

Abstract
Despite recent advances in cancer therapies, metastatic renal cell carcinoma (RCC) remains difficult to treat. Most RCCs result from inactivation of the von Hippel Lindau (VHL) tumor suppressor, leading to stable expression of Hypoxia-Inducible Factor-α (HIF-1α, -2α, -3α) and the induction of downstream target genes, including those responsible for angiogenesis and metastasis. While VHL is inactivated in the majority of RCC cases, expression of the PTEN tumor suppressor is reduced in about 30% of cases. PTEN functions to antagonize PI3K/Akt/mTOR signaling, thereby controlling cell growth and survival. Activation of PI3K/Akt/mTOR leads to increased HIF-1α expression in certain cancer cells, supporting the rationale of using mTOR inhibitors as anti-cancer agents. Notably, HIF-2α, rather than HIF-1α, has been shown to play a critical role in renal tumorigenesis. To investigate whether HIF-2α is similarly regulated by the PI3K pathway in VHL−/− RCC cells, we manipulated PI3K signaling using PTEN overexpression and siRNA knockdown studies and pharmacologic inhibition of PI3K or Akt. Our data support a novel role for wild-type PTEN in promoting HIF-2α activity in VHL null RCC cells. This mechanism is unique to the cellular environment in which HIF-2α expression is deregulated, resulting from the loss of VHL function. Our data show that PTEN induces HIF-2α transcriptional activity by inhibiting expression of Yin Yang 1 (YY1), which acts as a novel corepressor of HIF-2α. Further, PTEN suppression of YY1 is mediated through antagonism of PI3K signaling. We conclude that wild-type PTEN relieves the repressive nature of YY1 at certain HIF-2α target promoters and that this mechanism may promote early renal tumorigenesis resulting from VHL inactivation by increasing HIF-2α activity.
Keywords: PTEN, YY1, VHL, HIF-2α, renal cell carcinoma, MT1-MMP
INTRODUCTION
Renal cell carcinoma (RCC) affects approximately 40,000 people in the United States per year (1). Clear cell RCC is genetically linked to loss of the von Hippel Lindau (VHL) tumor suppressor gene in both familial and sporadic cases (2). About 30% of RCC patients present with metastatic disease at the time of diagnosis (3–5), and although targeted therapies for RCC are being developed and have shown some improvement in patient care, the majority of patients with advanced disease remain refractory to treatment (1, 6, 7). Thus, understanding the molecular mechanisms of this disease is important for the design of better patient-based therapies and the selection of patients for the appropriate therapy regimen.
The most well-characterized function of VHL is controlling the oxygen-sensing mechanism of the cell through negative regulation of the transcription factor, Hypoxia Inducible Factor alpha (HIF-α) (8, 9). VHL is part of an ubiquitin ligase complex that targets HIF-α protein for proteosomal degradation. Thus, inactivation of VHL in RCC is associated with overexpression of HIF-α and an increase in hypoxia-inducible genes, such as those involved in angiogenesis (VEGF, PDGF), glycolysis (Glut1), cell growth and survival (Cyclin G2, EGFR, TGF-α), and tumor invasion and metastasis (MT1-MMP, CXCR4), suggesting that the genes upregulated by the VHL-HIF pathway are involved in the progression of renal cell carcinoma (10–12). Three isoforms of HIF-α protein exist. HIF-1α and HIF-2α are the most well-described, and their dysregulation has been linked to cancer progression, namely in the regulation of a tumor’s survival response to hypoxia and/or changes in metabolism (13). Notably, successful tumor suppression in renal cells depends on the proper regulation of HIF-2α rather than on the presence of wild-type VHL or on the proper regulation of HIF-1α (14–17). These findings suggest that the HIF-2α isoform behaves as a renal oncogene in the context of VHL null RCC and illustrate the importance of understanding the molecular mechanisms of HIF-2α regulation in renal cell tumorigenesis.
During normoxia, HIF-α protein is specifically hydroxylated on two proline residues by oxygen regulated prolyl hydroxlases, a modification that permits the interaction with VHL and results in rapid turnover of the protein (18). When VHL function is lost, or when a cell enters a hypoxic state, hydroxylation of HIF-α does not occur, and the isoforms are stabilized. Stabilized HIF-α then binds DNA after heterodimerization with the constitutively expressed HIF-β/ARNT subunit. Although both HIF-1α and HIF-2α dimerize to HIF-β/ARNT, recognize the same DNA consensus sequence (RCGTG), and share similar mechanisms of regulation, HIF-1α and HIF-2α display non-redundancy both in target gene pools as well as in phenotypes of knockout mice (19, 20). Determination of HIF-1α versus HIF-2α specific targets does not appear to be dictated by the binding of the proteins to the consensus hypoxia responsive element (HRE), but rather by differences in cofactor recruitment as determined by cell type as well as promoter context (21–23).
While VHL is inactivated in the majority of RCC cases, PTEN, another tumor suppressor, is rarely mutated (24–26). The PTEN tumor suppressor normally functions to antagonize phosphoinositide 3-kinase (PI3K)/Akt signaling through its lipid phosphatase activity, thereby controlling cell growth, survival, and metabolic processes (27). While mutations in the PTEN gene are rarely found in RCC, loss of heterozygosity (LOH) at or around the PTEN locus has been observed in approximately 30–40% of RCC tumors (24, 28). Recently, the PI3K/Akt/mTOR signaling pathway has been shown to positively regulate HIF-1α protein in certain cancer cells (29–31), making mTOR inhibitors attractive anti-cancer drugs. However, the role of PTEN in the regulation of the HIF-2α isoform, which has been implicated in the development of renal tumors, has not been clearly defined in RCC cells harboring genetic loss of VHL (32–34).
To our knowledge, no study has addressed the effect of PTEN antagonism of PI3K/Akt signaling on stabilized HIF-2α protein resulting from VHL inactivation in RCC cells. The human 786-0 RCC cell line, which is null for VHL expression, has been extensively used as a model to study the tumor suppressor function of VHL (14–17, 35). Interestingly, the 786-0 cell line also lacks expression of the PTEN tumor suppressor and has been used as a PTEN null model cell line for studies of PTEN function and regulation (36–38). Of note, the 786-0 cell line only expresses the HIF-2α isoform (9). Thus, these tumor cells have a cellular environment of stabilized HIF-2α protein and constitutively active Akt signaling, making it an ideal model to study the role of PTEN/PI3K/Akt on HIF-2α regulation in the context of stabilized HIF-2α expression due to VHL loss. We compared the 786-0 cells to another VHL null line, the A498 cells, which retains expression of wild-type PTEN.
We investigated whether restoration of PTEN function, and antagonism of PI3K/Akt signaling in the 786-0 cells, regulates HIF-2α, given that inhibition of this pathway has been shown to regulate expression of the HIF-1α isoform (29–31). While PTEN properly antagonized PI3K/Akt signaling in our experiments, no effect on HIF-2α protein levels was observed, suggesting that HIF-2α expression is not regulated by PI3K/Akt in VHL−/− RCC cells. However, to our surprise, restoration of PTEN expression in the 786-0 RCC cells induced HIF-2α activity and downstream target gene expression in VHL−/− RCC cells. Reciprocally, knockdown of PTEN expression in the A498 (VHL−/−, PTENwt) cells resulted in decreased HIF-2α activity.
