

Abstract
We have recently identified two promoters, distal and proximal for rat mitochondrial glycerophosphate acyltransferase (mtGPAT). Here we are reporting further characterization of the promoters. Insulin and epidermal growth factor (EGF) stimulated while leptin and glucagon inhibited the luciferase activity of the distal promoter and the amounts of the distal transcript. Conversely, luciferase activity of the proximal promoter and proximal transcript remained unchanged due to these treatments. Only the distal promoter has binding sites for carbohydrate response element binding protein (ChREBP) and sterol regulatory element binding protein-1 (SREBP-1). Electromobility shift assays and chromatin immunoprecipitation assays demonstrated that ChREBP and SREBP-1 bind to the mtGPAT distal promoter. Insulin and EGF increased while glucagon and leptin decreased the binding of SREBP-1 and ChREBP to the distal promoter. Thus, the distal promoter is the regulatory promoter while the proximal promoter acts constitutively for rat mtGPAT gene under the influence of hormones and growth factor.
Introduction
Glycerol-3-phosphate acyltransferase (GPAT)1 converts sn-glycerol-3-phosphate to 1-acyl-sn-glycerol-3-phosphate, the first step in the biosynthetic pathway of all glycerolipids. Mitochondrial GPAT (mtGPAT) is an enzyme that can switch the fate of fatty acids from β-oxidation to glycerolipid synthesis [1, 2]. The mtGPAT isoform is known to control the asymmetric distribution of fatty acids in cellular glycerolipids. The asymmetric distribution of fatty acids plays an important role in maintaining structure and function of phospholipids present in cellular membranes [3]. The mitochondrial acyltransferase is known to comprise ~10% of total GPAT activity in most tissues except in the liver where it comprises ~50% of the total GPAT activity [4]. Hepatic mtGPAT mRNA level is up-regulated by a high carbohydrate, fat-free diet and by insulin administration to streptozotocin-diabetic mice. Also, mtGPAT activity is higher in the livers from both diet-induced obese mice and leptin-deficient ob/ob mice when compared with their lean controls [5].
It has been shown that elevated hepatic mtGPAT is associated with obesity since the enzyme can divert activated fatty acids from oxidation to glycerolipid synthesis. Thus, the increase in mtGPAT activity could be at least partly responsible for obesity and lipid disorders associated with obesity [2]. Therefore, understanding the molecular mechanism of mtGPAT transcriptional regulation is important.
We have previously shown the presence of two promoters for rat mtGPAT: a distal promoter which is ~ 30kb upstream of the first translational codon and a proximal promoter which is 63 bp upstream of the second translational codon. It is known that lipogenic enzymes such as acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS), like mtGPAT, are hormonally and nutritionally regulated - properties consistent with their role in triacylglycerol synthesis [6]. Studies show that insulin and EGF stimulate lipogenesis and ACC activity in isolated adipocytes [7]. On the other hand, leptin and glucagon are known to inhibit lipogenic enzyme synthesis by acting through decrease of the amounts of the lipogenic transcription factor, SREBP-1c and intracellular mediator cAMP, respectively [2, 6]. In an effort to determine whether the distal or the proximal promoter is responsible for the transcriptional regulation of rat mtGPAT, two stimulating agents (insulin and EGF) and two inhibiting agents (glucagon and leptin) for lipogenesis were used in this investigation.
The transcriptional factors such as ChREBP (100 kDa) and SREBP-1c (68 kDa) are known to be major regulators of lipogenic enzymes [8, 9]. ChREBP is predominantly expressed in liver, kidney, white and brown adipose tissues and is known to recognize E box sequences in the promoters of target genes and is predominantly present in inactive phosphorylated form in the cytoplasm [10]. The regulation of ChREBP is relatively simple and efficient, and involves phosphorylation-dependent mechanisms responsive to feeding (glucose and fatty acids) and fasting (glucagon) [8]. The extent of phosphorylation of ChREBP determines its nuclear or cytoplasmic location. For example, when overnight-fasted mice were refed a high-carbohydrate diet for 18 h, there was low ChREBP phosphorylation on Ser196 causing it to be predominantly located in the nucleus. When glucagon was injected (0.5 U/kg) into the portal vein of refed mice, ChREBP phosphorylation on Ser196 was increased and ChREBP protein was exported from the nucleus [11]. Three SREBPs have been identified so far, SREBP-1a and SREBP-1c are encoded from the same gene while SREBP-2 is encoded from a separate gene. The predominant SREBP-1 isoform in liver and adipose tissue is 1c rather than 1a [12]. Hence in all likelihood, SREBP-1c actually binds to the distal promoter due to its abundance in liver cells. Promoter inspector analysis showed the presence of the binding sites for SREBP-1 and ChREBP only in the distal promoter of rat mtGPAT gene. There is no such presence of lipogenic transcription factor binding sites in the proximal promoter region [13]. Leptin is known to decrease expression of lipogenic enzyme gene through modification in the SREBP-1c gene expression [14]. EGF stimulates SREBP-1c mRNA levels in prostate cancer cells and causes an increase in mRNA levels of the lipogenic enzyme FAS [15]. Studies have shown that the negative target for leptin is SREBP-1, suggesting that this transcription factor may be involved in mediating the inhibitory effect of leptin on lipogenic gene expression [16, 17,]. Insulin was found to increase the levels of ChREBP mRNA in 3T3 L1 adipocytes and to promote lipogenesis in newly formed adipocytes [18].
In this study, we have investigated the role of the two promoters in transcriptional regulation of rat mtGPAT gene expression. We present evidence that the distal promoter is the regulatory promoter under the influence of hormones and growth factors while the proximal promoter acts like a constitutive promoter. To gain further insight into the molecular mechanism of how the distal promoter regulates the mtGPAT gene transcription, we have assessed the role of lipogenic transcription factors, ChREBP and SREBP-1c by means of electromobility shift assay (EMSA) and chromatin immunoprecipitation (ChIP).
Materials and Methods
Cell culture
Buffalo rat liver cells (BRL) which are hepatocytes were bought from ATCC (CRL-1442) and were cultured in Eagle’s minimum essential medium (ATCC) supplemented with 10% fetal bovine serum (ATCC) and 50 U/ml of penicillin-streptomycin at 37°C in a humidified atmosphere of 5% CO2.
Chemicals
Insulin, EGF, glucagon, leptin, formaldehyde and protease inhibitors were purchased from Sigma - Aldrich; penicillin-streptomycin was from Gibco; wortmannin and 1--(5-isoquinolinylsulfonyl)-2-methylpiperazine (H7) were from Calbiochem.
Plasmid constructs
Serial deletion constructs for distal promoter regions: D 520, D 340 and D 215, were generated using D 932 [13] as the template and the primers as shown in Table 1 and Fig. 1A. These fragments were then subcloned into pGL3-basic vector into NheI and KpnI sites. After subcloning, the PCR amplified fragments were sequenced (Genewiz) to exclude the possibility of mutations caused by PCR. For the proximal promoter region, “Proximal 500” from the previous study was used [13]. The plasmid pRLTK containing renilla luciferase reporter gene (Promega), was used as an internal control for normalizing firefly luciferase activity.
Table 1. Oligonucleotide PCR primers.
List of primers used for serial deletion constructs, ChIP assay, EMSA and semiquantitative PCR, respectively.
| Primer | Primer sequence (5′→ 3′) | Orientation | Plasmid name | Product size (bp) |
|---|---|---|---|---|
| Serial deletion constructs: | ||||
| F1 | AAACAGTTGCTAGCGCGTACCTG | Forward | D 932 | 932 |
| R1 | CGTGTAGCTAGCGAGGCTGGAGG | Reverse | ||
| F2 | GCCACGAGCCTAGAGCTCCATGTGAC | Forward | D 520 | 520 |
| R1 | CGTGTAGCTAGCGAGGCTGGAGG | Reverse | ||
| F3 | TAAATTATAACGAGCCCTAAAACTGGC | Forward | D 340 | 340 |
| R1 | CGTGTAGCTAGCGAGGCTGGAGG | Reverse | ||
| F4 | TGAAAGAGCCTTCCTCCTCCTCCTCCAC | Forward | D 215 | 215 |
| R1 | CGTGTAGCTAGCGAGGCTGGAGG | Reverse | ||
| ChIP primers: | Primer name | |||
| F5 | GCCCCACTCAAGTCTCTTTCCTAGC | Forward | ChF | 200 |
| R5 | GTAGAGATTTTTCTAACGGGGCAGGG | Reverse | ChR | |
| F6 | TCGAGCTTCGCAACCCTCCCCCTC | Forward | SREF | 164 |
| R6 | AAGCATGAGAAGAGGCAGACCCAGGC | Reverse | SRER | |
| RT PCR primers: | ||||
| F7 | GGCATTTCTCCGGGGTTACAGCCA | Forward | 124 | |
| R7 | ATCCCCAGACAGCTTCAGAAGACAGCCT | Reverse | ||
| F8 | TTGCTGACAGGATGCAGAAGGAG | Forward | 120 | |
| R8 | GTGAGGCCAGGATAGAGCCACCAATCC | Reverse | ||
| EMSA primers: | ||||
| SREBP-1 | GCCAAGAAGACACCCCACCCCCCACTCACCCCTGTG | |||
| mutSREBP-1 | GCCAAGAAGACATTTTGTTCCCCACTCACCCCTGTG | |||
| ChREBP | CGAGCCTAGAGCTCCATGTGACCTCTGGATGCT | |||
| mutChREBP | CGAGCCTAGAGCTCCCGAGGACCTCTGGATGCT | |||
| Semiquantitative PCR primers: | ||||
| AAP | GGCCACGCGTCGACTAGTA CGGGIIGGGII GGGIIG | |||
| NGSP2 | GCTGGGCAGATCCATCCGGGTTCAA | |||
Fig. 1. Deletion analysis of rat mtGPAT distal promoter by using Luciferase reporter gene constructs.
