suffices to induce homology-directed gene targeting at AAVS1 and its endonucleolytic activity is fundamental to this process
The smaller Rep68 protein contains all the structural information required for binding to and nicking at RBE and trs sequences, respectively. Thus, we postulated that the HR-inducing activity of Rep78/68 discovered above would be recapitulated by expressing exclusively Rep68. To test this hypothesis, we generated the expression plasmid pGAPDH.Rep68. This construct encodes Rep68 only. Moreover, to establish whether the stimulatory effect of the large AAV Rep proteins on HR-mediated gene targeting depends on their nicking activity, expression plasmid pGAPDH.Rep68(Y156F) was made. This construct is identical to pGAPDH.Rep68 except for a point mutation within the rep68 ORF that confers a nicking-defective phenotype to the encoded protein [designated Rep68(Y156F)] due to substitution of the tyrosine residue at amino acid position 156 by phenylalanine (28,29). Both AAV rep68 expression plasmids were independently co-transfected into HeLa cells together with the targeting vector pA1.GFP.A2 and, as before, cells stably transduced with pA1.GFP.A2 sequences were identified by hrGFP-specific flow cytometry following extensive subculturing. As shown in Figure 4 A, the frequency of stably transduced cells was higher in cell cultures initially exposed to pGAPDH.Rep68 than in those transfected with pGAPDH.Rep68(Y156F). Next, the PCR-based assay illustrated in Figure 2A was deployed to investigate homology-directed gene targeting at AAVS1 in cells that received either Rep68 or Rep68(Y156F). Genomic DNA extracted from non-transfected HeLa cells and from HeLa cell clone 25 (Figure 2B, upper two panels) served as negative and positive controls, respectively. Specific 2.7-kb PCR products were only detected in samples of cell populations derived from cultures initially provided with Rep68 molecules. These data demonstrate that Rep68 alone can induce HR-mediated gene targeting at AAVS1 and that its nicking activity is involved in this process (Figure 4B). The confirmation that the 2.7-kb PCR products resulted from HR-mediated gene targeting at AAVS1 was obtained by the detection of diagnostic 2.3-kb DNA fragments (Figure 4B) following semi-nested PCR using primers #649 and #186 (Figure 2A). To determine the amount of rep68 expression plasmid required to induce detectable HR-mediated chromosomal gene targeting, we performed a dose-response experiment in HeLa cells using as read-out the PCR assay depicted in Figure 2A. The HeLa cells were transfected with a constant amount of targeting vector pA1.GFP.A2 mixed with increasing quantities of pGAPDH.Rep68 ranging from 1 to 100 ng. Light microscopy analyzes revealed that transfection efficiencies were similar for each experimental condition (Figure 5 A). The PCR assay detected AAV Rep68-dependent HR events at AAVS1 in cells exposed to 33.3 and 100 ng of pGAPDH.Rep68 (Figure 5B). Moreover, these events could not be detected in cells receiving 100 ng of pGAPDH.Rep68(Y156F) confirming that Rep68-dependent nicking is necessary for the observed stimulation of homology-directed gene targeting at chromosomal DNA in human cells (Figure 5B).
Figure 4.

Testing the capacity of endonuclease-proficient Rep68 and endonuclease-deficient Rep68(Y156F) proteins to stimulate homology-directed gene targeting at AAVS1. (A) Quantification by flow cytometry of stably transduced HeLa cells following co-transfection with pA1.GFP.A2 and pGAPDH.Rep68(Y156F) or with pA1.GFP.A2 and pGAPDH.Rep68 (Rep68). The frequency of genetically modified cells was determined at 27 days post-transfection through hrGFP-based flow cytometric analysis of 10 000 events per sample. Bars represent mean ± SEM of three independent experiments. (B) Agarose gel electrophoresis of PCR products resulting from amplifications carried out on chromosomal DNA from HeLa cell clone 25 (Figure 2B, upper two panels) (lane 1), parental HeLa cells (lane 2) and HeLa cell populations 1 month after co-transfection with pA1.GFP.A2 and pGAPDH.Rep68 (lane 3) or with pA1.GFP.A2 and pGAPDH.Rep68(Y156F) (lane 4). Lanes M, Gene Ruler DNA Ladder Mix molecular weight marker. Upper panel, internal control PCR products resulting from amplifications performed with the human HPRT1-specific primers hHPRT.1 (#49) and hHPRT.2 (#50). Middle panel, PCR species obtained after amplifications carried out with oligodeoxyribonucleotides #649 and #651 (Figure 2A, upper panel). Lower panel, semi-nested PCR products resulting from PCR reactions using primers #649 and #186 (Figure 2A, upper panel) on 0.004% of the DNA synthesized with the aid of oligodeoxyribonucleotides #649 and #651. The positions and sizes (in kilo base pairs) of the various amplicons are indicated by solid arrows.