Next, we show that the mechanism of PTEN induction of HIF-2α activity is mediated through PTEN suppression of Yin Yang 1 (YY1). Using pharmacologic and siRNA mediated inhibition of PI3K/Akt, we further demonstrate that PTEN suppression of YY1 is mediated through antagonism of PI3K/Akt signaling. YY1 (also known as δ, NF-E1, UCRBP, and CF1) is a ubiquitously expressed, highly conserved, multi-functional protein belonging to the GLI-kruppel family of zinc finger transcription factors. YY1 acts as either a corepressor or coactivator of transcription of certain target promoters. Whether YY1 is a corepressor or coactivator depends largely on promoter context and cell type (39–41). The importance of YY1 is beginning to be recognized in cancer biology in the regulation of several transcription factors, including p53 and c-myc, and it has been postulated that the loss of YY1 expression or activity may be an early event in tumorigenesis by regulating cell cycle control and/or apoptotic pathways. We identified the PTEN tumor suppressor as an inhibitor of YY1 expression and characterized YY1 as a corepressor of HIF-2α activity. Further, our study demonstrates that the wild-type functions of the tumor suppressors, VHL and PTEN, have opposing functions in the regulation of HIF-2α in RCC cells. Specifically, when VHL function is lost and HIF-2α levels are stabilized, wild-type PTEN functions to enhance HIF-2α activity through the negative regulation of the YY1 corepressor.
RESULTS
PTEN expression in VHL−/− RCC cells correlates with increased HIF-2α transcriptional activity and MT1-MMP expression
The human 786-0 RCC cell line is null for both VHL and PTEN expression and has been extensively used as a cellular model to study the tumor suppressor functions of both of these proteins (14–17, 35–38). The A498 line, another human RCC cell line widely used as a model to study VHL function (9, 42–44), is null for VHL and expresses wild-type PTEN (Figure 1A). Of note, both of these cell lines only express the HIF-2α isoform (9). To begin our investigation of PI3K/Akt regulation of HIF-2α in the context of VHL inactivation, we characterized the 786-0 and A498 RCC cells for expression of VHL, HIF-2α, PTEN, and P-Akt. As shown in Figure 1A, HIF-2α protein levels are stabilized in both cell lines due to VHL deficiency. The A498 cells express PTEN, resulting in reduced levels of phosphorylated Akt (ser473), whereas the 786-0 cells, lacking PTEN expression, have high levels of phosphorylated Akt under the same conditions. Notably, the presence of PTEN and lower levels of active Akt in the A498 line had no effect on HIF-2α protein levels, suggesting that antagonism of PI3K/Akt signaling in these cells may not regulate HIF-2α protein expression the same way this pathway has been shown to regulate the HIF-1α isoform in other cell types (29–31).
Figure 1. PTEN expression in VHL null cells correlates with increased expression of the HIF-2α target, MT1-MMP.
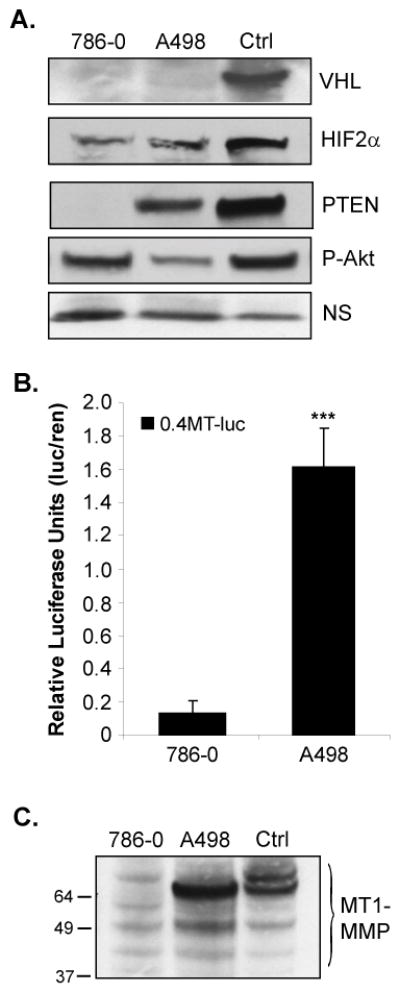
A. Whole cell lysates of the human RCC cell lines, 786-0 and A498, were analyzed by western blotting for expression of VHL, HIF-2α, PTEN, and P-Akt (ser 473). As a positive control (Ctrl) for immunoblotting purposes only, the 786-0 derivative line (WT8) was transfected to express VHL, HIF-2α, or PTEN. The WT8 cells have endogenously high levels of P-Akt, due to lack of PTEN expression. A non-specific (NS) band was used as a loading control. B. 786-0 and A498 cells were transiently co-transfected with 0.4kb of the human MT1-MMP promoter upstream of luciferase (0.4MT-luc) and pCMV-renilla. Cells were harvested and analyzed by the Dual Luciferase Assay®. Results represent three independent experiments of triplicates samples and are presented as relative luciferase units (luciferase normalized to renilla). C. MT1-MMP protein expression in whole cell lysates of the 786-0 and A498 lines as analyzed by western blotting. As a control, WT8 cells were transfected with the MTpc3SE construct to express MT1-MMP as previously described (12). Note that MT1-MMP protein appears as several cleavage products, which is not unusual for this protein (70). Results represent the average of three independent experiments of triplicates samples. Protein expression studies were repeated at least three times. Columns, mean; bars, SD; p<0.0001.
To investigate whether PTEN antagonism of Akt activation had any effect on HIF-2α target gene expression, we measured MT1-MMP expression, a HIF-2α target gene we characterized as playing an important role in RCC tumor cell invasion (11, 12). MT1-MMP promoter activity was measured in the 786-0 (VHL−/−, PTEN−/−) and A498 (VHL−/−, PTENwt) cells by transient transfection of a 0.4kb region of the MT1-MMP promoter containing a HIF-2α responsive element (HRE) we identified at −125 (11), upstream of luciferase (Figure 1B). MT1-MMP promoter activity was increased in the A498 cells compared to the 786-0 cells by approximately ten-fold. This increase was mirrored by high levels of MT1-MMP protein expressed in the A498 cells compared to the 786-0 cells (Figure 1C). Collectively, these data suggest that VHL null RCC cells that retain wild-type PTEN (such as the A498s) have increased HIF-2α activity and increased expression of certain downstream targets of HIF-2α. Furthermore, because HIF-2α protein levels are comparable between the cell lines, these data suggest that PTEN may regulate HIF-2α on the level of activity rather than expression.
Wild-type PTEN induces HIF-2α activity in VHL−/− RCC cells
We next asked if restoration of PTEN expression in the 786-0 cells would affect HIF-2α activity, as measured by the transactivation of a luciferase reporter construct containing three copies of the hypoxia-inducible enhancer region of the erythropoietin gene (HRE-luc) (45). Transient transfection of the 786-0 cells with a wild-type PTEN expression construct (pc/PTEN) inhibited Akt phosphorylation (Figure 2A), demonstrating that PTEN tumor suppressor function was properly restored in these cells. While PTEN expression did not affect HIF-2α protein levels (Figure 2A), PTEN induced HRE-luc transcription by approximately five-fold when co-transfected (Figure 2B), indicating that in VHL null RCC cells, PTEN induces HIF-2α transcriptional activity without influencing HIF-2α protein levels. To measure whether PTEN induction of HIF-2α activity affected a downstream target of HIF-2α, the PTEN null 786-0 cells were co-transfected with the PTEN expression construct and the proximal MT1-MMP promoter (0.4MT-luc) (Figure 2C). Reintroduction of PTEN expression in the 786-0 cells induced the MT1-MMP promoter about seven-fold, indicating that the PTEN tumor suppressor promotes transcriptional activity of a HIF-2α target gene in VHL−/− RCC cells.