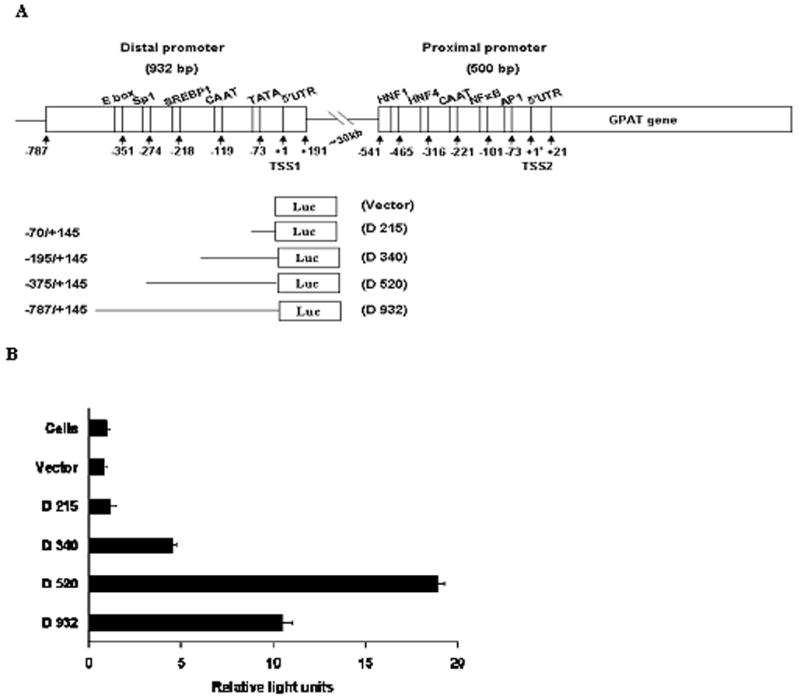
A. Schematic presentation of the distal and proximal promoters. TSS1 is represented as +1 and TSS2 as +1*. The distal promoter is ~30kb upstream of the proximal promoter region. Transcription factor binding sites, TATA box, inverted CAAT box and 5′UTR of both the distal and the proximal promoters relative to their individual TSSs are shown. Luciferase constructs of the distal promoter are shown with the numbers indicating the number of nucleotides upstream of TSS1. B. Cells were cotransfected with serial deletion constructs and pRLTK (internal control) or with the promoterless vector (pGL3-basic) and pRLTK. Luciferase activity was measured 24 hours post-transfection. Promoter activity is expressed in relative light units. Results are the averages of three independent experiments performed in triplicates ± S.E.
Transient transfections, treatments and luciferase activity assays
For 5′-end serial deletion analysis, all the plasmid DNA constructs were transfected into cells in 24 well plates using Lipofectamine (Invitrogen) following the manufacturer’s protocol. Twenty hours before transfection, 8 × 104 cells were plated in 0.5 ml of medium/well. For each well, 2 μl of lipofectamine was mixed with plasmid DNA (2 μg) in serum-free media to allow the complex to form. The complex was added to each of the wells and incubated for 24 hours and then lysed with 250 μl of 1X passive lysis buffer (Promega). Luciferase activity was measured using equal amounts of protein with dual luciferase kit (Promega) with Berthold multiplate reader LB140. Proximal 500 and D 520 constructs were used for luciferase assays to determine the effect of insulin, EGF, glucagon and leptin on proximal and distal promoters, respectively. Transfections were done as mentioned above and 24 hours later cells were treated with either 1, 10, 100 or 1000 nM of insulin, EGF, glucagon or leptin [19–22]. To determine the effects of the inhibitors, the cells were pretreated with 20 μM wortmannin or with 50 μM H7 (1-(5-isoquinolinylsulfonyl)-2-methylpiperazine) for 2 hours and then treated with the respective agents as mentioned under “Results”. Cells were lysed 24 hours post treatment and luciferase activity was measured as mentioned above on equal amounts of proteins for each sample. Data (mean ± S.E.) from triplicate assays from three independent experiments were computed.
Isolation of rat mitochondria and nuclear extracts from treated cells
Mitochondria were isolated from cell culture by differential centrifugation at 600×g and 11000×g using Mitochondria Isolation kit (Sigma - Aldrich). Intactness of isolated mitochondrial membrane was determined by measuring the latency of the cytochrome c-oxidase activity by measuring the enzyme in the presence and absence of the detergent, n-Dodecyl β-D-Maltoside (~95% intact). Nuclear extracts were prepared according to the manufacturer’s protocol using NXTRACT CelLytic NuCLEAR Extraction Kit (Sigma-Aldrich).
Western blotting of mtGPAT, SREBP-1c and ChREBP
Whole mitochondria (5 μg) or nuclear extract (4 μg) isolated from cells was used for western blotting as explained elsewhere [13]. IM1GAT (24) for whole mitochondria and anti SREBP-1 (Santa Cruz) which detects both SREBP-1a and SREBP-1c or anti-ChREBP (Novus Biologicals) antibodies were used for nuclear extracts. The membranes were stripped and reprobed for β-actin (Sigma - Aldrich) to ensure equal loading of proteins as described previously [13]. As a control to show that the increase or decrease in the nuclear levels of ChREBP or SREBP-1c is caused by the four agents, cells were pretreated for 2 hours either with 20 μM wortmannin and then treated with 100 nM of EGF or leptin, or with 50 μM H7 followed by treatment with 100 nM of insulin and glucagon.
GPAT Assay
GPAT assay was performed as described previously by measuring sn-[2-3H]-glycerol-3-phosphate incorporated in 1-butanol-extractable phospholipids, lysophosphatidic acid, and phosphatidic acid [23]. Each assay was performed in the presence of NEM (5 mM) to inhibit microsomal GPAT activity and was initiated by the addition of 50μl of isolated mitochondrial sample. GPAT specific activity was measured as nmole/min/mg [13].
RNA isolation of treated cells, semiquantitative RTPCR and quantitative real-time PCR
Cells were treated with 100 nM or 1000 nM of either insulin, glucagon, leptin or EGF and incubated for 24 hours. Total RNA was isolated using RNeasy minikit (Qiagen) and treated with RNase-free DNase. The first-strand cDNA was synthesized using GSP1 primer (5′GCTTTAGTAACACCCAGCCAGTCAGCC3′) and Superscript II reverse transcriptase (Invitrogen) using 2 μg of total RNA for insulin or EGF treated cells and 4 μg of total RNA for glucagon or leptin treated cells. Five μl of each sample cDNA was subsequently used for PCR. cDNA amplifications were performed in 40 cycles for mtGPAT and 25 cycles for β-actin (loading control), to get detectable amounts of DNA and to keep the reactions in the exponential phase. The thermal profile of PCR was as follows: denaturation at 94°C for 1 min, annealing at 65°C for 1 min, extension at 72°C for 2 min and final extension at 72°C for 7 mins. The primers F7 and R7 (Table 1) were used to amplify 124 bp region of mtGPAT while primers F8 and R8 were used to amplify 120 bp of rat β-actin. The PCR products were resolved on 2% agarose gel, stained with ethidium bromide and photographed. Semiquantitative analysis of the amplicons was performed by measuring the band intensities using Un-scan-it gel software (Silk Scientific). Quantitative real time PCR (Biorad) was performed on the first strand cDNA that was synthesized from total RNA (3μg). The same sets of primers and PCR conditions were used as mentioned above.
Semiquantification of the two transcripts
The first strand was synthesized using 2 μg of total RNA from insulin and EGF treated cells and 4 μg of total RNA from cells treated with glucagon and leptin to get detectable bands using gene specific primer of rat mtGPAT cDNA, GSP2 (5′GGAGACTGTAGCAACCATTTCCTGGAGG3′). Homopolymeric (C) tails were added to the 3′ end of the first strand cDNA using dCTP and terminal deoxynucleotidyl transferase (Invitrogen). First round of PCR was performed for amplification of the target first strand cDNA using AAP (Abridged Anchor Primer) 5′GGCCACGCGTCGACTAGTA CGGGIIGGGII GGGIIG-3′, where I is inosine) (Invitrogen) and nested gene specific primer, NGSP2 5′GCTGGGCAGATCCATCCGGGTTCAA3′ for 40 cycles as shown in table 1. β-actin was used as a loading control. The PCR cycles were optimized for both the transcripts. The same PCR program as mentioned above was used for amplification of mtGPAT transcripts and for β-actin. Both GSP2 and NGSP2 were designed from exon 6 of mtGPAT gene as shown in Fig. 4A. The amplified DNA was run on 1.5% agarose gel. Two amplified bands were excised from the gel. The DNA was isolated following gel extraction using MinElute gel extraction kit (Qiagen) and subsequently cloned into pCR4-TOPO cloning vector (Invitrogen) and sequenced by Genewiz to confirm the presence of the two transcripts.
Fig. 4. Effect of the agents on the distal and proximal promoter transcript of mtGPAT gene.