Figure 5.

Rep68 dose dependency of HR-mediated gene targeting at AAVS1. (A) Phase-contrast microscopy (upper panel) and hrGFP direct fluorescence microscopy (lower panel) of HeLa cells that were mock-transfected (mock) and of HeLa cells that were transfected with targeting vector pA1.GFP.A2 mixed with ‘empty plasmid’ (−Rep) or mixed with plasmid pGAPDH.Rep68(Y156F) encoding the endonuclease-deficient version of Rep68 Y156F. In parallel, phase-contrast microscopy (upper panel) and hrGFP direct fluorescence microscopy (lower panel) was carried out on HeLa cells transfected with pA1.GFP.A2 together with increasing amounts of rep68 expression construct pGAPDH.Rep68 (i.e. 1, 3.3, 10, 33.3 and 100 ng). The total amount of transfected recombinant DNA was kept constant by adding, whenever required, extra ‘empty plasmid’. Micrographs were acquired at 72-h post-transfection. Numerals below the various columns correspond to the deployed amounts (in nanograms) of the rep68 expression plasmids and of the pA1.GFP.A2 targeting DNA. Original magnification: 100×. (B) Agarose gel electrophoresis of PCR products resulting from amplifications performed on genomic DNA samples with the aid of primers #649 and #651 plus primers #649 and #186 (upper panel), as diagrammed in Figure 2A. Genomic DNA was isolated at 4 days post-transfection from mock-transfected HeLa cells (mock), from HeLa cells co-transfected with pA1.GFP.A2 and ‘empty plasmid’ (−Rep), from HeLa cells co-transfected with pA1.GFP.A2 and pGAPDH.Rep68(Y156F) (Y156F), and from HeLa cells transfected with pA1.GFP.A2 mixed with increasing amounts of pGAPDH.Rep68 (i.e. 1, 3.3, 10, 33.3 and 100 ng) (upper panel). The negative (−) and positive (+) controls were obtained using nuclease-free water and genomic DNA isolated from HeLa cell clone 25 (Figure 2B, upper two panels) as starting material. Lanes M, Gene Ruler DNA Ladder Mix molecular weight marker. Agarose gel electrophoresis of PCR species resulting from amplifications performed with the aid of the human HPRT1-specific primers hHPRT.1 (#49) and hHPRT.2 (#50) (lower panel). These reactions were carried out in parallel and served as an internal control to monitor chromosomal DNA integrity. The positions and sizes (in kilo base pairs) of the amplicons are indicated by solid arrows.
Chromosomal junctions between endogenous and exogenous DNA generated by AAV Rep78/68-mediated HR are accurate
Finally, we asked whether the junctions between AAVS1 and foreign DNA in cells stably transduced through AAV Rep-induced HR were precise at the nucleotide level. To this end, we performed PCR amplifications on genomic DNA from cell clones 4, 7 and 9 with the aid of primer sets #649 plus #651 and #635 plus #650 to amplify, respectively, left (i.e. telomeric) and right (i.e. centromeric) junctions between endogenous and exogenous DNA (Figure 6 A). The resulting diagnostic 2.7- and 5.4-kb PCR fragments were subsequently cloned and the cloning products were used to molecularly characterize all 12 AAVS1-foreign DNA junctions. Nucleotide sequence analysis demonstrated the absence of point mutations and/or microrearrangements (e.g. small deletions and/or insertions) at both sides of the two homology arms (Figure 6B). These results together with those of the PCR (Figure 2) and Southern blot (Figure 3) experiments firmly establish the capacity of an enzyme with sequence- and strand-specific endonuclease activity to trigger homology-directed insertion of a functional expression unit at a native locus in the human genome.
