

Abstract
Tumor necrosis factor-alpha (TNF-α) is a cytokine that may contribute to the pathogenesis of septic shock, rheumatoid arthritis, cancer and diabetes. Prostaglandins endogenously produced by macrophages act in an autocrine fashion to limit TNF-α production. We investigated the timing and signaling pathway of prostaglandin-mediated inhibition of TNF-α production in RAW 264.7 and J774 macrophages. TNF-α mRNA levels were rapidly modulated by PGE2 or carbaprostacylin. PGE2 or carbaprostacyclin prevented and rapidly terminated ongoing TNF-α gene transcription within 15 min of prostaglandin treatment. Selective activation of PKA type I, but not PKA type II or Epac, with chemical analogs of cAMP was sufficient to inhibit LPS-induced TNF-α mRNA levels. The mechanisms by which prostaglandins limit TNF-α mRNA levels may underlie endogenous regulatory mechanisms that limit inflammation, and may have important implications for understanding chronic inflammatory disease pathogenesis.
Keywords: RAW 264.7 cells, inflammation, cAMP, Epac, PGE2, prostacyclin, macrophage, tumor necrosis factor-alpha, lipopolysaccharide
INTRODUCTION
Induction of inflammatory genes in macrophages is necessary for effective immune responses. Equally important, inflammation must be down-regulated to prevent excess tissue damage and chronic inflammatory states. One key pro-inflammatory cytokine produced by macrophages is tumor necrosis factor alpha (TNF-α). Dysregulated production of TNF-α may lead to human diseases including septic shock, rheumatoid arthritis, inflammatory bowel disease, insulin resistance (diabetes) and cancer [1; 2]. These examples of human disease may stem from a lack of appropriate down-regulation of the inflammation.
Macrophages release TNF-α, and activate cyclooxygenase-2 (COX-2) in response to stimulation with lipopolysaccharide (LPS). The subsequent release of prostaglandins (PGs) have well-known pro-inflammatory effects [3]. The limiting effects of PGs on inflammation are less well-appreciated. Prostaglandin E2 (PGE2) and prostacyclin (PGI2) may endogenously limit stimulus-induced cytokine secretion [4; 5]. Inhibition of constitutive COX-1 activity and PG synthesis in resident peritoneal macrophages (RPM) enhances TNF-α secretion from RPM responding to LPS. Adding back PGE2 or the stable PGI2 analog, carbaprostacyclin (cPGI2) returns TNF-α secretion to levels seen with endogenously produced PGs. Thus, endogenous PG biosynthesis plays a key role in regulating TNF-α secretion from LPS-stimulated RPM [4].
In contrast to RPM, the murine macrophage cell line RAW264.7 (Raw) does not produce PGs to auto-regulate TNF-α but maintains sensitivity to TNF-α suppression by exogenous PGs [6]. Lack of auto-regulation makes the Raw cell line well-suited to study the dynamics and mechanisms by which PGs suppress TNF-α biosynthesis in macrophages, both incompletely characterized in these cells.
PGE2 and PGI2 increase cytosolic cAMP through G-Protein coupled receptors (EP2, EP4 and IP). PGE2 inhibits LPS-induced TNF-α primarily at the level of gene transcription [7]. PGE2 or dibutyryl-cAMP also inhibit TNF-α protein release and mRNA levels when added to macrophages after 90-minute LPS induction [7; 8]. The major cellular effectors of increased cAMP include activation of cAMP-sensitive ion channels, exchange protein activated by cAMP (Epac) [9], and cAMP-dependent protein kinase (PKA) [10]. The two forms of PKA (types I and II) differ in regulatory subunit composition (RI or RII). In alveolar macrophages, cAMP analogs that non-specifically activate PKA, inhibit TNF-α release after LPS stimulation [11; 12]. The specific roles of PKA types I and II and the dynamics of TNF-α mRNA regulation by PGs is not known. We extended our characterization of macrophage responses to inflammatory stimuli and investigated the mechanisms that limit TNF-α biosynthesis.
MATERIALS AND METHODS
Cell culture and treatments
Raw and J774 cells (ATCC) were propagated in DMEM-GlutaMax (Gibco) and 10% heat-inactivated FCS (Atlas). E. Coli 0111:B4 LPS (Calbiochem) and cAMP analogs (Axxora) were delivered as 100x concentrates in sterile H2O. For cell treatments, macrophages were plated 6-well plates (60-80% confluence) and cultured overnight prior to the experiments. Cells were treated in serum free DMEM with a maximum DMSO vehicle content of 0.1%.