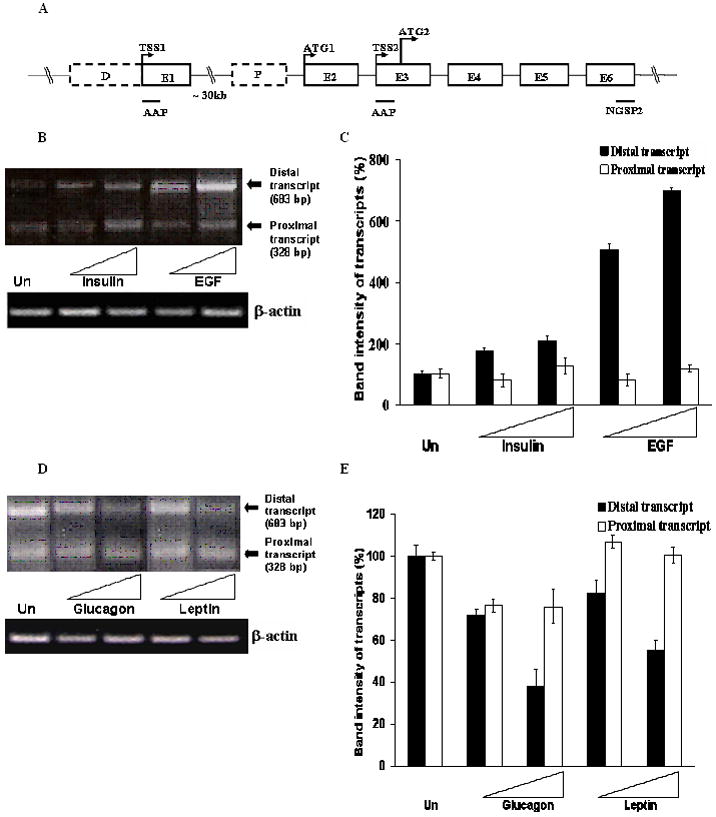
A. Schematic presentation of the genomic region of mtGPAT gene with the arrangement of exons and introns and their respective sizes. Dotted boxes indicate promoters (D –distal promoter, P – proximal promoter), solid boxes indicate exons and lines indicate the introns. B, D. Total RNA, 2 μg from cells treated with insulin and EGF (stimulators) and 4 μg from cells treated with glucagon and leptin (inhibitors), was used to synthesize the first strand cDNA. PCR was performed for 40 cycles and the products were run on 1.5% agarose gel. Upper band is from the distal promoter transcript while the lower band is from the proximal promoter transcript. β-actin was used as a loading control. C, E. Densitometric analysis of the band intensities. Black bars and white bars represent average values obtained from the densitometric analysis of the distal and proximal products obtained from four different experiments. Error bars indicate ± S.E. The intensities of each band in the top panel were normalized to the intensity of the corresponding actin band for each lane in the bottom panel to correct for differences in loading. The values are expressed as a percentage of the band intensities of the untreated cells.
Electromobiltiy shift assays (EMSA)
The double stranded oligonucleotides were used as probes for EMSA as shown in Table 1. The consensus sequences for SREBP-1c and ChREBP binding regions of rat mtGPAT distal promoter are underlined and the mutated nucleotides are shown in bold. End labeling was performed using T4 kinase (Promega) in the presence of [γ-32P]ATP. The labeled oligonucleotide was purified on a Sephadex G-25 column, as described elsewhere [24]. Nuclear extracts (about 6 μg of protein in 5 μl) from untreated cells were incubated for 20 min at room temperature with 10 fmol of either radiolabeled probe (SREBP-1/ChREBP) or with radiolabeled mutated probe (mutSREBP-1/mutChREBP) in 20 μl of binding buffer (20 mM Tris-Cl, pH 7.5, 150 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, 0.1% Nonidet P-40, 6% glycerol) supplemented with 20 μg of acetylated bovine serum albumin and 2 μg of poly(dI-dC). For competition analysis, the indicated amount of wild type or mutated double-stranded oligonucleotides was added to the binding mixture as indicated in the figure legends or 1 μg of specific polyclonal antibody of SREBP-1 (Santa Cruz) and ChREBP (Santa Cruz), respectively, and incubated 20 min at room temperature before adding 32P-labeled oligonucleotide (SREBP-1/ChREBP). The resulting complexes were resolved on 5% nondenaturing polyacrylamide gels, the gel was dried and analyzed by Phosphorimager (Packard Cyclone) as described elsewhere [24].
Chromatin immunoprecipitation assays
ChIP analyses were performed using an assay kit (Upstate Biotechnology) as described previously [13]. An aliquot (50 μl) was removed to use as an input DNA control. Immunoprecipitation was performed with antibodies against ChREBP (Novus Biologicals) or SREBP-1 (Santa Cruz Biotechnology) or with normal rabbit serum (Calbiochem) that served as a control for 12–18 hours at 4°C. For a negative control, no-antibody immunoprecipitation was performed by incubating the supernatant fraction with 50% slurry of Salmon Sperm DNA/Protein A agarose for 1 hour at 4°C with rotation. The immunoprecipitated DNA was analyzed by real-time PCR (25 μl reaction mixture) using the iQ SYBR Green Supermix and the Bio-Rad MyIQ Single Color Real-Time PCR Detection System (Bio-Rad). Each PCR reaction was used to determine the enrichment of ChREBP or SREBP-1c at its respective binding sites. The occupancy of ChREBP and SREBP-1c was calculated using β-actin coding sequence as a negative control and corrected for the efficiency of primers. The primers F5 and R5 were designed to flank the E box region and primers F6 and R6 were designed to flank the SREBP-1c region of the distal promoter, respectively (Table 1 and Fig. 1A). Primers F8 and R8 were used for rat β-actin coding sequence as a negative control (Table 1). Briefly, to quantify the association of ChREBP or SREBP-1c with the distal promoter, a ΔCt value was calculated for immunoprecipitated sample (IP) or input sample (total DNA) by subtracting the Ct value for the β-actin from the Ct value obtained with the corresponding immunoprecipitated sample as template. The obtained ΔCt value for the samples immunoprecipitated with specific antibodies was subtracted from the ΔCt value of samples immunoprecipitated with rabbit IgG as control to give Δ ΔCt. Fold differences were calculated using the formula 2ΔΔCt, since one cycle difference is equivalent to a 2-fold difference in amplification. After PCR amplification, melt curve analysis was performed for each sample to ensure that the melting curve characteristics were consistent with the presence of a single PCR product. ChIP assays were performed on at least 3 independent occasions and for each ChIP assay, promoter samples were quantified in triplicates. Thus, for every sample, data represents the average of at least 9 determinations ± S.D., and are presented as fold-differences relative to control conditions (untreated cells). The relative fold change among the ChIP DNA samples was calculated following normalization with the input DNA. To ensure that ChREBP and SREBP-1 bind to the putative binding sites in the distal promoter region, PCR was performed using the same sets of primers as mentioned before for SREBP-1c, ChREBP and rat β-actin. The DNA isolated from input, immunoprecipitated (ChREBP/SREBP-1/IgG) and negative control samples were used and the PCR products were run on a 2% agarose gel [13].
Band intensity quantification
Semiquantitative analysis of the band intensities were performed by measuring the band intensities using Un-scan-it gel software (Silk Scientific). The intensities of each band were normalized to the intensity of the corresponding actin band for each lane in the bottom panel to correct for differences in loading.
Results
The distal 520bp mtGPAT promoter is essential for its regulatory effect
A schematic diagram of the distal and the proximal promoters with putative transcription factor binding sites, TATA box and inverted CAAT box is presented in Fig. 1A. We have previously shown that 500 bp of the proximal promoter region has about 2 fold luciferase activity over the full-length 1050 bp region [13]. Similarly, transfection experiments were performed to define the region of the distal promoter that is required for its maximum promoter activity. A series of 5′-nested serial deletion constructs of rat mtGPAT distal promoter were cloned into pGL3-basic vector that contained firefly luciferase as the reporter gene as shown in Fig. 1A and as explained under “Materials and Methods”. The constructs made for luciferase activity using the primers as shown in Table 1 and the inserts shown in Fig 1A were as follows: (a) plasmid containing the entire distal promoter region upstream of luciferase (D 932), (b) plasmid containing all the transcription factor binding sites of distal promoter upstream of luciferase (D 520), (c) plasmid containing only inverted CAAT box and TATA box of distal promoter upstream of luciferase (D 340), and (d) plasmid excluding transcription factor binding sites, and inverted CAAT and TATA boxes (D 215). Comparative analysis of the luciferase activities of the serial deletion constructs revealed that the 520 bp region of the distal promoter displayed stronger transcriptional activity than that of the 932 bp region. This is probably due to the presence of all the transcription factor binding sites in the 520 bp region (Fig. 1B) and the presence of inhibitory domains of the active promoter in the upstream sequence. This situation is similar to the presence of the inhibitory domains in the proximal promoter region of the rat Cathepsin L gene [25].
The distal, but not the proximal promoter is regulatory in BRL cells
To investigate the roles that the distal and the proximal promoters play in the transcriptional regulation of rat mtGPAT gene, cells were transfected with either D 520 (distal promoter) or with Proximal 500 (proximal promoter) constructs along with pRLTK as an internal control and treated with varying concentrations of insulin, glucagon, leptin or EGF as explained in “Materials and Methods”. As shown in Fig. 2A, the luciferase activity of the cells transfected with D 520 construct and treated with insulin or EGF was up to 175% and 200% of the luciferase activity of the untreated cells, respectively. The luciferase activity of the cells transfected with D 520 construct and treated with either glucagon or leptin, was only 83% and 77% of the untreated cells’ luciferase activity at the highest concentration, respectively (Fig. 2B). On the other hand, the luciferase activity of cells transfected with Proximal 500 and treated with any one of the four agents remained at about 110% to 140% of the untreated cells’ luciferase activity at the highest concentration (Fig 2C, D). Cells were also pretreated with H7 as explained in “Materials and Methods”, a PKA/PKC inhibitor, and is known to block insulin and glucagon mediated pathways [26, 27]. These cells were used as a control. H7 reversed insulin mediated stimulation of luciferase assay completely and glucagon mediated inhibition of luciferase assay of the distal promoter marginally (Fig. 2A, B). Leptin and EGF are known to work through PI3K pathway and hence wortmannin, a PI-3K inhibitor [29] was used as a control. Wortmannin blocked EGF and leptin mediated change in luciferase activity (Fig. 2A, B). Later experiments showed that the inhibition of protein synthesis of SREBP-1c and ChREBP by 100 nM glucagon was reversed by treating cells with H7 (Fig. 6A, B). This treatment of cells with inhibitors served as a control to show that the stimulation or inhibition of the luciferase activity is hormone or EGF specific and is not due to some other stimuli in the cell.