Figure 6.

Nucleotide sequence analysis of centromeric and telomeric junctions between endogenous and exogenous DNA corresponding to three independent Rep78/68-induced HR events. (A) Diagrammatic representation of the hrGFP transcription unit (red, green and pink boxes) inserted at 19q13.3-qter (thick horizontal yellow lines) following homology-directed gene targeting using as donor template pA1.GFP.A2 DNA. Primers used to amplify the left- and right-hand junctions (dark and light blue half arrows, respectively) are drawn in relation to their recognition sequences. The 2.7- and 5.4-kb PCR products specific for the telomeric and centromeric junctions are indicated by dark and light blue bars, respectively. (B) Primary nucleotide sequence data corresponding to transition regions between the homology arms of pA1.GFP.A2 and outward host chromosomal DNA and between the homology arms of pA1.GFP.A2 and inward transgene DNA.
DISCUSSION
We found that a nicking endonuclease can stimulate by several orders of magnitude homology-directed insertion of at least 4.1 kb of foreign genetic information into a native human chromosomal locus. Stable gain-of-function through homology-directed DNA targeting was demonstrated by combining flow cytometric, PCR, Southern blot and DNA sequence analyses. Consistent with the exceedingly low level of spontaneous HR in mammalian cells in general and in HeLa cells in particular, no targeted gene addition events were detected in pA1.GFP.A2-transfected HeLa cells in the absence of the AAV Rep78/68 proteins. Importantly, the HR stimulatory effect could be attributed to the single-strand endonucleolytic activity of these proteins as demonstrated in experiments deploying Rep68 and its nicking-defective mutant Rep68(Y156F). Thus, on the basis of these proof-of-principle experiments, we propose that protein engineering strategies for gene targeting purposes are expanded to include not only enzymes or enzyme complexes, which induce double-strand DNA breaks, but also those that simply produce single-strand DNA lesions. Finally, our results on harnessing the cellular HR pathway using the Rep78/68 endonucleases increases the range of AAV-based approaches to bring about stable genetic modification of human cells (16,17,30,31). A possible advantage of exploiting the cellular HR machinery during Rep78/68-mediated targeted gene addition is the insertion of exogenous DNA devoid of AAV cis-acting elements. This feature should prevent the integrated DNA from becoming a target for Rep78/68-induced rearrangements. In addition, if carefully designed, a targeting construct containing within its arms of homology PPP1R12C (also known as MBS85) sequences should, in principle, lead to the reconstitution of this gene following its Rep78/68-mediated allelic disruption (see below).
It is possible that Rep78/68-induced HR is caused by single-strand breaks that evolve into larger and thus more recombinogenic single-strand gaps. Recent experiments in the recombination-prone chicken lymphoma cell line DT40 suggest, at least in the case of immunoglobulin V gene conversion, that such intermediates can directly trigger HR (32). As our experiments were performed with highly proliferating cell populations, it is also possible that Rep78/68 promotes gene targeting in a chromosomal context via host cell DNA replication fork stalling at single-strand DNA breaks or gaps. Various studies performed in prokaryotic and yeast systems have indicated that protein-DNA barriers such as stalled replisomes can be bypassed by HR-dependent processes (33). Interestingly, in Saccharomyces cerevisiae, intrachromosomal recombination between direct repeats containing in the intervening sequence a site-specific nicking endonuclease gene II protein (gIIp) recognition site, was not only dependent on gIIp expression (34,35) but also on cell cycle progression (35). Of note, Rep78/68 and gIIp share several key features including the ability to cleave double-strand replicative DNA intermediates of their cognate viruses in a sequence- and strand-specific manner and the presence of evolutionarily conserved motifs belonging to a superfamily of rolling circle replication initiator proteins (36).