Real-Time RT-PCR
Total cellular RNA was primed with Oligo-dT and reverse-transcribed using Taq Man RT Reagents (Applied Biosystems). Real-time PCR reactions contained 0.1 μg cDNA, 200 nM primers, 25 μL of 2x Syber Green icycler supermix (Biorad), in a total volume of 50 μL and were run on a Biorad iCycler with melting point determination. RNA starting quantities were calculated using the Ct method (Biorad iCycler) based on known concentrations of linearized cDNA standard (PGEM-TNF or PGEM-GAPDH). Primers sequences were: TNF-α, 5′-cgtagcaaaccaccaagtgga-3′ and 5′-gctggcaccactagttggttgt-3′, GAPDH, were 5′-atggcaaagtggagattgttgg-3′ and 5′-tgccattgaatttgccgtg-3′ (Operon/Qiagen).
Plasmids
Cyclophillin cDNA was obtained from M. Magnuson, Vanderbilt University. TNF-α cDNA (GenBank accession no. X02611) was PCR-cloned from LPS-induced Raw cells and ligated into the PGEM easy-T vector. The amplified regions of the TNF-α mRNA and GAPDH mRNA were ligated into the PGEM easy-T vector and linearized to generate PGEM-TNF and PGEM-GAPDH. All DNA was sequenced prior to use.
Nuclear run-on transcription assay
108 Raw cells were washed in ice-cold PBS. Cells were lysed (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40), nuclei were pelleted, washed with lysis buffer and resuspended in 200 μL glycerol storage buffer (50 mM Tris-HCl, pH 8.3, 40% (v/v) glycerol, 5 mM MgCl2, 0.1 mM EDTA). 200 μL of 2x Reaction Buffer (10 mM Tris-HCl, pH 8.0, 5 mM MgCl2, 5 mM DTT, 0.3 M KCl, 1 mM ATP, 1 mM GTP, 1 mM CTP) and 10 μL of [α32-P]-UTP (3000 mCi/mmol) were added to nuclei and incubated for 30 min at 30 C with shaking. Aliquots (750,000 cpm) of nuclear RNA were hybridized for 60 h at 65 C in QuickHyb to cDNA probes on Hybond N+ membranes (7 μg linearized, denatured cDNA per slot). Blots were washed (twice 2xSSC/65 C/60 min, 2xSSC/10mg/mL RNAse A/37 C/30 min, 2xSSC/37 C), dried and quantitated by phosphorimager analysis. Transcription was determined relative to murine cyclophilin A. Values were normalized to values obtained from untreated controls.
Data analysis and statistical methods
Data were analyzed in Graph Pad Prism. Error bars represent the standard error of the mean of at least three experiments with duplicate determinations. P-values were determined by paired student’s T-test.
RESULTS
PGE2 rapidly reduces TNF-α mRNA levels
To gain insight into mechanisms responsible for inhibition of TNF-α mRNA accumulation, PGE2 or cPGI2 was added to Raw cells pretreated with LPS for 1 or 6 h (Figure 1). After 1 h of LPS pretreatment, Raw cells are maximally transcribing TNF-α mRNA and after 6 h of LPS pretreatment secreted TNF-α protein levels are highest (Figure S1). Addition of PGE2 to 1 h-pretreated cells abrogated the LPS-stimulated accumulation of TNF-α mRNA, and TNF-α mRNA levels declined by 40% within 1 h following PGE2 addition. cPGI2 also initiated a rapid decline in TNF-α mRNA levels when added to macrophages pretreated with LPS for 6 h (Figure 1B). TNF-α mRNA levels declined by 50% within 30 min of cPGI2 addition, and by 80% within 1 h after cPGI2 addition (Figure 1B). Thus, PGE2 and cPGI2 both rapidly lower TNF-α mRNA levels in macrophages during the early and late response to LPS (Figure 1A and 1B). These results indicate that the cellular signaling events that initiate PGE2-mediated reductions in LPS-induced TNF-α mRNA levels must also be rapid.
Figure 1. PGE2 and cPGI2 rapidly decrease TNF-α mRNA levels.

Raw cells were pretreated with 1 μg/mL LPS for 1 (A) or 6 (B) hours. PGE2 (A, σ) or cPGI2 (B, σ) or DMSO (ν) was added and TNF-α mRNA levels were measured at 15 minute intervals following PG addition and normalized to GAPDH.