Fig. 2. Effect of the hormones and EGF on the luciferase activity of distal and proximal promoters.
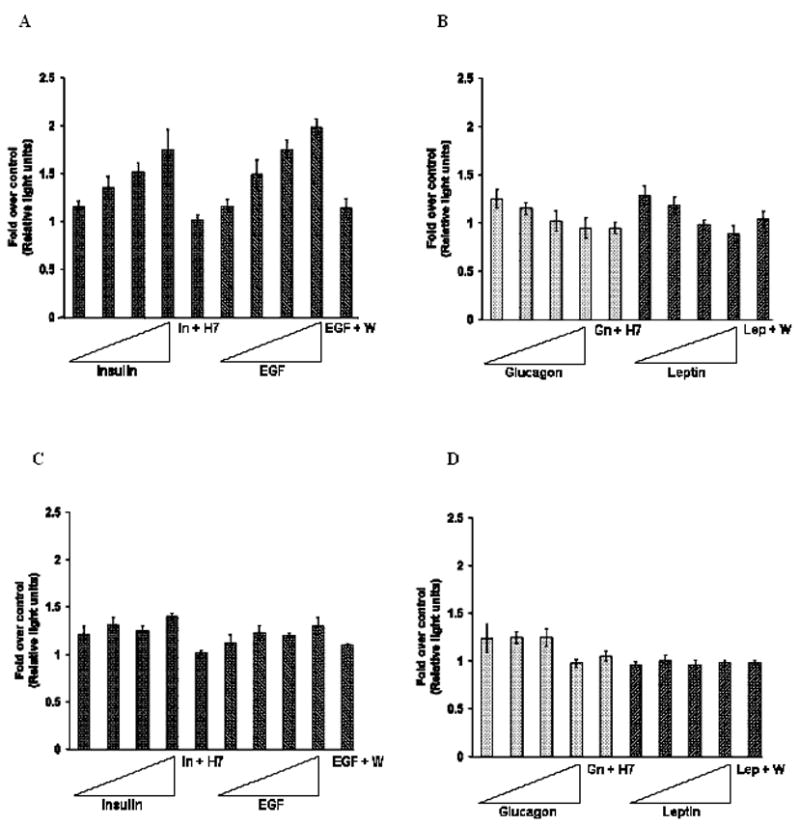
A, B.Plasmid D 520 was cotransfected with pRLTK (internal control) into cells and treated with increasing concentrations (1 nM–1000 nM) of insulin, EGF, glucagon or leptin for 24 hours. Luciferase assay of cells treated with 20 μM Wortmannin (W) or 50 μM of H7 for 2 hours followed by treatment with 100 nM of EGF, leptin, insulin or glucagon for 24 hours are denoted as E+W, L+W, I+H7 and Gn+H7, respectively. Results are expressed as fold over the luciferase activity of the untreated cells’ (control). The results are the averages of three independent experiments performed in triplicates ± S.E. C, D. Plasmid Proximal 500 was cotransfected with pRLTK (internal control) into cells. Cells were treated or untreated with increasing concentrations (1nM–1000 nM) of insulin, EGF, glucagon or leptin for 24 hours. Luciferase assay of cells treated with 20 μM Wortmannin (W) or 50 μM of H7 for 2 hours followed by treatment with 100 nM of EGF, leptin, insulin or glucagon for 24 hours are denoted as E+W, L+W, I+H7 and Gn+H7, respectively. Results are expressed as fold over the luciferase activity of the untreated cells’ (control). The results are the averages of three independent experiments performed in triplicates ± S.E.
Fig. 6. Protein levels of ChREBP and SREBP-1 in the nucleus of the treated cells.
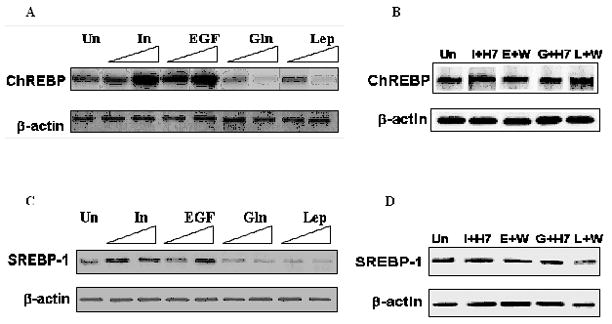
A, C Nuclear extracts from cells treated with 100 nM or 1000 nM of the agents were used to perform Western blotting to detect the amount of ChREBP and SREBP-1 available for binding to the promoter. B, D. Nuclear extracts from cells pretreated with H7 or wortmannin (W) for 2 hours followed by treatment with 100 nM of the respective four agents viz. insulin (I), EGF (E), glucagon (G) and leptin (L) as explained in “Materials and Methods” were used to perform Western blotting to detect the amount of ChREBP and SREBP-1.
mtGPAT protein expression is upregulated by insulin and EGF and downregulated by leptin and glucagon
To determine whether there was increased or decreased level of mtGPAT expression due to the agents, measuring GPAT specific activity as well as Western blotting were performed on mitochondria isolated from the treated cells. Cells were treated with the higher two concentrations (100 nM or 1000 nM) for all the agents. Cells treated for 24 hours with insulin or EGF at the highest concentrations, showed over 200% of the GPAT specific activity in comparison to that of the untreated cells (Fig. 3A). On the other hand, the cells treated for 24 hours with glucagon or leptin at the highest concentrations, showed only 78% and 62.5% of control GPAT specific activity, respectively (Fig 3A). Western blotting of mitochondria isolated from cells treated with 100 nM or 1000 nM of each of the four agents and the bands intensities of mtGPAT were quantified after normalizing with the respective β-actin bands. Cells treated with either insulin or EGF, was up to about 150% of the untreated cell’s band intensity. Similarly, for cells treated with glucagon and leptin, the band intensities were only 45% and 35% of the band intensities of the untreated cells, respectively. Thus, there is a strong correlation between the changes in the protein level and specific activities of the treated cells (Fig. 3A, B).
Fig. 3. Effect of the four agents on mtGPAT protein expression and specific activity.
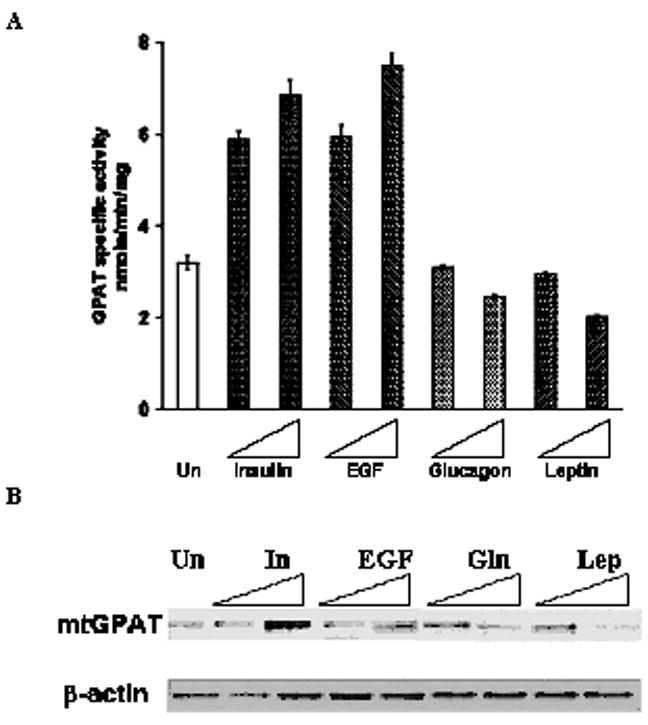
A. GPAT specific activity (nmole/min/mg) was measured for mitochondria isolated from the untreated cells and cells treated cells as described in “Materials and Methods”. Results are average of three independent experiments performed in triplicates. Error bars indicate ± S.E. B. Whole mitochondria (5μg) were subjected to 10% SDS-PAGE and immunoprecipitated with IM1GAT antibody. Equal amount of loading was confirmed by stripping the membrane and reprobing with β-actin antibody.
Effect of the agents on distal and proximal promoter transcripts
To determine the effect of the four agents on the two transcripts, semiquantitative PCR was performed. PCR cycle was optimized at 40 for both the distal and proximal transcript after confirming the linearity of the PCR reaction. Total RNA was isolated from cells treated with 100 nM or 1000 nM of each of the agents. Two μg of total RNA from cells treated with insulin or EGF and 4 μg of total RNA from cells treated with glucagon and leptin were used to synthesize the first strand cDNA. Thus, double the amount of total RNA was used for cells treated with glucagon and leptin to get detectable bands for the same number of cycles.