Very recently, Smith et al. (37) used a site-directed mutagenesis approach to convert the homing endonuclease I-AniI into an enzyme (designated I-AniI K227M) that preferentially nicks instead of cleaves its cognate target site. Using GFP expression rescue assays they measured gene repair frequencies in 293T cells induced by I-AniI K227M. Because in vitro experiments showed that I-AniI K227M also generates double-strand breaks in a dose-dependent manner (37) and as it is very difficult to control the intranuclear concentration of an enzyme following plasmid-driven constitutive overexpression, the possibility remains that double-strand DNA break formation contributed to GFP repair. Of note, contrary to current nick-inducing forms of RAG (18) and homing endonucleases (37,38), AAV Rep78/68 proteins strictly catalyzes the introduction of nicks in double-strand DNA molecules (39). This feature might be useful in studies into the mechanism(s) of DNA repair and recombination initiated by single-strand DNA breaks.
Upon nicking at the trs of AAVS1, Rep78/68 initiates local DNA rearrangements (26,27) that can lead to duplications of chromosomal DNA sequences (40). It has been postulated that this is caused by DNA polymerization initiating at the free 3′ hydroxyl group of the nicked DNA strand. This process is thought to reconstitute the downstream RBE that can subsequently serve as de novo target for Rep78/68-mediated nicking (e.g. 40,41). This process may explain the presence of rearranged AAVS1 loci in at least some of the HeLa cell clones that underwent targeted DNA insertion (Figure 3B). Consistent with this model, a previous study has shown that transient expression of rep68 can lead to canonical Rep78/68-mediated targeted DNA integration apparently in the absence of AAVS1 rearrangements (42). Random exogenous DNA insertions in cells that underwent Rep78/68-dependent AAVS1 disruption with or without targeted DNA insertion could also be identified (Figure 3B, clones 26 and 16, respectively).
The effects of Rep78/68 on target cells, including AAVS1 rearrangements, warrants further discussion in light of the potential utility of AAV Rep endonucleases for the targeted genetic modification of human cells either through the canonical pathway (30,31,40,41) or through the HR pathway reported herein. Critically, the AAVS1 locus is located in a gene-rich region and overlaps with the MBS85 gene (43). A very recent study indicates that Rep78/68 induces partial duplication of MBS85 that, intriguingly, preserves normal MBS85 transcript levels. These authors failed to detect functional consequences to host cells following Rep78/68-dependent wild-type and recombinant AAV DNA integration (40). However, it will be important to analyze larger numbers of cells from different cell types undergoing Rep78/68-induced AAVS1 disruptions and scrutinize in detail the consequences of monoallelic and, possibly, biallelic MBS85 knock-out. Moreover, studies on the effects of Rep78/68 on global karyotypic stability like those carried out for cells exposed to the bacteriophage P1 Cre recombinase (44) or the Streptomyces lividans ϕC31 integrase (45), will need to be performed. In addition, it is known that AAV Rep78/68 can induce cell cycle arrest and that constitutive high-level synthesis of these proteins trigger, especially in non-transformed cells, cytotoxicity and apoptosis (46). Finally, although an RBE and a properly spaced trs occur at AAVS1, the Rep78/68 DNA-binding consensus is, to some extent, degenerate. As a consequence, Rep78/68 can bind to other chromosomal sites as well (47) and, possibly, interfere with the transcriptional activity of local genes.
Given the above, it will be imperative to limit the duration of Rep78/68 activity in target cells so that the aforementioned unwanted side effects are minimized. Ongoing work in our laboratory is focusing on generating ligand-responsive Rep68 variants whose activity is dependent on the clinically applicable synthetic small-molecule drug 4-hydroxytamoxifen (4-OHT). Preliminary results deploying functional assays indicate that a new fusion protein between Rep68 and 4-OHT-responsive estrogen receptor domains retains drug-dependent biological activity in human cells.