PGE2 and cPGI2 rapidly halt TNF-α gene transcription
In response to inducing stimuli, TNF-α mRNA levels are increased by alterations in gene transcription and mRNA stability [1; 13]. Nuclear run-on transcription assays were conducted to directly measure TNF-α gene transcription (Figure 2). After 6 h of exposure to LPS, TNF-α gene transcription was 3.5 times higher in LPS-stimulated cells than untreated macrophages (Figure 2 bars A, B). Both PGE2 and cPGI2 completely prevented transcription when co-administered with LPS (bars C, F). To determine the effect of PGE2 and cPGI2 on ongoing transcription, macrophages were pretreated with LPS for 6 h, and PGE2 or cPGI2 was added to the cell culture medium for 15 or 30 min after the pretreatment and nuclear run-on analysis was performed (Figure 2 Bars D, E, G). Within 15 min of adding either prostanoid, the level of TNF-α transcription returned to unstimulated levels; PGE2 and cPGI2 completely halted ongoing TNF-α transcription.
Figure 2. PGE2 and cPGI2 prevent TNF-α transcription and halt ongoing TNF-α transcription.
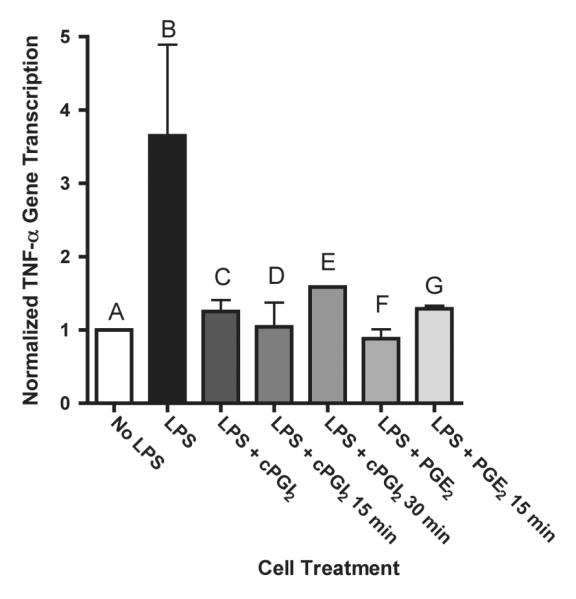
Nuclear run-on transcription was measured in Raw cells treated with 1 μg/mL LPS in the presence of DMSO/vehicle (B), 1 μM cPGI2 (C) or 1 μM PGE2 (F) for 6 h. Bars D, E, G: Raw cells were pretreated with1 μg/mL LPS for 6 h followed by addition of 1 μM cPGI2 (D, E) or 1 μM PGE2 (G) for 15 or 30 min.
The effects of PGE2 to inhibit TNF-α gene transcription are rapid and occur immediately following the addition of PG to LPS-pretreated macrophages. These findings indicate that the cellular mechanism leading to inhibition of TNF-α gene transcription likely involves rapid intracellular signaling and post-translational modification of cellular protein function rather than new protein synthesis. Therefore, we decided to investigate proximal PGE2 signaling to determine the cellular effectors of PGE2-mediated inhibition of TNF-α mRNA levels in Raw cells.
Activation of PKA type I inhibits LPS-induced TNF-α mRNA levels
PKA is activated by cAMP binding to regulatory subunits and subsequent dissociation of the regulatory and catalytic subunits. There are two types of PKA (I, II) classified based on regulatory subunit isoform. Each regulatory subunit contains two cAMP binding sites (A, B). Analogs of cAMP have been successfully employed to determine PKA type specificity for biologic actions [14]. Cyclic-AMP also binds to the exchange protein activated by cAMP (Epac) and activates Rap1. cPGI2 stimulation of Raw cells rapidly increases intracellular cAMP corresponding temporally to decreases in TNF-α mRNA levels following PGs (Figure 1, S2) It is not known whether PGs inhibit TNF-α through PKA type I or II. Synergistic pairs of cAMP analogs were employed to determine the involvement of PKA type I, PKA type II or Epac to mediate the inhibitory action on TNF-α mRNA levels of agents that increase cellular cAMP. The binding specificities and combinations of cAMP analogs used in these studies are listed in Table 1. J774 and Raw cells were stimulated with LPS and 250 μM of the respective cAMP analogs for 1 h. In both Raw cells and J774 cells, individual cAMP analogs did not significantly inhibit LPS-induced TNF-α mRNA levels (Figure 3A, 3C). Selective activation of PKA type I with 6-Bnz and 8-HA inhibited LPS-induced TNF-α mRNA levels by 50% in Raw and J774 macrophages (Figure 3A, 3C). Activation of PKA type II with 6-Bnz and 8-PIP did not significantly inhibit TNF-α mRNA levels induced by LPS stimulus in Raw or J774 macrophages (Figure 3A, 3C). Likewise, activation of Epac with 8-CPT did not inhibit TNF-α mRNA levels in either Raw or J774 macrophages (Figure 3B, 3D). In both cell lines, the inhibitory effect of PKA type I activation was constant in the presence of the Epac activator (Figure 3B, 3D).