PCR cycle was optimized at 40 cycles to ensure linearity of the reaction for both the stimulated and inhibited mRNA productions. PCR was performed with the first strand cDNA using reverse primers from exon 6 (Fig. 4A) and then the PCR product was run on 1.5% agarose gel. For cells treated with 100 nM or 1000 nM of insulin or EGF, there was up to 200% and 700% increase in band intensities of distal transcript (684 bp), respectively as compared with the untreated cells. But there was no such change in the proximal transcript (328 bp) for cells treated with 100 nM or 1000 nM of insulin or EGF (Fig. 4B, C). Similarly, for cells treated with 100 nM or 1000 nM of glucagon or leptin, the band intensities were about 40% and 55% of the band intensities of the untreated cells, respectively (Fig. 4D, E). No such pattern of decrease was observed for the proximal transcript for cells treated with 100 nM or 1000 nM of glucagon or leptin (Fig. 4D, E). Semiquantitative as well as quantitative real time PCR was performed to evaluate the effect of the agents on the distal transcript using the primers as shown in Table 1. The forward primer was designed from 5′UTR of the distal transcript to make it specific to the distal transcript. An increase in mRNA levels was observed when cells were treated with 100 nM or 1000 nM of insulin or EGF while a pattern of decrease was observed for cells treated with 100 nM or 1000 nM of glucagon or leptin (Fig. 5A). β-actin was used as the loading control for both the semiquantitative RT-PCR and quantitative real-time PCR results. Fig. 5B shows the expression levels of distal transcript of mtGPAT gene determined by real-time quantitative RT-PCR on 2 μg of total RNA isolated from untreated as well as treated cells. Amounts of mtGPAT distal transcripts obtained from cells treated with the higher concentrations of insulin or EGF were found to be 200% and 300% of those obtained from the untreated cells, respectively (Fig. 5B). mRNA obtained from cells treated with the higher concentrations of glucagon or leptin was 58% and 50% of those obtained from the untreated cells, respectively (Fig. 5B).
Fig. 5. Effect of 100 nM and 1000 nM of each of the agents on distal transcript of rat mtGPAT gene.
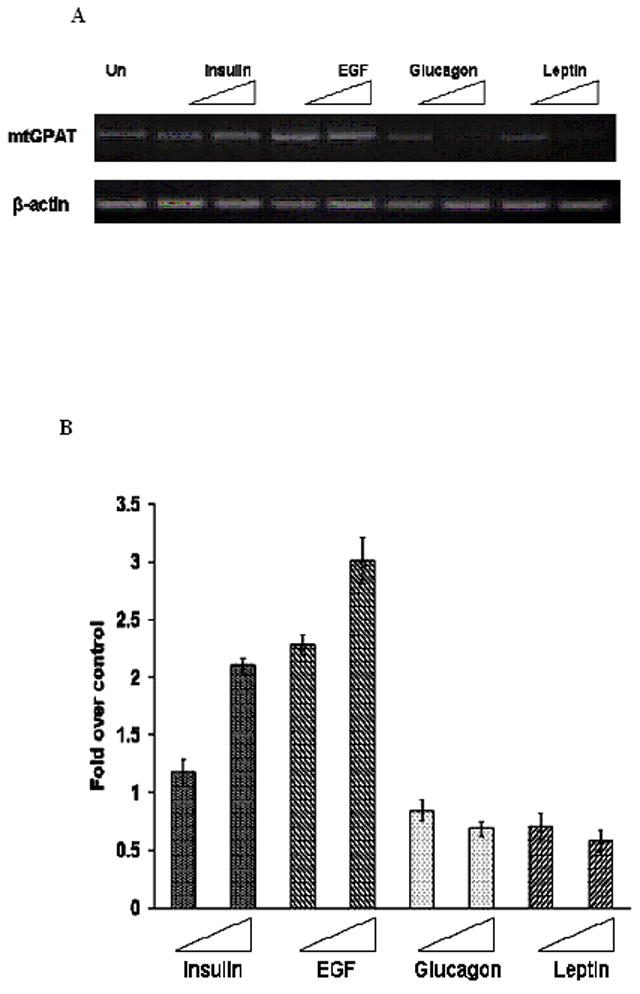
Cells were treated with either 100 nM or 1000 nM of each of insulin, EGF, glucagon or leptin. A. After 24 hours of treatment, total RNA was isolated and distal transcript (683 bp) and β-actin (120 bp) mRNA expression levels were analyzed by semiquantitative RT-PCR as described under “Materials and Methods”. The RT-PCR products were electrophoresed in 2% agarose gel. B. Quantification of distal promoter transcript by real-time PCR in untreated and treated cells. To correct for variations in input RNA, the data were normalized using β-actin. Data are represented as fold over control (untreated cells) mRNA levels as determined by ΔΔCt method. Data are represented as the average of four different experiments ± S.E.
Role of SREBP-1c and ChREBP in the transcriptional regulation of the mtGPAT gene
Promoter inspector analysis of the distal promoter has shown the presence of potential binding sites for lipogenic transcription factors such as SREBP-1c and ChREBP [13] which are known to play a role in the transcriptional regulation of lipogenic enzymes [8, 29]. Western blotting was performed to analyze how the four agents affect the nuclear level of SREBP-1c and ChREBP. Insulin or EGF caused an increase of ChREBP in the nucleus to about 215% of the band intensity of the untreated cells (Fig. 6A). For cells treated with glucagon or leptin the band intensities of ChREBP decreased to up to 45% and 35% of the untreated cell’s band intensities, respectively (Fig. 6A). Similarly, the amount of SREBP-1c in the nucleus for cells treated with insulin or EGF was up to 180% of that of the untreated cell’s band intensity (Fig. 6C). For cells treated with glucagon or leptin the amounts of SREBP-1c in nucleus was about 70% and 60% of those of the untreated cells, respectively (Fig. 6C). As a control to show that the increase or decrease in the nuclear levels of ChREBP or SREBP-1c is caused by the four agents, cells were pretreated with H7 or wortmannin for 2 hours followed by treating the cells with 100 nM of the respective four agents as explained in “Materials and Methods” (Fig. B, D). To identify ChREBP and SREBP-1c binding regions in the distal promoter, we used EMSA with 32P-labeled consensus sequence specific probes (SREBP-1/ChREBP) and mutated probes (mutSREBP-1/mutChREBP). The results shown in Fig. 7A, B, indicate that ChREBP and SREBP-1c bind to the respective putative binding sites on the distal promoter. There was no binding observed when the nuclear extract was incubated with radiolabeled mutated probes (Fig. 7A, B). Competition and supershift analysis revealed the specific binding of the radiolabeled oligo with the respective transcription factors viz. ChREBP and SREBP-1c (Fig. 7A, B). Since supershift assay performed by ChREBP and SREBP-1 antibodies produced a distinct single gel-shifted band with each probe, ChIP assay was further performed as mentioned in “Materials and Methods” to confirm the specificity of the binding of ChREBP and SREBP-1c to the putative binding sites. No antibody treatment (-IP) and precipitation with IgG (IgG control) were performed as negative controls. Immunoprecipitated bands were observed only when ChIP was performed in the presence of specific antibodies, not in the absence. Rat β-actin was detected only in input DNA sample (Fig. 7C). To further establish the role of ChREBP and SREBP-1c in the transcriptional regulation of mtGPAT gene, in vivo ChIP assays were performed in which DNA-binding proteins were covalently linked to genomic DNA by treating the cells with formaldehyde. Cross-linked chromatin was fragmented and immunoprecipitated with either specific (ChREBP or SREBP-1) or non specific antibodies (normal rabbit serum IgG). To determine whether ChREBP and SREBP-1c bind to rat mtGPAT gene in vivo, ChIP assays coupled with real time PCR analysis was performed on the precipitated DNA as explained in “Materials and Methods”. Results show that ChREBP recruitment in the distal promoter for cells treated with insulin was about 300% of the untreated cells’ recruitment; the cells treated with EGF showed no significant change in the recruitment of the ChREBP (Fig. 8B). The ChREBP recruitment in the glucagon or leptin treated cells was about 30% of that of the untreated cells (Fig. 8B). Recruitment of SREBP-1c for cells treated with insulin or EGF was up to 750% and 1400% of the untreated cells’ recruitment while in the cells treated with glucagon or leptin the SREBP-1c recruitment was about 30% of that of the untreated cells (Fig. 8C). When cells were treated with 50μM H7 for 2 hours followed by treatment with 1000 nM of insulin or glucagon there was no increase or decrease of both ChREBP and SREBP-1c recruitment and was almost similar to the SREBP-1c recruitment of the untreated cells (Fig. 8B, C). Similarly, when cells were treated with 20 μM wortmannin for 2 hours followed by treatment with 1000 nM of glucagon or leptin, they showed no such increase or decrease of recruitment of SREBP-1c (Fig. 8B, C).
Fig. 7. Electromobility shift assay for SREBP-1c and ChREBP binding to distal promoter.
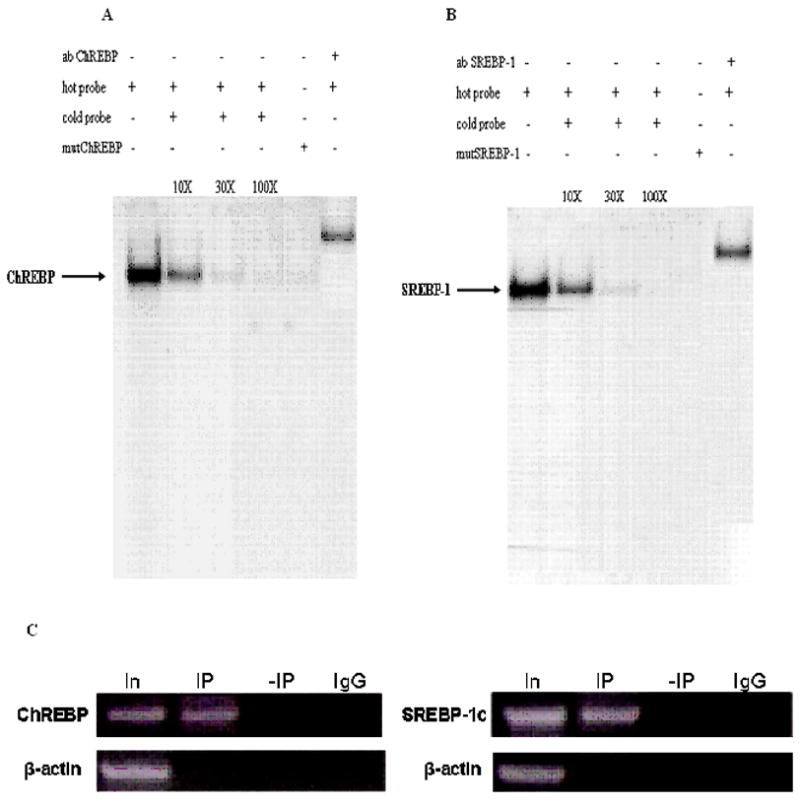
EMSAs were performed using radiolabeled oligonucleotide as mentioned in “Materials and Methods” for either the distal promoter containing the ChREBP (hot probe) binding site (A) or the SREBP-1 (hot probe) binding site (B), and nuclear extracts from untreated cells. The arrows denote the migration of SREBP-1 or ChREBP complex. Specificity of binding of ChREBP and SREBP-1c is demonstrated by competition by the respective non-radiolabeled probes (cold probe) added in excess (30 M) and the failure of the radiolabeled mutated probes to bind with the respective transcription factors. Competition assay was performed in the absence or presence of the indicated molar excess of unlabeled oligonucleotides. Supershift assay performed with the respective antibodies completely abolished the migration of the respective complexes. C. Chromatin immunoprecipitation of distal promoter region for binding of ChREBP and SREBP-1 to their respective binding sites. Input DNA or DNA immunoprecipitated with ChREBP or SREBP-1 antibodies or IgG were amplified as explained in “Materials and Methods”. In, input DNA; IP, immunoprecipitated DNA;-IP, no immunoprecipitation; IgG, immunoprecipitated with normal rabbit IgG.