Figure 2. Overexpression of PTEN in 786-0 cells induces HIF-2α activity.
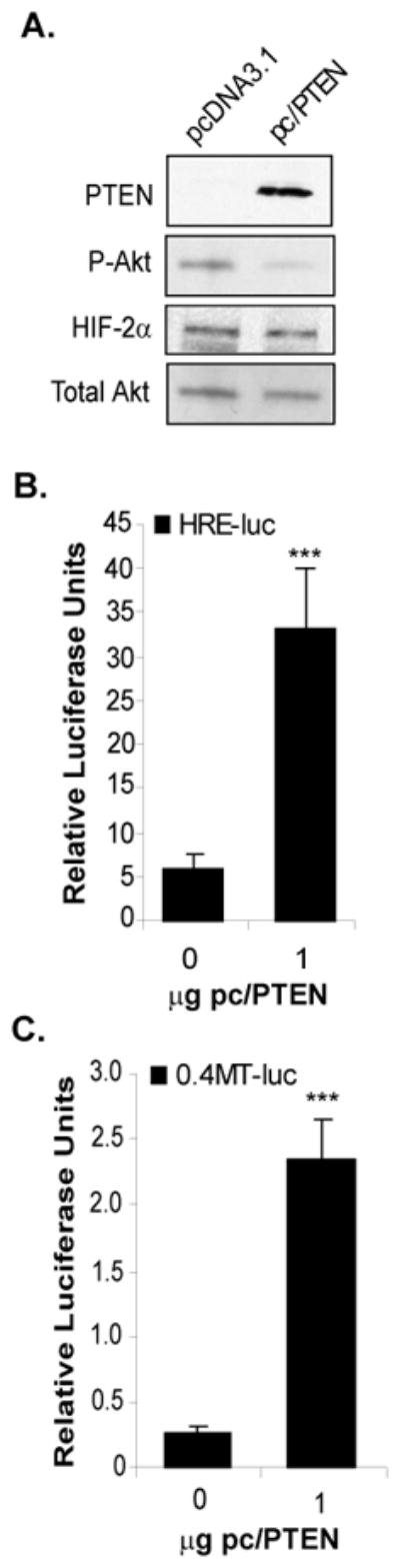
A. Western blot analysis of whole cell lysates from 786-0 cells transiently transfected to express wild-type PTEN using a PTEN expression construct (pc/PTEN) or an empty vector control (pcDNA3.1). PTEN protein expression was detected in pc/PTEN transfected cells and resulted in reduced P-Akt levels. Overexpression of PTEN had no effect on HIF-2α protein levels. Total Akt was used as a loading control. 786-0 cells transiently co-transfected to express either empty vector (pcDNA3.1) or wild-type PTEN (pc/PTEN) with either the HRE-luc (B) or 0.4-MT-luc (C) reporter constructs and assayed for luciferase activity. Results represent three independent experiments of triplicate samples. Columns, mean; bars, SD; p<0.0001.
Conversely, we next tested whether silencing PTEN expression in the A498 cells (VHL−/−, PTENwt) with siRNAs regulated HIF-2α activity. The A498 cells were transfected with either control or PTEN-specific SMARTpool® siRNAs. Endogenous PTEN mRNA and protein expression was reduced by >50% in the A498 cells in response to the PTEN siRNAs compared to the control (Figures 3A, 3B). As expected, knockdown of PTEN expression lead to an increase in phosphorylated Akt (Figure 3B). Importantly, HIF-2α protein levels were not affected by PTEN silencing (Figure 3B), agreeing with our above studies and suggesting that PTEN does not regulate the expression of HIF-2α in these cells. To measure the effects of PTEN knockdown on HIF-2α activity, the A498 cells were co-transfected with either control or PTEN-specific siRNAs together with the HRE-luc or the MT1-MMP promoter (0.4MT-luc). Silencing PTEN expression in the A498 cells inhibited activity of the HRE-luc (Figure 3C) as well as the 0.4MT-luc (Figure 3D) by approximately five-fold. Taken together, we conclude that wild-type PTEN positively regulates the activity of HIF-2α in VHL−/− RCC cells rather than the expression of HIF-2α.
Figure 3. Knockdown of PTEN expression by siRNA in A498 cells inhibits HIF-2α activity.
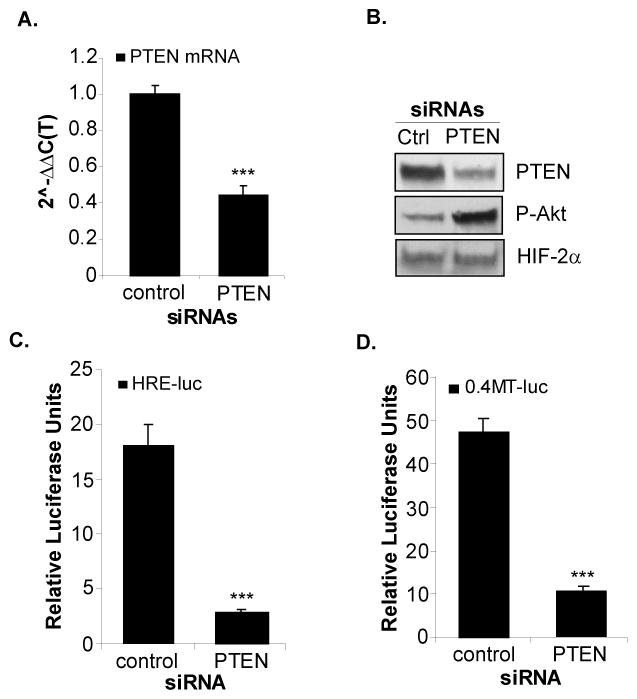
A498 cells were transiently transfected with control or PTEN specific SMARTpool® siRNAs [25nM] for 72hrs. Total RNA and protein was purified and analyzed for PTEN mRNA expression (A) and protein levels (B) as assayed by real-time RT-PCR and western blotting, respectively. B. Western blot analysis of A498 cells transfected with control or PTEN siRNAs measuring PTEN, P-Akt (ser473) and HIF-2α protein levels. Transient co-transfection of the A498 cells with control or PTEN siRNAs with either the HRE-luc (C) or 0.4MT-luc (D) and assayed for luciferase activity. Results represent three independent experiments of triplicate samples. Columns, mean; bars, SD; p<0.0001.
VHL tumor suppressor antagonizes PTEN induction of HIF-2α activity
Since VHL destabilizes HIF-2α protein levels (9, 11), we next hypothesized that the VHL tumor suppressor may counteract PTEN induction of HIF-2α activity. To test this, we co-transfected the 786-0 cells with the HRE-luc construct and either a PTEN expression construct (pc/PTEN), a VHL expression construct (pRc/VHL), or the two expression constructs together. As measured by the expression of the HRE-luc, PTEN induced HIF-2α activity about five-fold (Figure 4A) as in Figure 2B. Importantly, however, VHL inhibited both endogenous activity and PTEN-induced activity of the HRE-luc (Figure 4A). Further, restoration of VHL function reduced the stabilized HIF-2α protein levels, as expected, whereas restoration of PTEN expression had no effect on HIF-2α protein (Figure 4B). Finally, compared to VHL alone, co-expression of VHL and PTEN had no further effect on HIF-2α protein levels. Thus, we postulate that VHL and PTEN appear to regulate HIF-2α by opposing mechanisms: VHL antagonizes HIF-2α by targeting the protein for proteosomal degradation, leading to rapid turnover of the protein (Figure 4B) (9), whereas PTEN induces HIF-2α activity when HIF-2α protein is stabilized from the loss of VHL function.