Table 1.
Binding selectivity of cAMP analogs used in cell culture treatments.
 |
Figure 3. Selective activation of PKA Type I inhibits TNF-α mRNA levels in both J774 and Raw 264.7 macrophages.

A, C) Synergistic combinations of cAMP analogs (250 μM) were used to activate PKA type I or PKA type II in Raw (A) and J774 (C) macrophages. B, D) The addition of the Epac activator 8-CPT-2-OME did not alter inhibition of TNF-α mRNA levels by PKA type I or PKA type II activator combinations in Raw (B) or J774 (D) cells. E) J774 macrophages were induced with LPS (1 μg/mL) for 1 h with increasing concentrations of Sp-8-Br-cAMPS and/or Rp-8-Br-cAMPS. Macrophages were pretreated for 30 min with cAMP analogs prior to the addition of LPS (1 μg/mL).
Dibutyryl-cAMP inhibits TNF-α secretion from LPS-stimulated murine peritoneal macrophages and Raw 264.7 cells [8]. To test whether increases in cAMP and subsequent dissociation of the PKA holoenzyme are sufficient to decrease TNF-α mRNA levels, J774 macrophages were stimulated with LPS for 1 h in the presence of a cell permeable cAMP analog and activator of PKA, Sp-8-Bromo-cAMPS. In macrophages treated for 1 h with LPS and 1 μM Sp-8-Bromo-cAMPS, TNF-α mRNA levels were significantly decreased to 70.9 +/- 15.1% of LPS-stimulated levels. Addition of 250 μM Sp-8-Bromo-cAMPS inhibited TNF-α mRNA levels to 41.5 +/- 20.6% of LPS-stimulated levels (Figure 3E). To test whether dissociation of PKA regulatory and catalytic subunits was necessary to reduce TNF-α mRNA levels, Rp-8-Bromo-cAMPS was used as a competitive antagonist of Sp-8-Bromo-cAMPS. Rp-8-Bromo-cAMPS binds to regulatory subunits but does not stimulate the dissociation of the regulatory and catalytic subunits, preventing activation of PKA [15]. Rp-8-Bromo-cAMPS fully reversed the inhibitory effect of Sp-8-Bromo-cAMPS on TNF-α mRNA levels in J774 macrophages (Figure 3E) indicating that dissociation of PKA regulatory and catalytic subunits was required for the inhibitory effect on TNF-α mRNA levels. Because Rp-8-Br-cAMPS and Sp-8-Br-cAMPS bind competitively to cAMP binding sites in the PKA regulatory subunits, use of 250 μM Rp-8-Br-cAMPS only partially alleviated the repression observed with 250 μM Sp-8-Br-cAMPS.