Fig. 8. Chromatin Immunoprecipitation analysis of ChREBP and SREBP-1c binding to the mtGPAT distal promoter.
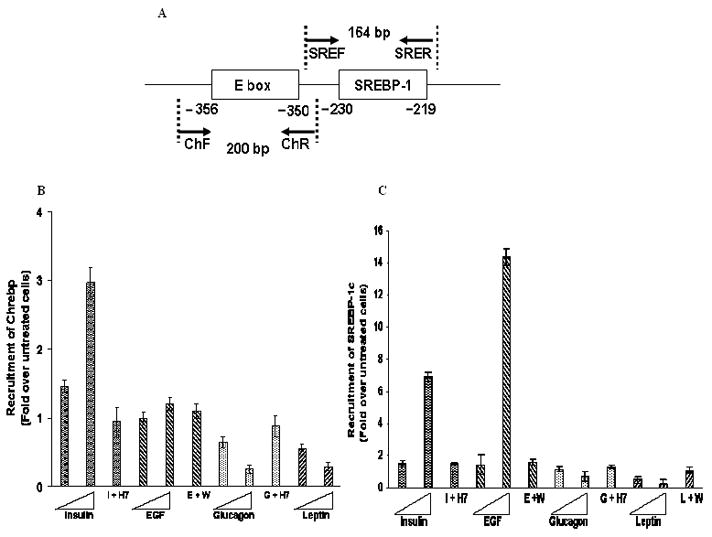
A. Schematic presentation of the putative ChREBP and SREBP-1c binding sites in the distal promoter region and the primers used for the ChREBP and SREBP-1c. The numbers represent the distance in nucleotides from the TSS1. B, C. Cells were treated with 100 nM or 1000 nM of insulin, EGF, glucagon or leptin for 24 hours. DNA binding proteins were crosslinked with formaldelhyde and chromatin was sonicated. Immunoprecipitation was performed using either specific antibodies such as ChREBP/SREBP-1 or with non specific antibody (normal rabbit serum). After reversing the cross links, immunoprecipitated DNA was isolated and analyzed. The primers that amplified 164 bp and 200 bp flanking the SREBP-1 and ChREBP binding sites in the distal promoter were used. Data represent the average of at least 9 determinations ± S.D., and are presented as fold-differences relative to control conditions (untreated cells).
Discussion
The present study provides insight into the mechanism of the transcriptional regulation of rat mtGPAT gene. We have demonstrated here that the distal promoter is the regulatory promoter under the influence of hormones and growth factor while the proximal promoter acts constitutively. We have further investigated the mechanism of the transcriptional regulation by the distal promoter and have determined that the distal promoter controls the transcriptional regulation by lipogenic transcription factors such as ChREBP and SREBP-1c.
We have previously established the presence of two promoter sequences controlling the transcription of rat mtGPAT gene [13]. The distal promoter is ~30 kb upstream (first intron) of the first translational codon while the proximal promoter is 63 bp upstream of second translational codon (Fig. 1A). The significance of these two promoter regions remains unclear. Previous study showed that when cells were starved and refed, luciferase activity regulated by the distal promoter was about three fold as compared to the cells grown in complete media. Conversely, the proximal promoter’s luciferase activity remained unchanged [13].
Fig 4A shows two initiating codons. Probably two proteins are produced under different conditions. We know, from our previous experiments (Onorato et al, 2005, Ref. 23; Balija and Haldar, Unpublished results) that the whole or a partially truncated (165 bp) mitochondrial GPAT cDNA can be used to express the active acyltransferase. However, these two proteins, even if they are produced simultaneously, cannot be detected as separate entities by Western blot. However, when expressed separately, each of them react to the same antibody.
In this study we found that the levels of expression of the distal transcript were more variable than that of the proximal transcript (Fig. 4B, D) after treating the cells with the agents and were more closely correlated with the expression and specific activity of mtGPAT protein (Fig. 3A, B). This was consistent with the idea that the distal promoter is the regulatory promoter which causes inducible/repressible level of expression of the distal transcript while the proximal promoter maintains a basal level of expression as shown by the semiquantitative RTPCR (Fig. 4 B, D). The distal transcript is synthesized from TSS1 present in exon 1 while the proximal transcript is synthesized from TSS2 present in exon 3. When usual range of concentrations of the four agents was used similar to previous studies [19–22], we found similar dose dependent changes caused in the luciferase activity of the distal promoter. However, it must be mentioned here that the higher concentrations of insulin might be working in part through the IGF-1(insulin-like growth factor I) receptor pathway [30]. Previous studies have shown that high-dose (1μM) of insulin activate IGF-1 receptor and induce an increase in SREBP-1 mRNA, protein and total lipid production in human SEB-1 sebocytes [31]. Thus, it seems possible that the high concentrations of insulin could also activate IGF-1 receptors and up-regulate SREBP-1 protein which then bind to the distal promoter of mtGPAT, thereby causing an increase in lipogenesis. In obesity, there is a development of insulin resistance causing an increase in insulin levels and cause an increase in bioavailable IGF-1. These two promote cell growth and inhibit apoptosis thereby contributing to tumorigenesis. However, the differential behavior of the two promoters under the influence of the four agents is obvious (Fig. 2A, B, C, D). There is a growing body of evidence that suggests that a number of mammalian genes are regulated by multiple promoters and have transcripts with different TSSs [32–35]. These multiple promoters behave differentially depending on the cellular conditions [36, 37]. For example, proximal promoter of the rat pyruvate carboxylase gene plays a major role in gluconeogenesis and lipogenesis, whereas the distal promoter is necessary for anaplerosis [38]. The general view about multiple promoters is that when a single gene is transcribed from multiple promoters, an organism gains additional flexibility in the control of the expression of that gene [39]. This could be the case with rat mtGPAT gene where the two promoters function differentially under the influence of nutritional or hormonal changes. Hence it is important to determine which of the two promoters is responsible for the transcriptional regulation of rat mtGPAT gene under the influence of the hormones and EGF. Although studies show that SREBP-1c and ChREBP act in synergy to induce lipogenic enzymes like ACC and FAS [18], the role of these transcription factors in the transcriptional regulation of the regulatory distal promoter have not been identified. In this study we have demonstrated that the up- or down-regulation of the distal promoter occurs due to these transcription factors (Fig. 8B, C). Previous studies have shown that exposure of insulin to hepatocytes causes an increase in lipogenic enzyme transcription mediated by an increase in SREBP-1c transcription.
Glucagon, cAMP and fatty acid lead to the phosphorylation of ChREBP and its subsequent exclusion from nucleus, whereas glucose causes dephosphorylation and subsequent nuclear localization of ChREBP. It is known that ChREBP is regulated at two levels, nuclear localization brought about by phosphorylation-dependent mechanism in response to glucagon (or hormones), cAMP, fatty acids and glucose and subsequent DNA binding. Hence in the presence of glucagon and leptin, ChREBP and SREBP-1c are expressed less and are probably in their inactive forms localized in the cytosol. It is known that in the presence of cAMP, a site near the nuclear localization signal is phosphorylated by PKA and ChREBP being unable to enter the nucleus, remains in the cytosol [11]. Also, the expression of SREBP-1c was also found to be regulated by insulin [40], and SREBP-1c abundance is tightly related to the nutritional state in liver and adipose tissue. SREBPs get activated by undergoing a sequential two-step cleavage process to release their NH2-terminal segments that can then translocate to the nucleus. This process is tightly regulated [40]. Hence when cells were treated with insulin or EGF, higher nuclear translocation of the active ChREBP and SREBP-1c was expected and observed (Fig. 6A, C). This might be responsible in the observed up-regulation of their bindings to the distal promoter region. Though EGF caused an increase in the nuclear translocation of ChREBP yet it failed to cause an increase in binding in the distal promoter. In contrast, glucagon and leptin caused a decrease in the nuclear translocation of both the transcription factors thereby causing a decreased binding to the distal promoter region. These observations were reversed when cells were treated with H7 or wortmannin (Fig. 6B, D).