Figure 4. VHL abrogates PTEN induction of HIF-2α activity.
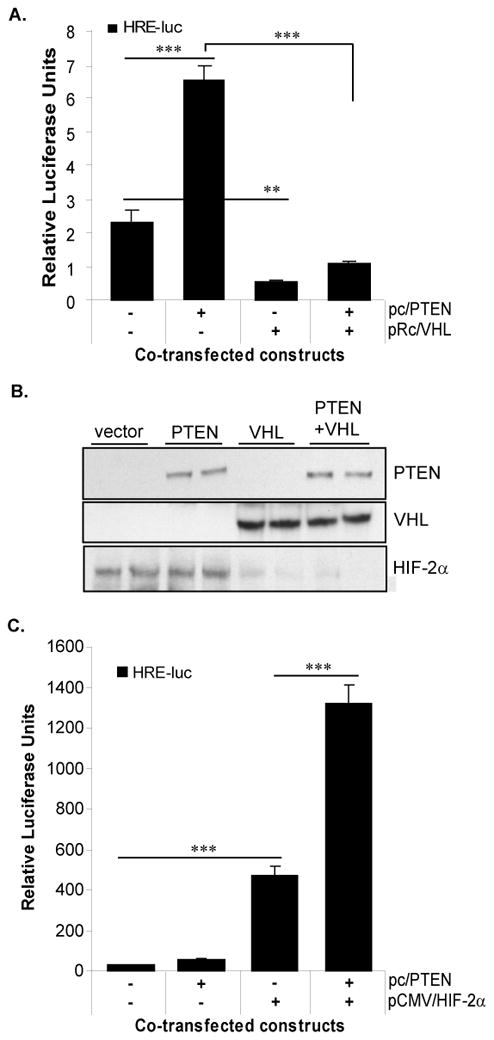
A. 786-0 cells were co-transfected with the HRE-luc and either an empty vector (pcDNA3.1), a PTEN expression construct (pc/PTEN), a VHL expression construct (pRc/VHL), or the PTEN and VHL expression constructs together, and assayed for luciferase activity. DNA content was balanced in all transfection conditions with empty vector. Results represent three independent experiments of triplicate samples. B. Western blot analysis for PTEN, VHL, and HIF-2α protein of transfectants in (A). C. WT8 RCC cells were co-transfected with the HRE-luc and either an empty vector (pcDNA3.1), a PTEN expression construct (pc/PTEN), a HIF-2α expression construct (pCMV-HIF2α), or both the PTEN and HIF-2α constructs and assayed for luciferase activity. DNA content in each transfection reaction was balanced by empty vector. Results represent three independent experiments of triplicate samples. Columns, mean; bars, SD; p<0.0001 (***), p<0.001 (**).
To investigate whether VHL antagonism of PTEN-induced HIF-2α activity (Figure 4A) resulted from direct regulation of HIF-2α by VHL and/or from HIF-independent functions of VHL, we employed the WT8 cells, a 786-0 derivative line stably transfected with wild-type VHL. It is known that the negative regulation of HIF-2α by VHL is saturable by overexpression of HIF-α (46, 47); thus, we reasoned that saturating the VHL/HIF-2α system would enable us to essentially eliminate this particular function of the VHL tumor suppressor without affecting other HIF-independent functions of VHL. Since the WT8 cells are also PTEN null, PTEN expression was reconstituted in these cells, and HIF-2α activity was measured by co-transfection with the HRE-luc reporter. As shown in Figure 4C, ectopic expression of PTEN had negligible effects on HRE-luc expression compared to the empty vector. This result is expected since these cells do not express detectable levels of HIF-2α due to the presence of VHL (11). In agreement with the notion that the VHL/HIF system is saturable, transfection of the WT8 cells with a HIF-2α expression construct (pCMV-HIF-2α) potently induced expression (~200 fold) of the HRE-luc construct (Figure 4C), as we have previously reported (11). To measure the effects of PTEN on overexpressed HIF-2α in a wild-type VHL background, we co-expressed HIF-2α and PTEN in the WT8 cells by transient transfection with the HRE-luc. PTEN synergized with HIF-2α in the induction of the HRE-luc reporter, resulting in a greater than two-fold increase above HIF-2α overexpression alone (Figure 4C). Together, these data show that when HIF-2α protein is overexpressed in the presence of wild-type VHL, PTEN functions to induce HIF-2α activity, indicating that this novel function of PTEN is apparent when HIF-2α expression is deregulated, rather than when the VHL tumor suppressor is inactivated.
PTEN tumor suppressor negatively regulates YY1 expression
We postulated that one mechanism for PTEN regulation of HIF-2α activity is through the regulation of a cofactor of HIF-2α. Recent studies have shown that the gene targets of HIF-1α and HIF-2α are partly dictated by cellular and promoter contexts, suggesting the role of cofactor recruitment in the regulation of the transcriptional activity of these isoforms (21–23, 43, 48). We hypothesized that PTEN induction of HIF-2α activity resulted from either the positive regulation of a coactivator or the negative regulation of a corepressor. One candidate factor is the transcriptional coregulator, Yin Yang 1 (YY1), which acts as either a transcriptional repressor or activator depending on cell type and promoter context (39–41). We hypothesized that YY1 acts as a cofactor of HIF-2α activity and that PTEN regulates YY1. To address this, we first compared YY1 expression levels between the 786-0 and A498 cells. Compared to the 786-0 cells, YY1 was expressed at very low levels in the A498 cells (Figure 5A), which express PTEN, suggesting that PTEN may negatively regulate YY1. To test whether PTEN regulates YY1, we overexpressed PTEN in the 786-0 cells and measured YY1 protein levels. Indeed, reintroduction of PTEN by transfection of the pc/PTEN expression construct drastically reduced YY1 protein levels (Figure 5B). Reciprocally, siRNA mediated knockdown of PTEN expression in the A498 cells led to an increase in YY1 protein levels (Figure 5C). Thus, PTEN tumor suppressor functions as an inhibitor of YY1 expression in VHL−/− RCC cells.
Figure 5. PTEN negatively regulates the YY1 cofactor.
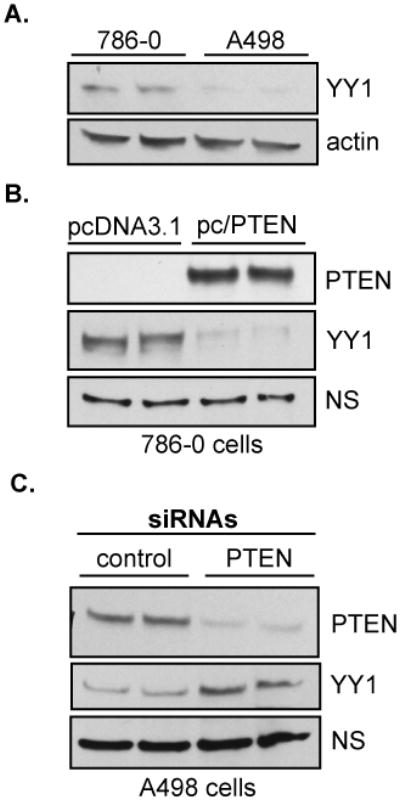
A. Whole cell lysates of 786-0 and A498 cells were immunoblotted for YY1 expression. Total protein was quantitated by Bradford Assay and equalized before loading. Actin was used as a loading control. Protein expression analysis was repeated independently three times. B. The 786-0 line was transiently transfected with either the empty vector, pcDNA3.1, or the pc/PTEN expression construct. Whole cell lysates were purified and immunoblotted for PTEN and YY1 protein expression. C. The A498 line was transfected with either control or PTEN-targeted SMARTpool® siRNAs [25nM] for 72hrs. Whole cell lysates were purified and analyzed for PTEN and YY1 protein levels by western blotting. A non-specific (NS) band was used as a loading control in B and C. Transfections were repeated three times in triplicate.