DISCUSSION
The generation of macrophage cytokine responses is important for coordinating the immune response to infection. Macrophage stimulation with LPS induces cytokines genes including TNF-α, concomitant with activation of cyclooxygenases and subseqent release of PGs. Although the pro-inflammatory effects of PGs are well-appreciated, it is clear that PGE2 and PGI2 also have important endogenous roles to limit or direct inflammatory responses [4; 6; 7; 16]. The limiting effects of PGE2 on macrophage TNF-α secretion are partially responsible for increased susceptibility to lung infection after bone marrow transplantation [5]. PGE2 or cAMP-mediated suppression of dendritic cell function may also be responsible for decreased tumor cell immunosurveilance and decreased dendritic cell cytotoxicity and TNF-α secretion [17; 18]. Since autocrine PG signaling does not affect TNF-α secretion by Raw cells [6], and the cell line is frequently used to study inflammatory signaling, we characterized TNF-α regulation in Raw cells stimulated with LPS and PGE2 or cPGI2. We report that regulation of TNF-α mRNA levels is a dynamic process that happens within minutes after the addition of the PG to LPS-stimulated macrophages (Figure 1). The rapid inhibition of TNF-α mRNA levels by PGE2 or cPGI2 happens at early and late time points in the response of macrophages to LPS (Figure 1). Our results are consistent with previous reports that delayed addition of PGE2 to LPS-stimulated macrophages inhibits TNF-α mRNA levels, when measured hours after the addition of the PG [7; 8; 16]. The biological half-lives of PGE2 and PGI2 are short (< 1.5 min in circulation) owing to rapid in vivo metabolism [19; 20]. Consequently, it is physiologically important that regulation of TNF-α mRNA levels by PGs requires neither copious concentrations [7; 8], nor pre-exposure to the inhibitory agent. Rather, PGE2 and PGI2 may regulate TNF-α mRNA levels at any point in the macrophage response to LPS, and on the same time scale as the PGs accumulate locally and are metabolized in vivo.
Our studies suggest key characteristics about the mechanism by which PGs and cAMP lower LPS-induced TNF-α secretion. PGE2 and cPGI2 rapidly alter TNF-α mRNA levels through a robust and abrupt halt of on-going TNF-α gene expression (Figure 2) leading to rapid reductions in TNF-α mRNA levels (Figure 1). The rapid kinetics of transcriptional inhibition by PGE2 and cPGI2 and the decrease in mRNA levels correspond temporally with observed increases in cAMP accumulation following cPGI2 treatment of LPS-stimulated macrophages (Figures 3, S2). In addition, cAMP analogs recapitulate the effects of PGE2 and cPGI2 to inhibit LPS-induced TNF-α mRNA levels (Figure 3). These results indicate that rapid increases in intracellular cAMP levels are sufficient to inhibit TNF-α transcription in response to PGE2 and cPGI2. The timing of cAMP formation and rapid inhibition of transcription and mRNA stability, indicate that new protein synthesis is probably not necessary for PG-mediated regulation of TNF-α mRNA levels in macrophages.
We investigated the downstream signaling of PGE2 and cAMP to determine the target of cAMP signaling in macrophages. Using synergistic pairs of cAMP analogs that preferentially activate PKA type I or PKA type II, we demonstrate that inhibition of TNF-α mRNA levels by cAMP occurs selectively through the activation of PKA type I in both Raw 264.7 and J774 macrophages (Figure 3). Inhibition of TNF-α mRNA levels by PKA type I was not altered by the co-activation of Epac (Figure 3). These results agree with recently published results in alveolar macrophages demonstrating a lack of Epac involvement and PKA-dependent inhibition of TNF-α secretion from alveolar macrophages [11]. Thus, selective activation of PKA type I with cAMP analogs recapitulated the effects of PGE2 and cPGI2 on TNF-α mRNA levels in two macrophage cell lines. These results provide an important link between the known mechanisms that stimulate TNF-α gene transcription in J774 macrophages [13] and the pathway for cAMP-dependent inhibition of TNF-α biosyntheses in Raw and J774 macrophages. Collectively, our results indicate that PGE2 and cPGI2 inhibit LPS-induced TNF-α transcription in macrophages through an immediate increase of intracellular cAMP levels that selectively activates PKA type I to inhibit TNF-α gene transcription.
Differences in the cellular effects of PKA type I and PKA type II also occur in other immune cells [18; 21]. For example, activation of PKA type I, but not PKA type II inhibits cytotoxic activity and cytokine secretion from lymphokine-activated killer cells [18]. Also, the most upstream mechanism for cAMP-mediated inhibition of T cell activation by T cell receptor stimuli requires the activation of PKA type I leading to the inhibition of Lck [22]. Signaling specificity occurs because PKA type I specifically co-localizes in lipid rafts with the T cell receptor and Lck. The LPS receptor complex, TLR4, may also reside within lipid raft membrane microdomains [23]. As with T cells, PKA type I could also be specifically targeted to the lipid raft domains in macrophages, and similar targeting may be responsible for the specific involvement of PKA type I in the inhibitory action of PG on TNF-α biosynthesis. However, we have observed that PGE2 does not globally or specifically inhibit LPS stimulated phosphorylation of MAP kinases, p38, or JNK (JBS, unpublished findings), suggesting that, unlike T cell receptor activation, inhibition of LPS-stimulated TNF-α transcription by PKA type I may not occur at the level of TLR4 receptor activation. In addition, recently reported dual ligand screening studies of cytokine secretion from Raw cells co-treated with LPS and PGE2 indicate that PGE2 does not globally inhibit cytokine secretion from Raw cells. Rather, PGE2 inhibits only LPS-induced TNF-α and MIP1α secretion, while stimulating LPS-induced GCSF and IL-10 secretion [24]. These results suggest that the mechanism by which PKA type I activation inhibits TNF-α transcription may not occur through upstream inhibition of TLR4 signaling.