More generically, but still from an applied research point of view, using natural and/or engineered site-specific nicking endonucleases may have advantages over enzymes or enzyme complexes that directly induce double-strand DNA cleavage especially if: (i) the complexity of enhanced gene targeting procedures can be reduced (e.g. the coordinated action of two different ZFNs is normally required to generate a specific double-strand DNA break) and (ii) making site-specific nicks will comprise a milder process by which to accomplish enhanced gene targeting (e.g. double-strand chromosomal breaks can be repaired by error-prone non-homologous end-joining or induce chromosomal translocations). In this regard, the very recently reported engineering of nick-inducing forms of the FokI restriction endonuclease constitutes an interesting development (48). However, ultimately, the merit of enzymes that generate single- instead of double-strand chromosomal DNA breaks will depend, to a large extent, on whether the former can per se stimulate HR, i.e. without replication fork-dependent double-strand chromosome break formation (18,32).
Future research aiming at defining optimal nick-inducing enzyme levels as well as transgene size constraints for single-strand DNA break-induced HR will profit from adopting naked DNA transfection-independent methods to efficiently introduce donor templates and endonuclease-encoding genes into both dividing and quiescent cells. Indeed, viral gene delivery vehicles, and in particular high-capacity adenovirus vectors, may constitute a suitable platform to examine the impact of transgene length and dose, extent of homology and cell cycle status on nicking endonuclease-stimulated genome editing. This relates to the fact that these adenovirus vectors can accommodate up to 37 kb of foreign DNA and allow for highly efficient cell cycle-independent gene transfer (49,50).
In conclusion, in this study we demonstrate for the first time that homology-directed gene targeting at an endogenous human locus can be triggered by a bona fide nicking enzyme and expand the number of strategies that make use of AAV elements to bring about long-term transgene expression in these cells. Finally, our findings provide a rationale to devise new approaches for targeted editing of the genomes of higher eukaryotes.
ACCESSION NUMBERS
FUNDING
Association Française contre les Myopathies (AFM) [12259-SR-GROUPE E].
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Robert Kotin (Laboratory of Biochemical Genetics, National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD) for providing plasmid pBRIA-N3 containing an 8.3-kb AAVS1 EcoRI fragment and Ietje van der Velde-van Dijke (Virus and Stem Cell Biology Laboratory, Department of Molecular Cell Biology, Leiden University Medical Center, the Netherlands) for technical assistance.
Footnotes
Present addresses: Gijsbert P. van Nierop, Department of Virology, Erasmus Medical Center, Dr. Molewaterplein 50, 3015 GE Rotterdam, The Netherlands.
Krijn R. Vrijsen, Laboratory of Experimental Cardiology, Department of Cardiology, University Medical Center Utrecht, Heidelberglaan 100, 3584 CX Utrecht, The Netherlands.