YY1 expression is regulated by PI3K/Akt signaling
PTEN induction of HIF-2α transcriptional activity and MT1-MMP promoter activity correlated with decreased activation of PI3K/Akt signaling and decreased expression of YY1 (Figures 2, 3, 5). To examine whether the mechanism of PTEN induction of HIF-2α activity and the MT1-MMP promoter was mediated by the ability of this tumor suppressor to antagonize PI3K/Akt signaling, we first treated the 786-0 cells with inhibitors of PI3K, Akt, or mTOR, and measured both HIF-2α and YY1 protein expression. Pharmacologic inhibition of PI3K using LY294002 [10μM] abolished phosphorylation of Akt (ser473), which corresponded with a 50% decrease in YY1 protein levels, indicating that YY1 expression is partly regulated by PI3K signaling (Figure 6A). To test this effect more specifically, we treated the 786-0 cells with the Akt inhibitor, triciribine [20μM, 40μM], as well as the mTOR inhibitor, rapamycin [20nM]. Triciribine treatment decreased P-Akt (ser473) and YY1 protein levels in a dose responsive manner, while rapamycin had no effect on P-Akt or on YY1 expression. The dose of rapamycin used [20nM] completely inhibited downstream activation of mTOR targets as measured by phosphorylation of p70/p85 and p4EBP1 (data not shown). Importantly, HIF-2α protein levels were unaffected by any of these treatments (Figure 6A), which is consistent with our finding that PTEN induces HIF-2α activity without altering HIF-2α protein expression (Figures 2–4). Finally, to directly test whether YY1 expression is dependent on Akt, we transfected the 786-0 cells with SignalSilence siRNAs that target both Akt 1 and 2 isoforms. Total Akt expression in the 786-0 cells was reduced by the target siRNAs by approximately 75%, leading to a modest decrease in YY1 levels, while HIF-2α protein levels were not affected by Akt knockdown (Figure 6B).
Figure 6. Akt regulates YY1 protein expression.
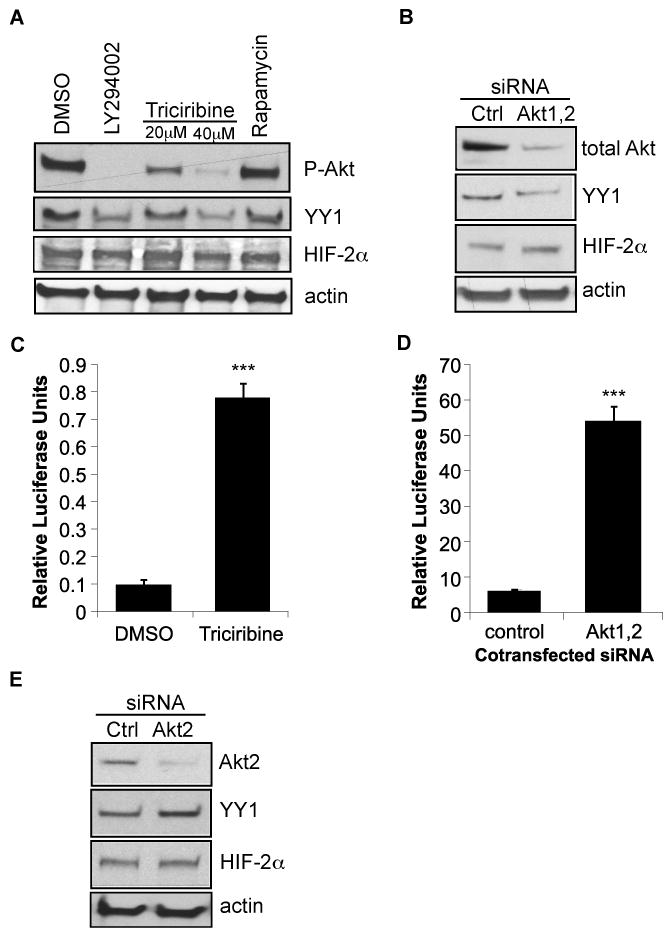
A. The 786-0 cells were treated with either vehicle (DMSO) alone, the PI3K inihibitor, LY294002 [10μM], the Akt inhibitor, triciribine [20μM, 40μM], or the mTOR inhibitor, rapamycin [20nM] in serum-free conditions overnight. Whole cell lysates were purified and immunoblotted for P-Akt (ser473), YY1, and HIF-2α. Actin was used as a loading control. B. The 786-0 cells were transfected with 50nM of control siRNA or siRNAs specifically targeting both Akt1 and Akt2 isoforms. Whole cell lysates were harvested 24 hrs later and immunoblotted for total Akt, YY1, HIF-2α and actin as a loading control. The 786-0 cells were co-transfected with the HRE-luc and either treated with 20μM triciribine (C) or co-transfected with 50nM of control or Akt1, 2 siRNAs (D). Transfectants were harvested after 24 hrs and analyzed for luciferase activity. Results represent three independent experiments of triplicate samples. Columns, mean; bars, SD; p<0.0001 (***). E. 786-0 cells were transfected with 50nM of control or Akt2 specific siRNAs for 24hrs. Whole cells lysates were then purified and analyzed by immunoblotting for total Akt2, YY1, HIF-2α, and actin as a loading control.
Next, to test whether inhibition of Akt signaling affected HIF-2α activity, we transfected the 786-0 cells with the HRE-luc construct and treated the transfectants with triciribine. Alternatively, we co-transfected the HRE-luc with either control or Akt siRNAs. Inhibition of Akt by either triciribine treatment (Figure 6C) or siRNA-mediated knockdown (Figure 6D) induced HIF-2α activity, consistent with our finding that PTEN antagonism of Akt signaling results induction of HIF-2α activity (Figure 2). Similar results were obtained using the MT1-MMP promoter in parallel experiments (data not shown). These data indicate that at least one mechanism by which PTEN regulates YY1 is through the inhibition of PI3KAkt signaling.
While this manuscript was being submitted for review, Toschi et al (34), published that HIF-2α protein expression in the 786-0 cells is dependent on Akt2 signaling, rather than on Akt1. In their study, Toschi and colleagues demonstrated that siRNA mediated knockdown of Akt2 levels in the 786-0 cells correlated with inhibition of HIF-2α protein expression. To test whether Akt2 was playing a role in the regulation of YY1 expression in the 786-0 cells, we transfected the 786-0 cells with Akt2 siRNAs and measured YY1 and HIF-2α protein levels. Total Akt2 protein was reduced by greater than 75% by the siRNAs in the 786-0 cells, but Akt2 inhibition had no measurable effect on YY1 or HIF-2α protein levels, suggesting YY1 may be predominately regulated by Akt1 rather than by Akt2 (Figure 6E). One explanation for the discrepancy between our findings and that of Toschi, et al, regarding Akt2 regulation of HIF-2α is that our siRNA studies were harvested 24 hours post-transfection, and Toschi, et al, harvested their experiments 72 hours post-transfection. Perhaps prolonged knockdown of Akt2 results in reduction of HIF-2α protein expression.