Previous studies have shown that ChREBP and SREBP-1c function coordinately to regulate lipogenesis. The purpose of having two factors to control transcriptional regulation is that the metabolic intermediates cannot be stored until all conditions such as glucose and insulin signals which work via ChREBP and SREBP-1c, respectively, are optimally set. Another explanation is that there is an interplay between ChREBP and SREBP-1c thereby giving a better means of integrated transcriptional regulation [41]. For example, mice with a deletion of the SREBP-1 gene have an impaired ability to fully respond to a high-carbohydrate diet. However, these mice do retain some significant dietary response, which is caused by ChREBP gene [41–43]. Hence the synergistic effect of the two transcription factors would provide an effective means to regulate transcription of lipogenic enzymes by integrating multiple nutritional or hormonal inputs. This integration is important to control the energy status in mammals [41]. Most lipogenic enzyme genes (e.g., fatty acid synthase) have response elements for binding ChREBP (ChoRE) and SREBP (SRE). These two factors work synergistically to induce transcription of the lipogenic enzyme genes in the presence of glucose and insulin. Glucagon, through its intracellular mediator cAMP, and polyunsaturated fatty acids (PUFAs) act to inhibit the activity of ChREBP and SREBP, respectively [44]. In this manner, the output of lipogenic enzyme gene production is integrated to multiple nutrient and hormonal signals [41].
So, the next question arises why rat mtGPAT gene requires two promoters to regulate its transcriptional activity? It is known that some eukaryotic genes such as rat ACC gene [45] and rat cAMP phosphodiesterase gene have more than one promoter, usually one that has a TATA box and one that does not [46]. Rat ACC promoter I has a TATA box and CAAT box similar to rat mtGPAT distal promoter – both are induced under lipogenic conditions. Rat ACC promoter II, like rat mtGPAT proximal promoter, is a TATA less promoter [46, 47]. Promoter II of rat ACC is expressed constitutively in several tissues while promoter I product is expressed in a tissue specific manner [47]. This could be the case with rat mtGPAT gene where the distal promoter is the regulatory promoter, whose regulation is brought about by the lipogenic transcription factors SREBP-1c and ChREBP. Furthermore, the proximal promoter of mtGPAT gene has the characteristics of the promoter of “housekeeping”-type genes [48]. Why? First, there is no TATA box within the promoter region. Secondly, the GC content of the promoter region is high (about 60%) and has potential Sp1 binding sites. This is similar to cystic fibrosis gene which too has a “housekeeping” promoter and the gene is expressed at low levels like the mtGPAT proximal promoter transcript [49].
The novel feature of the work presented here is that it further reveals a remarkable similarity how ACC and mtGPAT are regulated. These are two important enzymes strategically located in the overall biosynthetic pathway of glycerolipids including triacylglycerols. Both enzymes are stimulated by ATP, citrate [3], insulin and EGF, but inhibited by AMPK, glucagon, leptin, adrenaline and casein kinase II [23]. Moreover, multiple promoters control the expression of ACC (45) and mtGPAT [13]. Only one of the promoters, that contains TATA box, appears to be affected by the hormones and growth factor, the other promoter(s) act(s) constitutively. Why is this similarity important? Increasing the rate of fatty acid synthesis by stimulating ACC may not guarantee that the fatty acids will be diverted towards triacylglycerol biosynthesis. Without the mtGPAT being similarly regulated as ACC, the fatty acids may be used either for biosynthesis of glycerolipids or for degradation via β-oxidation in the mitochondrial matrix.
In summary, we have investigated the role of the two promoters in the transcriptional regulation of rat mtGPAT gene. Our results indicate that the distal promoter is the regulatory promoter and is capable of up- or down- regulating its transcript (distal transcript) via key lipogenic transcription factors such as ChREBP and SREBP-1c. The proximal promoter, on the other hand, acts like a “housekeeping”-type promoter and maintains a low basal level of transcription of its product (proximal transcript). Knowledge of the ways in which these promoters control the transcription may prove useful in targeting mtGPAT gene for obesity related drugs.
Acknowledgments
We would like to thank Dr. Ales Vancura for his help. This research was supported by the National Institutes of Health Grant GM-57643 (DH) and by Clare Boothe Luce fellowship (KKA). This work forms a portion of a doctoral thesis (PG) submitted to the faculty of Biological Sciences, St. John’s University. The costs of publication of this article were defrayed in part by the payment of page charges. This article must, therefore, be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are
- ACC
acetyl-CoA carboxylase
- ChIP
chromatin Immunoprecipitation
- ChREBP
Carbohydrate response element binding protein
- EGF
epidermal growth factor
- EMSA
electromobility shift assay
- FAS
fatty acid synthase
- GPAT
glycerol-3-phosphate acyltransferase
- H7
1--(5-isoquinolinylsulfonyl)-2-methylpiperazine
- mtGPAT
mitochondrial glycerophosphate acyltransferase
- NEM
N-ethylmaleimide
- PI3K
Phosphoinositide 3-kinases
- SREBP-1
Sterol regulatory element binding protein-1
- TSS
transcription start site
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Haldar D, Kelker HC, Pullman ME. Properties of acyl coenzyme A: 1-acyl-sn-glycerol 3-phosphate acyltransferase from rat liver mitochondria and microsomes. Trans N Y Acad Sci. 1983;41:173–182. doi: 10.1111/j.2164-0947.1983.tb02799.x. [DOI] [PubMed] [Google Scholar]
- 2.Linden D, William-Olsson L, Rhedin M, Asztely AK, Clapham JC, Schreyer S. Overexpression of mitochondrial GPAT in rat hepatocytes leads to decreased fatty acid oxidation and increased glycerolipid biosynthesis. J Lipid Res. 2004;45:1279–1288. doi: 10.1194/jlr.M400010-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Onorato TM, Chakraborty S, Balija VS, Haldar D. In: LIPIDS: Glycerolipid Metabolizing Enzymes. Haldar D, Das SK, editors. Research Signpost; Trivandrum: 2002. [Google Scholar]
- 4.Hammond LE, Gallagher PA, Wang S, Hiller S, Kluckman KD, Posey-Marcos EL, Maeda N, Coleman RA. Mitochondrial glycerol-3-phosphate acyltransferase-deficient mice have reduced weight and liver triacylglycerol content and altered glycerolipid fatty acid composition. Mol Cell Biol. 2002;22:8204–8214. doi: 10.1128/MCB.22.23.8204-8214.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindén D, William-Olsson L, Ahnmark A, Ekroos K, Hallberg C, Sjögren HP, Becker B, Svensson L, Clapham JC, Oscarsson J, Schreyer S. Liver-directed overexpression of mitochondrial glycerol-3-phosphate acyltransferase results in hepatic steatosis, increased triacylglycerol secretion and reduced fatty acid oxidation. FASEB J. 2006;20:434–443. doi: 10.1096/fj.05-4568com. [DOI] [PubMed] [Google Scholar]
- 6.Coleman RA, Lee DP. Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res. 2004;43:134–176. doi: 10.1016/s0163-7827(03)00051-1. [DOI] [PubMed] [Google Scholar]
- 7.Haystead TA, Hardie DG. Both insulin and epidermal growth factor stimulate lipogenesis and acetyl-CoA carboxylase activity in isolated adipocytes. Importance of homogenization procedure in avoiding artefacts in acetyl-CoA carboxylase assay. Biochem J. 1986;234:279–284. doi: 10.1042/bj2340279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Menendez JA, Colomerc R, Lupu R. Why does tumor-associated fatty acid synthase (oncogenic antigen-519) ignore dietary fatty acids? Med Hypotheses. 2005;64:342–349. doi: 10.1016/j.mehy.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 9.Ishii S, Iizuka K, Miller BC, Uyeda K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc Natl Acad Sci U S A. 2004;101:15597–15602. doi: 10.1073/pnas.0405238101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci USA. 2004;101:7281–7286. doi: 10.1073/pnas.0401516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimomura I, Shimano H, Horton JD, Goldstein JL, Brown MS. Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J Clin Invest. 1997;99:838–845. doi: 10.1172/JCI119247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denechaud PD, Bossard P, Lobaccaro JMA, Millatt L, Staels B, Girard J, Postic C. ChREBP, but not LXRs, is required for the induction of glucose-regulated genes in mouse liver. J Clin Invest. 2008;118:956–964. doi: 10.1172/JCI34314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aneja KK, Guha P, Shilpi RY, Chakraborty S, Schramm LM, Haldar D. The presence of distal and proximal promoters for rat mitochondrial glycerol-3-phosphate acyltransferase. Arch Biochem Biophys. 2008;470:35–43. doi: 10.1016/j.abb.2007.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nogalska A, Sucajtys-Szulc E, Swierczynski J. Leptin decreases lipogenic enzyme gene expression through modification of SREBP-1c gene expression in white adipose tissue of aging rats. Metabolism. 2005;54:1041–1047. doi: 10.1016/j.metabol.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 15.Swinnen JV, Heemers H, Deboel L, Foufelle F, Heyns W, Verhoeven G. Stimulation of tumor-associated fatty acid synthase expression by growth factor activation of the sterol regulatory element-binding protein pathway. Oncogene. 2000;19:5173–5181. doi: 10.1038/sj.onc.1203889. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z, Zhou YT, Kakuma T. Leptin resistance of adipocytes in obesity: role of suppressors of cytokine signaling. Biochem Biophys Res Commun. 2000;277:20–26. doi: 10.1006/bbrc.2000.3615. [DOI] [PubMed] [Google Scholar]