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
REFERENCES
- 1.van Gent DC, Hoeijmakers JH, Kanaar R. Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet. 2001;2:196–206. doi: 10.1038/35056049. [DOI] [PubMed] [Google Scholar]
- 2.Itzhaki JE, Porter AC. Targeted disruption of a human interferon-inducible gene detected by secretion of human growth hormone. Nucleic Acids Res. 1991;19:3835–3842. doi: 10.1093/nar/19.14.3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porter AC, Itzhaki JE. Gene targeting in human somatic cells. Complete inactivation of an interferon-inducible gene. Eur. J. Biochem. 1993;218:273–281. doi: 10.1111/j.1432-1033.1993.tb18375.x. [DOI] [PubMed] [Google Scholar]
- 4.Ganguly A, Smelt S, Mewar R, Fertala A, Sieron AL, Overhauser J, Prockop DJ. Targeted insertions of two exogenous collagen genes into both alleles of their endogenous loci in cultures human cells: the insertions are directed by relatively short fragments containing the promoters and the 5′ ends of the genes. Proc. Natl Acad. Sci. USA. 1994;91:7365–7369. doi: 10.1073/pnas.91.15.7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thyagarajan B, Johnson BL, Campbell C. The effect of target site transcription on gene targeting in human cells in vitro. Nucleic Acids Res. 1995;23:2784–2790. doi: 10.1093/nar/23.14.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown JP, Wei W, Sedivy JM. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science. 1997;277:831–834. doi: 10.1126/science.277.5327.831. [DOI] [PubMed] [Google Scholar]
- 7.Capecchi MR. Altering the genome by homologous recombination. Science. 1989;244:1288–1292. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- 8.Lukacsovich T, Yang D, Waldman AS. Repair of a specific double-strand break generated within a mammalian chromosome by yeast endonuclease I-SceI. Nucleic Acids Res. 1994;22:5649–5657. doi: 10.1093/nar/22.25.5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rouet P, Smih F, Jasin M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl Acad. Sci. USA. 1994;91:6064–6068. doi: 10.1073/pnas.91.13.6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl Acad. Sci. USA. 1996;93:1156–1160. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bivikova M, Carroll D, Segal DJ, Trautman JK, Smith J, Kim Y-G, Chandrasegaran S. Stimulation of homologous recombination through targeted cleavage of chimeric nucleases. Mol. Cell Biol. 2001;21:289–297. doi: 10.1128/MCB.21.1.289-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porteus MH, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300:763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- 13.Durai S, Mani M, Kandavelou K, Wu J, Porteus MH, Chandrasegaran S. Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res. 2005;33:5978–5990. doi: 10.1093/nar/gki912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cavazzana-Calvo M, Fisher A. Gene therapy for severe combined immunodeficiency: are we there yet? J. Clin. Invest. 2007;117:1456–1465. doi: 10.1172/JCI30953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cornu TI, Thibodeau-Beganny S, Guhl E, Alwin S, Eichtinger M, Joung JK, Cathomen T. DNA-binding specificity is a major determinant of the activity and toxicity of zinc-finger nucleases. Mol. Ther. 2007;16:352–358. doi: 10.1038/sj.mt.6300357. [DOI] [PubMed] [Google Scholar]
- 16.Russell DW, Hirata RK. Human gene targeting by viral vectors. Nat. Genet. 1998;18:325–330. doi: 10.1038/ng0498-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vasileva A, Jessberger R. Precise hit: adeno-associated virus in gene targeting. Nat. Rev. Microbiol. 2005;3:837–847. doi: 10.1038/nrmicro1266. [DOI] [PubMed] [Google Scholar]
- 18.Lee GS, Neiditch MB, Salus SS, Roth DB. RAG proteins shepherd double-strand breaks to a specific pathway, suppressing error-prone repair, but RAG nicking initiates homologous recombination. Cell. 2004;117:171–184. doi: 10.1016/s0092-8674(04)00301-0. [DOI] [PubMed] [Google Scholar]