YY1 is a corepressor of HIF-2α activity
Since our data demonstrate that PTEN induces HIF-2α activity and that PTEN inhibits the expression of YY1, we hypothesized that YY1 acts as a corepressor of HIF-2α activity in VHL−/− RCC cells. To our knowledge, whether YY1 regulates HIF-2α transcriptional activity has not yet been investigated. To test this, we used siRNA mediated knockdown of YY1 in the 786-0 derivative line, pRc-9, and measured the effects of YY1 silencing on HIF-2α activity. Like the 786-0 line, the pRc-9 cells are null for both VHL and PTEN expression and have comparable YY1 levels (data not shown). However, the pRc-9 cells have higher transfection efficiency (~30%) compared to the 786-0 cells (~15%), thus, we were able to consistently achieve greater knockdown of YY1 in these cells, compared to the 786-0 line (data not shown). Transfection of the pRc-9 cells with YY1 siRNAs resulted in a reduction of mRNA levels (~40%) (data not shown) and protein levels (>75%) (Figure 7A), as compared to the control siRNA. Co-transfection of the pRc-9 cells with the HRE-luc and either the control or YY1 siRNAs increased HIF-2α transcriptional activity four-fold (Figure 7B), suggesting that YY1 is a corepressor of HIF-2α. siRNA mediated knockdown of YY1 had similar effects on MT1-MMP promoter activity (Figure 7C). Next, to measure the effect of YY1 overexpression on HIF-2α activity, we co-transfected the A498 cells, which express low levels of YY1, with the HRE-luc and either an empty vector control (pCEP4) or a YY1 expression construct (pCEP4/YY1). Overexpression of YY1 decreased HIF-2α transcriptional activity, further supporting the role of YY1 as a corepressor of HIF-2α (Figure 7D). YY1 overexpression also decreased MT1-MMP promoter activity in similar co-transfection experiments (Figure 7E). We conclude from these studies that PTEN induction of HIF-2α transcriptional activity is mediated by the inhibition of YY1, which acts as a corepressor of HIF-2α at certain target promoters.
Figure 7. YY1 acts as a corepressor of HIF-2α activity.
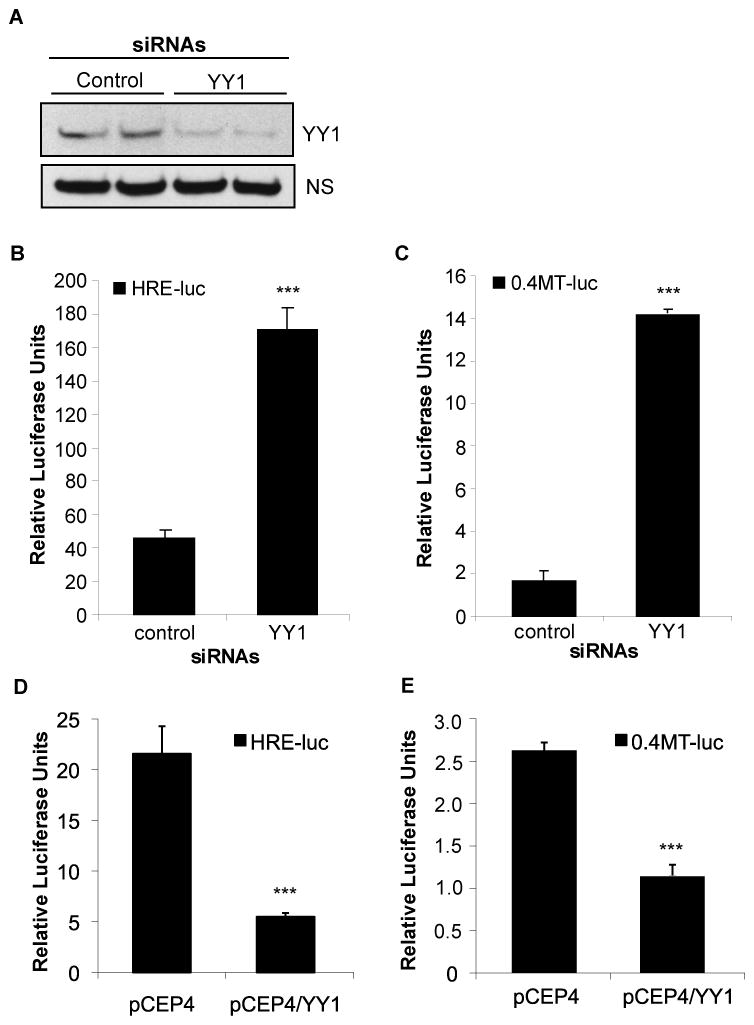
The 786-0 derivative, pRc-9, was transfected with control or pooled YY1 siRNAs [25nM] for 24hrs. Total protein was purified and analyzed for YY1 expression by western blot analysis (A). A non-specific (NS) band was used as a loading control. The pRc-9 cells were co-transfected with either the HRE-luc reporter (B) or 0.4MT-luc (C) and either control or YY1 siRNAs [25nM] for 24hrs and assayed for luciferase activity. Results represent two independent experiments of triplicate samples. The A498 cells were co-transfected with either the HRE-luc reporter (D) or 0.4MT-luc (E) and either pCEP4 empty vector control or the pCEP4/YY1 expression construct. Transfectants were harvested 24hrs later and analyzed for luciferase activity. Results represent three independent experiments of triplicate samples. Columns, mean; bars, SD; p<0.0001.
YY1 may corepress HIF-2α activity through the recruitment of HDACs
YY1 associates with the general transcriptional machinery and with coregulators, such as histone acetyl transferases (HATs) and histone deacetylases (HDACs) (49). HATs and HDACs were first identified to regulate general transcription through the modification of histone proteins by acetylation and deacetylation; however, recently, both HATs and HDACs were identified to modify non-histone proteins as well, including transcription factors, such as YY1 (49, 50). Full repressive activity of YY1 requires direct acetylation of YY1, and once acetylated, HDACs bind YY1 more readily, leading to transcriptional corepression of target genes (49). We reasoned that if YY1 corepresses HIF-2α activity through the recruitment of HDACs, then treatment with Trichostatin A (TSA), an HDAC inhibitor, should relieve that repression. To test this hypothesis, we first transfected the 786-0 cells with the HRE-luc or 0.4MT-luc reporter constructs and then treated the transfectants with increasing concentrations of TSA. Inhibition of HDACs with TSA induced both the HRE-luc (Figure 8A) and the MT1-MMP promoter constructs (Figure 8B) in a dose responsive manner, suggesting that HDACs are involved in the regulation of certain HIF-2α target promoters.
Figure 8. Inhibition of HDACs induces HIF-2α activity.

The 786-0 cells were transfected with the HRE-luc (A) or 0.4MT-luc (B) and then treated with increasing concentrations of TSA overnight before being assayed for luciferase activity. (C) 786-0 cells were co-transfected with the HRE-luc and either an empty vector (pcDNA3.1) or the PTEN expression construct (pc/PTEN). Transfectants were then treated with TSA [25nM] or DMSO vehicle control in DMEM/10%FBS for 18 hrs. Cells were harvested and assayed for luciferase activity. (D) A498 cells were co-transfected with the HRE-luc reporter and either an empty vector (pCEP4) or the YY1 expression construct (pCEP4/YY1). After 6 hours of transfection, cells were treated with TSA [200nM] or DMSO vehicle control for an additional 18hrs. Cells were harvested and analyzed for luciferase activity. Results represent three independent experiments of triplicate samples. Columns, mean; bars, SD; p<0.0001 (***).