- 17.Soukas A, Cohen P, Socci ND, Friedman JM. Leptin-specific patterns of gene expression in white adipose tissue. Genes Dev. 2000;14:963–980. [PMC free article] [PubMed] [Google Scholar]
- 18.He Z, Jiang T, Wang Z, Levi M, Li J. Modulation of carbohydrate response element-binding protein gene expression in 3T3-L1 adipocytes and rat adipose tissue. Am J Physiol Endocrinol Metab. 2004;287:E424–430. doi: 10.1152/ajpendo.00568.2003. [DOI] [PubMed] [Google Scholar]
- 19.Bachran C, Heisler I, Fuchs H, Sutherland M. Influence of protein transduction domains on target-specific chimeric proteins. Biochem Biophys Res Commun. 2005;337:602–609. doi: 10.1016/j.bbrc.2005.09.095. [DOI] [PubMed] [Google Scholar]
- 20.Yada T, Nakata M, Shioda S. Insulinotropin PACAP potentiates insulin action. Stimulation of glucose uptake in 3T3-LI adipocytes. Ann N Y Acad Sci. 2000;921:473–477. doi: 10.1111/j.1749-6632.2000.tb07018.x. [DOI] [PubMed] [Google Scholar]
- 21.Baratta M, Saleri R, Mainardi GL, Valle D, Giustina A, Tamanini C. Leptin regulates GH gene expression and secretion and nitric oxide production in pig pituitary cells. Endocrinology. 2002;143:551–557. doi: 10.1210/endo.143.2.8653. [DOI] [PubMed] [Google Scholar]
- 22.Belina VD, Mableya JG, Jamesa RFL, Swift SM, Clayton HA, Titheradge MA, Green IC. Glucagon decreases cytokine induction of nitric oxide synthase and action on insulin secretion in RIN5F cells and rat and human islets of Langerhans. Cytokine. 1999;11:585–592. doi: 10.1006/cyto.1998.0486. [DOI] [PubMed] [Google Scholar]
- 23.Onorato TM, Chakraborty S, Haldar D. Phosphorylation of rat liver mitochondrial glycerol-3-phosphate acyltransferase by casein kinase 2. J Biol Chem. 2005;280:19527–19534. doi: 10.1074/jbc.M410422200. [DOI] [PubMed] [Google Scholar]
- 24.Ghosh CC, Vu HY, Mujo T, Vancurova I. Analysis of nucleocytoplasmic shuttling of NF kappa B proteins in human leukocytes. Methods Mol Biol. 2008;457:279–92. doi: 10.1007/978-1-59745-261-8_21. [DOI] [PubMed] [Google Scholar]
- 25.Charron M, DeCerbo JN, Wright WW. A GC-box within the proximal promoter region of the rat cathepsin L gene activates transcription in Sertoli cells of sexually mature rats. Biol Reprod. 2003;68:1649–1656. doi: 10.1095/biolreprod.102.012328. [DOI] [PubMed] [Google Scholar]
- 26.Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry. 1984;23:5036–5041. doi: 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- 27.Kumahara E, Ebihara T, Saffen D. Protein kinase inhibitor H7 blocks the induction of immediate-early genes zif268 and c-fos by a mechanism unrelated to inhibition of protein kinase C but possibly related to inhibition of phosphorylation of RNA polymerase II. J Biol Chem. 1999;274:10430–10438. doi: 10.1074/jbc.274.15.10430. [DOI] [PubMed] [Google Scholar]
- 28.Mabuchi S, Ohmichi M, Kimura A, Hisamoto K, Hayakawa J, Nishio Y, Adachi K, Takahashi K, Arimoto-Ishida E, Nakatsuji Y, Tasaka K, Murata Y. Inhibition of phosphorylation of BAD and Raf-1 by Akt sensitizes human ovarian cancer cells to paclitaxel. J Biol Chem. 2002;277:33490–33500. doi: 10.1074/jbc.M204042200. [DOI] [PubMed] [Google Scholar]
- 29.Dentin R, Pégorier JP, Benhamed F, Foufelle F, Ferré P, Fauveau V, Magnuson MA, Girard J, Postic C. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem. 2004;279:20314–20326. doi: 10.1074/jbc.M312475200. [DOI] [PubMed] [Google Scholar]
- 30.Li G, Barrett EJ, Wang H, Chai W, Liu Z. Insulin at physiological concentrations selectively activates insulin but not insulin-like growth factor I (IGF-I) or insulin/IGF-I hybrid receptors in endothelial cells. Endocrinology. 2005;146:4690–4696. doi: 10.1210/en.2005-0505. [DOI] [PubMed] [Google Scholar]
- 31.Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CAM, Taylor MS, Engstrom PG, Frith MC, Forrest ARR, Alkema WB, Tan SL, Plessy C, Kodzius R, Ravasi T, Kasukawa T, Fukuda S, Kanamori-Katayama M, Kitazume Y, Kawaji H, Kai C, Nakamura M, Konno H, Nakano K, Mottagui-Tabar S, Arner P, Chesi A, Gustincich S, Persichetti F, Suzuki H, Grimmond SM, Wells CA, Orlando V, Wahlestedt C, Liu ET, Harbers M, Kawai J, Bajic VB, Hume DA, Hayashizaki Y. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet. 2006;38:626–635. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- 32.Schmid CD, Perier R, Praz V, Bucher P. EPD in its twentieth year: towards complete promoter coverage of selected model organisms. Nucleic Acids Res. 2006;34:D82–D85. doi: 10.1093/nar/gkj146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klinge CM, Jernigan SC, Smith SL, Tyulmenkov VV, Kulakosky PC. Estrogen response element sequence impacts the conformation and transcriptional activity of estrogen receptor alpha. Mol Cell Endocrinol. 2001;174:151–166. doi: 10.1016/s0303-7207(01)00382-3. [DOI] [PubMed] [Google Scholar]
- 34.Hall JM, McDonnell DP, Korach KS. Allosteric regulation of estrogen receptor structure, function, and coactivator recruitment by different estrogen response elements. Mol Endocrinol. 2002;16:469–486. doi: 10.1210/mend.16.3.0814. [DOI] [PubMed] [Google Scholar]
- 35.Chang WM, Bohm RA, Strauss JC, Kwan T, Thomas T, Cowmeadow RB, Atkinson NS. Muscle-specific transcriptional regulation of the slowpoke Ca(2+)-activated K(+) channel gene. J Biol Chem. 2000;275:3991–3998. doi: 10.1074/jbc.275.6.3991. [DOI] [PubMed] [Google Scholar]
- 36.Bohm RA, Wang B, Brenner R, Atkinson NS. Transcriptional control of Ca(2+)-activated K(+) channel expression: identification of a second, evolutionarily conserved, neuronal promoter. J Exp Biol. 2000;203:693–704. doi: 10.1242/jeb.203.4.693. [DOI] [PubMed] [Google Scholar]
- 37.Jitrapakdee S, Gong Q, MacDonald MJ, Wallace JC. Regulation of rat pyruvate carboxylase gene expression by alternate promoters during development, in genetically obese rats and in insulin-secreting cells. Multiple transcripts with 5′-end heterogeneity modulate translation. J Biol Chem. 1998;273:34422–34428. doi: 10.1074/jbc.273.51.34422. [DOI] [PubMed] [Google Scholar]
- 38.Sehibler J, Hagenhuehle U, Wcllauer PK, Pittet AC. A single gene encodes multiple neuropeptides mediating a stereotyped behavior. Cell. 1983;33:501–508. doi: 10.1016/0092-8674(83)90492-0. [DOI] [PubMed] [Google Scholar]
- 39.Gosmain Y, Dif N, Berbe V, Loizon E, Rieusset J, Vidal H, Lefai E. Regulation of SREBP-1 expression and transcriptional action on HKII and FAS genes during fasting and refeeding in rat tissues. J Lipid Res. 2005;46:697–705. doi: 10.1194/jlr.M400261-JLR200. [DOI] [PubMed] [Google Scholar]
- 40.Towle HC. Glucose and cAMP: adversaries in the regulation of hepatic gene expression. Proc Natl Acad Sci. 2001;98:13476–13478. doi: 10.1073/pnas.251530798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu J, Nakamura MT, Cho HP, Clarke SD. Sterol regulatory element binding protein-1 expression is suppressed by dietary polyunsaturated fatty acids. A mechanism for the coordinate suppression of lipogenic genes by polyunsaturated fats. J Biol Chem. 1999;274:23577–23583. doi: 10.1074/jbc.274.33.23577. [DOI] [PubMed] [Google Scholar]
- 42.Mater MK, Thelen AP, Pan DA, Jump DB. Sterol response element-binding protein 1c (SREBP1c) is involved in the polyunsaturated fatty acid suppression of hepatic S14 gene transcription. J Biol Chem. 1999;274:32725–32732. doi: 10.1074/jbc.274.46.32725. [DOI] [PubMed] [Google Scholar]
- 43.Foretz M, Pacot C, Dugail I, Lemarchand P, Guichard C, Le Liepvre X, Berthelier-Lubrano C, Spiegelman B, Kim JB, Ferre P, Foufelle F. ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol Cell Biol. 1999;19:3760–3768. doi: 10.1128/mcb.19.5.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Monaco L, Vicini E, Conti M. Structure of two rat genes coding for closely related rolipram-sensitive cAMP phosphodiesterases. Multiple mRNA variants originate from alternative splicing and multiple start sites. J Biol Chem. 1994;269:347–357. [PubMed] [Google Scholar]
- 45.Tae HJ, Luo X, Kim KH. Roles of CCAAT/enhancer-binding protein and its binding site on repression and derepression of acetyl-CoA carboxylase gene. J Biol Chem. 1994;269:10475–10484. [PubMed] [Google Scholar]
- 46.Lopez-Casillas F, Kim KH. Heterogeneity at the 5′ end of rat acetyl-coenzyme A carboxylase mRNA. Lipogenic conditions enhance synthesis of a unique mRNA in liver. J Biol Chem. 1989;264:7176–7184. [PubMed] [Google Scholar]
- 47.Dynan WS. Promoters for housekeeping genes. Trends Genet. 1986;2:196–197. [Google Scholar]
- 48.Yoshimura K, Nakamura H, Trapnell BC, Dalemans W, Pavirani A, Lecocq JP, Crystal RG. The cystic fibrosis gene has a “housekeeping”-type promoter and is expressed at low levels in cells of epithelial origin. J Biol Chem. 1991;266:9140–9144. [PubMed] [Google Scholar]
- 49.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;10:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]