- 19.Gonçalves MAFV, van Nierop GP, Tijssen MR, Lefesvre P, Knaän-Shanzer S, van der Velde I, van Bekkum D, Valerio D, de Vries AAF. Transfer of the full-length dystrophin-coding sequence into muscle cells by a dual high-capacity hybrid viral vector with site-specific integration ability. J. Virol. 2005;79:3146–3162. doi: 10.1128/JVI.79.5.3146-3162.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brielmeier M, Béchet J-M, Falk MH, Pawlita M, Polack A, Bornkamm GW. Improving stable transfection efficiency: antioxidants dramatically improve the outgrowth of clones under dominant marker selection. Nucleic Acids Res. 1998;26:2082–2085. doi: 10.1093/nar/26.9.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laird PW, Zijderveld A, Linders K, Rudnicki MA, Jaenisch R, Berns A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Tuyn J, Pijnappels DA, de Vries AAF, de Vries I, van der Velde-van Dijke I, Knaän-Shanzer S, van der Laarse A, Schalij MJ, Atsma DE. Fibroblasts from human postmyocardial infarction scars acquire properties of cardiomyocytes after transduction with a recombinant myocardin gene. FASEB J. 2007;21:3369–3379. doi: 10.1096/fj.07-8211com. [DOI] [PubMed] [Google Scholar]
- 23.Gonçalves MAFV, van der Velde I, Knaän-Shanzer S, Valerio D, de Vries AAF. Stable transduction of large DNA by high-capacity adeno-associated virus/adenovirus hybrid vectors. Virology. 2004;321:287–296. doi: 10.1016/j.virol.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Ni T-H, Zhou X, McCarty DM, Zolotukhin I, Muzyczka N. In vitro replication of adeno-associated virus DNA. J. Virol. 1994;68:1128–1138. doi: 10.1128/jvi.68.2.1128-1138.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kotin RM, Siniscalco M, Samulski RJ, Zhu XD, Hunter L, Laughlin CA, McLaughlin S, Muzyczka N, Rocchi M, Berns KI. Site-specific integration by adeno-associated virus. Proc. Natl Acad. Sci. USA. 1990;87:2211–2215. doi: 10.1073/pnas.87.6.2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonçalves MAFV. Adeno-associated virus: from defective virus to effective vector. Virology J. 2005;e2:e43. doi: 10.1186/1743-422X-2-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Young SM, Jr, McCarty DM, Degtyareva N, Samulski RJ. Roles of adeno-associated virus Rep protein and human chromosome 19 in site-specific recombination. J. Virol. 2000;74:3953–3966. doi: 10.1128/jvi.74.9.3953-3966.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis MD, Wu J, Owens RA. Mutational analysis of adeno-associated virus type 2 Rep68 protein endonuclease activity on partially single-stranded substrates. J. Virol. 2000;74:2936–2942. doi: 10.1128/jvi.74.6.2936-2942.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith RH, Kotin RM. An adeno-associated virus (AAV) initiator protein, Rep78, catalyzes the cleavage and ligation of single-stranded AAV ori DNA. J. Virol. 2000;74:3122–3129. doi: 10.1128/jvi.74.7.3122-3129.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Owens RA. Second generation adeno-associated virus type 2-based gene therapy systems with the potential for preferential integration into AAVS1. Curr. Gene Ther. 2002;2:145–159. doi: 10.2174/1566523024605627. [DOI] [PubMed] [Google Scholar]
- 31.Smith RH. Adeno-associated virus integration: virus versus vector. Gene Ther. 2008;15:817–822. doi: 10.1038/gt.2008.55. [DOI] [PubMed] [Google Scholar]
- 32.Nakahara M, Sonoda E, Nojima K, Sale JE, Takenaka K, Kikuchi K, Taniguchi Y, Nakamura K, Sumitomo Y, Bree RT, et al. Genetic evidence for single-strand lesions initiating Nbs1-dependent homologous recombination in diversification of Ig V in chicken B lymphocytes. PLoS Genet. 2009;5:e1000356. doi: 10.1371/journal.pgen.1000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Labib K, Hodgson B. Replication fork barriers: pausing or stalling for time? EMBO Rep. 2007;8:346–353. doi: 10.1038/sj.embor.7400940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strathern JN, Weinstock KG, Higgins DR, McGill CB. A novel recombinator in yeast based on gene II protein from bacteriophage f1. Genetics. 1991;127:61–73. doi: 10.1093/genetics/127.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galli A, Schiestl RH. Effects of DNA double-strand and single-strand breaks on intrachromosomal recombination events in cell-cycle-arrested yeast cells. Genetics. 1998;149:1235–1250. doi: 10.1093/genetics/149.3.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hickman AB, Ronning DR, Kotin RM, Dyda F. Structural unity among viral origin binding proteins: crystal structure of the nuclease domain of adeno-associated virus Rep. Mol. Cell. 2002;10:327–337. doi: 10.1016/s1097-2765(02)00592-0. [DOI] [PubMed] [Google Scholar]