Next, to ask whether PTEN regulation of HIF-2α involved HDACs, we co-transfected the 786-0 cells with the HRE-luc reporter and either an empty vector (pcDNA3.1) or the PTEN expression construct (pc/PTEN) and cultured the cells in the presence or absence of TSA (Figure 8C). TSA treatment alone induced HIF-2α activity to levels similarly achieved with PTEN expression alone, indicating that HIF-2α activity in these cells is regulated by HDACs and suggesting that PTEN may function to induce HIF-2α activity by modulating HDAC involvement (through the suppression of YY1). To test whether inhibition of HDACs and PTEN expression regulated HIF-2α activity in the same pathway, PTEN was expressed in the 786-0 cells, and the cells were treated with TSA. TSA treatment and PTEN expression enhanced the induction of HRE-luc activity, suggesting that HDACs and PTEN may be working in the same mechanism (Figure 8C). The regulation of HIF-2α activity by HDACs and PTEN was mirrored in the regulation of MT1-MMP transcription as measured by the 0.4MT-luc promoter construct (data not shown).
To next determine whether HDAC inhibition reversed YY1 repression of HIF-2α activity, we cotransfected the HRE-luc with either the pCEP4 empty vector or the pCEP4/YY1 expression construct in the A498 cells and then treated the cells with or without TSA. Inhibition of HDAC activity in the A498 cells resulted in an induction of HIF-2α activity (Figure 8D). HIF- 2α activity was less responsive to TSA treatment in the A498 cells than the 786-0 cells, which may be due to the low levels of YY1 in the A498 cells. Overexpression of YY1 in the A498 cells suppressed the HRE-luc as shown in Figure 7, and inhibition of HDACs by TSA partially reversed YY1 repression of HIF-2α activity (Figure 8D). Similar results were obtained using the MT1-MMP promoter (data not shown). These data suggest that one mechanism of YY1 corepression of HIF-2α activity may be through the recruitment of HDACs. In contrast, other studies have shown that HDACs positively regulate the HIF-1α isoform protein and stability, leading to increased activity (51–53). As such, treatment with HDAC inhibitors, such as TSA, down-regulate HIF-1α levels and activity. Our results showing TSA treatment increases HIF-2α activity indicate that HIF-2α may not share this mechanism of regulation by HDACs with HIF-1α.
DISCUSSION
Loss of VHL function results in the stabilization of HIF-α isoforms, and consequently, VHL inactivation in renal cell carcinoma is associated with highly vascularized renal tumors. As such, an anti-angiogenic approach to the treatment of RCC has been under intense investigation over the last several years, aimed to inhibit the classic angiogenesis growth factor, VEGF. In fact, sunitinib, a receptor tyrosine kinase inhibitor that blocks VEGF signaling, has recently been approved as a first-line therapy for metastatic RCC (54). While targeted therapies are showing signs of success, the improved response over standard care is often minimal, with most patients suffering from advanced disease remaining refractory or resistant to treatment (1, 6, 7). Thus, understanding the intricacies of the molecular mechanisms driving disease progression may not only identify new targets, but also identify patients that will be more responsive than others to a given therapy.
Our goal in the current investigation was to explore the role of PTEN antagonism of the PI3K pathway in the regulation of HIF-2α in VHL−/− renal cell carcinoma cells. To our surprise, when wild-type PTEN was ectopically expressed in the 786-0 (VHL−/−, PTEN−/−) cells, it inhibited the activation of Akt and induced HIF-2α activity and the MT1-MMP promoter, a downstream target of HIF-2α (Figures 1, 2), while having no effect on HIF-2α protein levels. In agreement, knockdown of PTEN expression in the A498 (VHL−/−, PTENwt) cells led to an increase in Akt activation and a decrease in HIF-2α activity (Figure 3) with no change in HIF-2α protein. In addition, our studies indicate that the wild-type functions of the tumor suppressors, VHL and PTEN, have opposing functions in the regulation of HIF-2α. Specifically, VHL down-regulates HIF-2α protein stability, whereas PTEN promotes HIF-2α activity in the context of VHL inactivation (Figure 4).
Collectively, these data suggest that HIF-2α is not positively regulated by PI3K/Akt signaling in these cells as has been shown for HIF-1α in other cells types (29–31). Recently, Toschi, et al, published that Akt2 is necessary for HIF-2α protein expression in the 786-0 cells (34). Our data did not support this mechanism, but did not exclude it either (Figure 6D). We measured no change in HIF-2α protein expression 24 hours after siRNA knockdown of Akt2, while Toschi, et al, measured decreased HIF-2α protein after 72 hours of siRNA knockdown of Akt2 [ref]. It is possible that HIF-2α protein becomes downregulated in response to prolonged Akt2 knockdown. The goal of our studies was to address the role of PI3K/Akt signaling in the regulation of HIF-2α activity. We used experimental approaches that affected all isoforms of Akt, including the manipulation of PTEN expression, pharmacologic inhibition of PI3K, Akt, and mTOR, and siRNAs against both Akt1 and Akt2. Our data consistently demonstrate that HIF-2α protein levels are unchanging under these conditions, while YY1 protein expression is reduced and HIF-2α transcriptional activity is increased (Figures 2, 3, 5, 6). Thus, we conclude that the mechanism of PTEN induction of HIF-2α activity is mediated by its ability to antagonize Akt signaling, which results in a decrease in expression of the YY1 corepressor (Figure 9A). When PTEN function is lost, Akt becomes activated, leading to YY1 expression and repression of HIF-2α activity (Figure 9B). Additionally, we postulate that PTEN may regulate YY1 through Akt independent mechanism(s) since complete inhibition of Akt signaling by LY294002 (Figure 6A) reduced YY1 levels by only about 50%, whereas overexpression of PTEN (Figure 5B) resulted in almost complete abrogation of YY1 expression.
Figure 9. Model of PTEN induction of HIF-2α transcriptional activity in VHL null RCC cells.
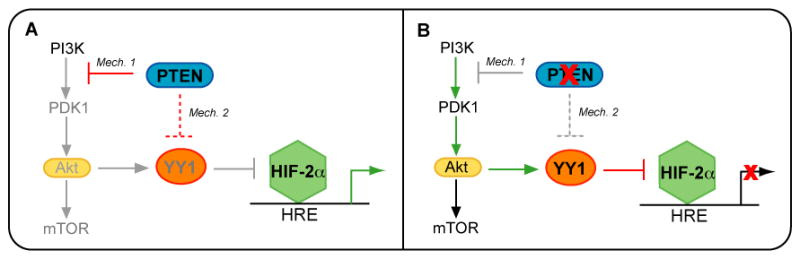
(A) The PTEN tumor suppressor functions to antagonize PI3K/Akt signaling, leading to the downregulation of YY1 expression (Mechanism 1). PTEN may also suppress YY1 levels via Akt independent mechanisms (Mechanism 2). YY1 acts as a corepressor of HIF-2α transcriptional activity; therefore, PTEN relieves YY1 corepression of HIF-2α, leading to an increase in certain HIF-2α target genes. (B) Inactivation of PTEN results in aberrant activation of PI3K/Akt signaling. YY1 expression is no longer suppressed under these conditions, thereby resulting in YY1 corepression of HIF-2α activity.