Collectively, these studies indicate that regulation of the TNF-α cytokine levels happens in part through rapid and dynamic alterations in mRNA levels. Owing to the short TNF-α mRNA half-life and the sensitivity of TNF-α gene transcription to repression, TNF-α mRNA levels are regulated rapidly in response to PG signaling. The proximal inhibitory mechanism involves the specific activation of PKA type I. Mediators that increase cAMP may serve to direct, modulate, or turn off TNF-α biosynthesis in response to inflammatory stimuli, or may be necessary for normal regulation of inflammatory pathways under physiologic conditions.
Supplementary Material
AKNOWLEDGEMENTS
This research was supported by a grant from the National Institutes of Health (GM015431).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity. 1996;4:445–54. doi: 10.1016/s1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- [2].Moller DE. Potential role of TNF-alpha in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol Metab. 2000;11:212–7. doi: 10.1016/s1043-2760(00)00272-1. [DOI] [PubMed] [Google Scholar]
- [3].Higgs GA. The role of eicosandoids in inflammation. Prog Lipid Res. 1986;25:555–61. doi: 10.1016/0163-7827(86)90113-x. [DOI] [PubMed] [Google Scholar]
- [4].Rouzer CA, Kingsley PJ, Wang H, Zhang H, Morrow JD, Dey SK, Marnett LJ. Cyclooxygenase-1-dependent prostaglandin synthesis modulates tumor necrosis factor-alpha secretion in lipopolysaccharide-challenged murine resident peritoneal macrophages. J Biol Chem. 2004;279:34256–68. doi: 10.1074/jbc.M402594200. [DOI] [PubMed] [Google Scholar]
- [5].Ballinger MN, Aronoff DM, McMillan TR, Cooke KR, Olkiewicz K, Toews GB, Peters-Golden M, Moore BB. Critical role of prostaglandin E2 overproduction in impaired pulmonary host response following bone marrow transplantation. J Immunol. 2006;177:5499–508. doi: 10.4049/jimmunol.177.8.5499. [DOI] [PubMed] [Google Scholar]
- [6].Rouzer CA, Jacobs AT, Nirodi CS, Kingsley PJ, Morrow JD, Marnett LJ. RAW264.7 cells lack prostaglandin-dependent autoregulation of tumor necrosis factor-alpha secretion. J Lipid Res. 2005;46:1027–37. doi: 10.1194/jlr.M500006-JLR200. [DOI] [PubMed] [Google Scholar]
- [7].Kunkel SL, Spengler M, May MA, Spengler R, Larrick J, Remick D. Prostaglandin E2 regulates macrophage-derived tumor necrosis factor gene expression. J Biol Chem. 1988;263:5380–4. [PubMed] [Google Scholar]
- [8].Spengler RN, Spengler ML, Lincoln P, Remick DG, Strieter RM, Kunkel SL. Dynamics of dibutyryl cyclic AMP-and prostaglandin E2-mediated suppression of lipopolysaccharide-induced tumor necrosis factor alpha gene expression. Infect Immun. 1989;57:2837–41. doi: 10.1128/iai.57.9.2837-2841.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–7. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- [10].Taylor SS, Knighton DR, Zheng J, Ten Eyck LF, Sowadski JM. cAMP-dependent protein kinase and the protein kinase family. Faraday Discuss. 1992:143–52. doi: 10.1039/fd9929300143. [DOI] [PubMed] [Google Scholar]
- [11].Aronoff DM, Canetti C, Serezani CH, Luo M, Peters-Golden M. Cutting edge: macrophage inhibition by cyclic AMP (cAMP): differential roles of protein kinase A and exchange protein directly activated by cAMP-1. J Immunol. 2005;174:595–9. doi: 10.4049/jimmunol.174.2.595. [DOI] [PubMed] [Google Scholar]