- 37.Smith AM, Takeuchi R, Pellenz S, Davis L, Maizels N, Monnat RJ, Jr, Stoddard BL. Generation of a nicking enzyme that stimulates site-specific gene conversion from the I-AniI LAGLIDADG homing endonuclease. Proc. Natl Acad. Sci. USA. 2009;106:5099–5104. doi: 10.1073/pnas.0810588106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niu Y, Tenney K, Li H, Gimble FS. Engineering variants of the I-SceI homing endonuclease with strand-specific and site-specific DNA-nicking activity. J. Mol. Biol. 2008;382:188–202. doi: 10.1016/j.jmb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Im DS, Muzyczka N. The AAV origin binding protein Rep68 is an ATP-dependent site-specific endonuclease with helicase activity. Cell. 1990;61:447–457. doi: 10.1016/0092-8674(90)90526-k. [DOI] [PubMed] [Google Scholar]
- 40.Henckaerts E, Dutheil N, Zeltner N, Kattman S, Kohlbrenner E, Ward P, Clément N, Rebollo P, Kennedy M, Keller GM, et al. Site-specific integration of adeno-associated virus involves partial duplication of the target locus. Proc. Natl Acad. Sci. USA. 2009;106:7571–7576. doi: 10.1073/pnas.0806821106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Young SM, Jr, Samulski RJ. Adeno-associated virus (AAV) site-specific recombination does not require a Rep-dependent origin of replication within the AAV terminal repeat. Proc. Natl Acad. Sci. USA. 2001;98:13525–13530. doi: 10.1073/pnas.241508998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rinaudo D, Lamartina S, Roscilli G, Ciliberto G, Toniatti C. Conditional site-specific integration into human chromosome 19 by using a ligand-dependent chimeric adeno-associated virus/Rep protein. J. Virol. 2000;74:281–294. doi: 10.1128/jvi.74.1.281-294.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan I, Ng CH, Lim L, Leung T. Phosphorylation of a novel myosin binding subunit of protein phosphatase 1 reveals a conserved mechanism in the regulation of actin cytoskeleton. J. Biol. Chem. 2001;276:21209–21216. doi: 10.1074/jbc.M102615200. [DOI] [PubMed] [Google Scholar]
- 44.Loonstra A, Vooijs M, Beverloo HB, Al Allak B, van Drunen E, Kanaar R, Berns A, Jonkers J. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc. Natl Acad. Sci. USA. 2001;98:9209–9214. doi: 10.1073/pnas.161269798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu J, Jeppesen I, Nielsen K, Jensen TG. ϕC31 integrase induces chromosomal aberrations in primary human fibroblasts. Gene Ther. 2006;13:1188–1190. doi: 10.1038/sj.gt.3302789. [DOI] [PubMed] [Google Scholar]
- 46.Schmidt M, Afione S, Kotin RM. Adeno-associated virus type 2 Rep78 induces apoptosis through caspase activation independently of p53. J. Virol. 2000;74:9441–9450. doi: 10.1128/jvi.74.20.9441-9450.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu J, Davis MD, Owens RA. A Rep recognition sequence is necessary but not sufficient for nicking of DNA by adeno-associated virus type-2 Rep proteins. Arch. Biochem. Biophys. 2001;389:271–277. doi: 10.1006/abbi.2001.2348. [DOI] [PubMed] [Google Scholar]
- 48.Sanders KL, Catto LE, Bellamy SRW, Halfort SE. Targeting individual subunits of the FokI restriction endonuclease to specific DNA strands. Nucleic Acids Res. 2009;37:2105–2115. doi: 10.1093/nar/gkp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gonçalves MAFV, de Vries AAF. Adenovirus: from foe to friend. Rev. Med. Virol. 2006;16:167–186. doi: 10.1002/rmv.494. [DOI] [PubMed] [Google Scholar]
- 50.Jager L, Ehrhardt A. Emerging adenoviral vectors for stable correction of genetic disorders. Curr. Gene Ther. 2007;7:272–283. doi: 10.2174/156652307781369074. [DOI] [PubMed] [Google Scholar]