- [12].Aronoff DM, Carstens JK, Chen GH, Toews GB, Peters-Golden M. Short communication: differences between macrophages and dendritic cells in the cyclic AMP-dependent regulation of lipopolysaccharide-induced cytokine and chemokine synthesis. J Interferon Cytokine Res. 2006;26:827–33. doi: 10.1089/jir.2006.26.827. [DOI] [PubMed] [Google Scholar]
- [13].Tsai EY, Falvo JV, Tsytsykova AV, Barczak AK, Reimold AM, Glimcher LH, Fenton MJ, Gordon DC, Dunn IF, Goldfeld AE. A lipopolysaccharide-specific enhancer complex involving Ets, Elk-1, Sp1, and CREB binding protein and p300 is recruited to the tumor necrosis factor alpha promoter in vivo. Mol Cell Biol. 2000;20:6084–94. doi: 10.1128/mcb.20.16.6084-6094.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ogreid D, Ekanger R, Suva RH, Miller JP, Sturm P, Corbin JD, Doskeland SO. Activation of protein kinase isozymes by cyclic nucleotide analogs used singly or in combination. Principles for optimizing the isozyme specificity of analog combinations. Eur J Biochem. 1985;150:219–27. doi: 10.1111/j.1432-1033.1985.tb09010.x. [DOI] [PubMed] [Google Scholar]
- [15].Botelho LH, Webster LC, Rothermel JD, Baraniak J, Stec WJ. Inhibition of cAMP-dependent protein kinase by adenosine cyclic 3′-, 5′-phosphorodithioate, a second cAMP antagonist. J Biol Chem. 1988;263:5301–5. [PubMed] [Google Scholar]
- [16].Kunkel SL, Wiggins RC, Chensue SW, Larrick J. Regulation of macrophage tumor necrosis factor production by prostaglandin E2. Biochem Biophys Res Commun. 1986;137:404–10. doi: 10.1016/0006-291x(86)91224-6. [DOI] [PubMed] [Google Scholar]
- [17].Yang L, Yamagata N, Yadav R, Brandon S, Courtney RL, Morrow JD, Shyr Y, Boothby M, Joyce S, Carbone DP, Breyer RM. Cancer-associated immunodeficiency and dendritic cell abnormalities mediated by the prostaglandin EP2 receptor. J Clin Invest. 2003;111:727–35. doi: 10.1172/JCI16492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lokshin A, Raskovalova T, Huang X, Zacharia LC, Jackson EK, Gorelik E. Adenosine-mediated inhibition of the cytotoxic activity and cytokine production by activated natural killer cells. Cancer Res. 2006;66:7758–65. doi: 10.1158/0008-5472.CAN-06-0478. [DOI] [PubMed] [Google Scholar]
- [19].Hamberg M, Samuelsson B. On the metabolism of prostaglandins E 1 and E 2 in man. J Biol Chem. 1971;246:6713–21. [PubMed] [Google Scholar]
- [20].Wong PY, Sun FF, McGiff JC. Metabolism of prostacyclin in blood vessels. J Biol Chem. 1978;253:5555–7. [PubMed] [Google Scholar]
- [21].Tasken K, Stokka AJ. The molecular machinery for cAMP-dependent immunomodulation in T-cells. Biochem Soc Trans. 2006;34:476–9. doi: 10.1042/BST0340476. [DOI] [PubMed] [Google Scholar]
- [22].Vang T, Torgersen KM, Sundvold V, Saxena M, Levy FO, Skalhegg BS, Hansson V, Mustelin T, Tasken K. Activation of the COOH-terminal Src kinase (Csk) by cAMP-dependent protein kinase inhibits signaling through the T cell receptor. J Exp Med. 2001;193:497–507. doi: 10.1084/jem.193.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Triantafilou M, Morath S, Mackie A, Hartung T, Triantafilou K. Lateral diffusion of Toll-like receptors reveals that they are transiently confined within lipid rafts on the plasma membrane. J Cell Sci. 2004;117:4007–14. doi: 10.1242/jcs.01270. [DOI] [PubMed] [Google Scholar]
- [24].Natarajan M, Lin KM, Hsueh RC, Sternweis PC, Ranganathan R. A global analysis of cross-talk in a mammalian cellular signalling network. Nat Cell Biol. 2006;8:571–80. doi: 10.1038/ncb1418. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.