Activation of PI3K/Akt signaling in RCC is associated with poor prognosis (55), although the PTEN tumor suppressor, an inhibitor of PI3K signaling, is rarely mutated in RCC. In fact, PTEN expression co-exists with high levels of P-Akt in a subset of RCC patients, suggesting that Akt activation may occur independently of PTEN loss (56, 57), and that the activation of Akt may due to other factors, such as the overexpression of VEGF, PDGF, and TGFα/EGFR, which exert their functions through this signaling cascade. Of note, PTEN haploinsufficiency appears to promote a tumor phenotype in the prostate, suggesting that this tumor suppressor might not strictly follow Knudsen’s two hit hypothesis of tumor suppression (58, 59). Supporting this notion, 30–40% of RCCs have LOH at or around the PTEN locus, suggesting that PTEN haploinsufficiency may contribute to renal tumorigenesis as well. Based on these and our results, we postulate that PTEN suppression of YY1 may contribute to early tumorigenesis by increasing HIF-2α activity when HIF-2α expression is deregulated.
Studies in mice demonstrate that renal tumorigenesis is dependent on activation of the HIF-2α isoform rather than HIF-1α (14–17), suggesting that HIF-2α may be a renal oncogene and that these isoforms may not promote tumorigenesis in the same manner. Many studies have identified differences in HIF-1α and HIF-2α regulation as well as differences in downstream targets (21, 22, 43, 48). For instance, HIF-1α has been shown to be regulated by Hsp90 as well as HDACs in VHL-independent mechanisms (51–53, 60), whereas HIF-2α activity has been shown to be specifically regulated by NEMO and Nox4 (61, 62). Further, recent studies have indicated that HIF-2α transcriptional activity and specificity for target genes depends on recruitment of cofactors and/or cooperation with other transcription factors (11, 21, 43, 48). We show that the mechanism of PTEN induction of HIF-2α activity is through the negative regulation of YY1, which acts as a corepressor of HIF-2α and its downstream target gene, MT1-MMP (Figures 5, 7). Further, our data indicate that YY1corepression of HIF-2α activity is mediated partly through the recruitment of HDACs (Figure 8). This finding may define another difference between the regulation of HIF-1α and HIF-2α. Several groups have shown that HIF-1α protein is stabilized and/or activated by the association with HDACs, and that treatment with HDAC inhibitors, such as TSA, resulted in HIF-1α inhibition (51–53). In contrast, our data show that HDAC inhibition in the 786-0 cells, which express YY1, increased transcriptional activity of HIF-2α.
Hu, et al. described the existence of a specific repressor of HIF-2α activity in embryonic stem (ES) cells that could be titrated by overexpression of HIF-2α DNA binding mutants (21). We propose that PTEN may regulate such a repressor, thus explaining the synergy seen with PTEN and HIF-2α overexpression in the induction of HIF-2α activity (Figure 4C). In support of this hypothesis, our data show that reducing YY1 expression by siRNAs in the 786-0 derivative, pRc-9, which have endogenously high HIF-2α protein levels (11), increased HIF-2α activity. Further, overexpression of YY1 in the A498 cells, which have endogenously low YY1 levels and high HIF-2α protein levels, decreased HIF-2α transcriptional activity (Figure 7). These results may be explained by an increase or decrease in the HIF:YY1 ratio, respectively.
In addition to its well-described function as a lipid phosphastase, PTEN has Akt-independent functions as well, namely, in the regulation of p53 stabilization and activity (63, 64). Of particular interest is the recent finding that wild-type PTEN positively regulates the stability of a p53 gain-of-function mutant, resulting in a tumor-promoting phenotype, contrary to what one would expect given that PTEN is a well-described tumor suppressor (65). Since VHL loss results in gain-of-function of the HIF-2α protein, we postulate that wild-type PTEN may similarly enhance the tumor-promoting functions of HIF-2α in VHL−/− RCC. Our current finding that wild-type PTEN induces HIF-2α activity in VHL−/− RCC cells supports this notion. In conclusion, we have shown that PTEN inhibition of YY1 relieves YY1 corepression of HIF-2α in VHL null RCC. Therefore, our data suggest that VHL null RCC tumors retaining PTEN expression may have increased levels of certain HIF-2α target genes, thereby contributing to early tumorigenesis.
MATERIALS AND METHODS
Cell lines and cell culture
Cell lines were maintained in Dulbecco’s Modified Eagle’s Medium (Mediatech, Inc.) supplemented with 10% fetal bovine serum (FBS) (Hyclone), penicillin [100U/mL], streptomycin [100μg/mL], and l-glutamine and cultured at 37°C, 5% CO2. The human A498 and 786-0 renal cell carcinoma cell lines were purchased from American Type Culture Collection (ATCC). Both lines are null for VHL expression (9). The A498 cells were cultured under the above conditions supplemented with 1x non-essential amino acids (NEAA) (Invitrogen, Carlsbad, CA), except during transfection experiments when the NEAA were not added. The WT8 and pRc-9 cell lines (kindly provided by William Kaelin, Dana-Farber Cancer Institute, Boston, MA) represent stable subclones of the 786-0 cell line transfected with either pRc/CMV-HA-VHL or pRc/CMV empty, respectively (35), and were cultured under continuous selection with the addition of [1mg/mL] G418 sulfate (11). For serum-free conditions, cells were thrice washed with Hank’s Buffered Saline Solution and cultured in DMEM supplemented with 0.2% lactalbumin hydrosylate (12).
Reagents and plasmids
The pHRE-tk-luc reporter plasmid contains 3 copies of a 50nt hypoxia-inducible enhancer from the erythropoietin gene and has been shown to be inducible by co-expression of HIF-2α (11, 45). pHRE-tk-luc and the pCMV-HIF-2α expression construct containing full-length cDNA of HIF-2α were generous gifts of Richard Bruick (University of Texas, Southwestern Medical Center, Dallas, TX). pGL3-Basic-0.4MT-luc contains 0.4kb of the 5′-flanking region of the MT1- MMP gene linked to the firefly luciferase gene (generously provided by Jouko Lohi, University of Helsinki, Helsinki, Finland) (66). We previously identified a HIF-2α responsive site at position -125 in this promoter (11). The human MT1-MMP expression construct, MTpc3SE, was another kind gift of Jouko Lohi, and contains full-length MT1-MMP cDNA downstream of a CMV promoter (67). The pRc/CMV-HA-VHL expression construct was generously provided by William Kaelin, Dana-Farber Cancer Institute, Boston, MA (35). pCMV-renilla was a kind gift of Mark Israel (Dartmouth Medical School, Lebanon, NH). The PTEN expression construct, pcDNA3.1/PTEN (pc/PTEN), was constructed by subcloning the full-length human PTEN cDNA from pOTB7/PTEN (ATCC, #MGC-11227) into pcDNA3.1/zeo(+) (Invitrogen) using XhoI and EcoRI restriction sites. The pCEP4/YY1 plasmid was generously provided by Edward Seto, H. Lee Moffit Cancer Center, Tampa, FL) (49). The pCEP4 empty vector control plasmid was constructed from pCEP4/YY1 by removing the YY1 cDNA with HindIII/BamHI digestion. HindIII-NheI-BamHI adaptors were used to religate the empty vector. Ligation was performed using the Roche Rapid DNA Ligation Kit. Successful cloning was verified by DNA sequencing. Trichostatin A (TSA) was purchased from Sigma-Aldrich. LY294002, triciribine, and rapamycin were purchased from Calbiochem.
Transient transfections
Cells were plated in triplicate at a density of 1.5–2×105 cells/well in 6-well plates. The following day, cells were transfected with 1–4μg of DNA per well using either Lipofectamine 2000™ Transfection Reagent (Invitrogen) or Effectene (Qiagen) according to manufacturer’s instructions and as described previously (11, 12). In co-transfection studies, empty vector controls balanced the concentration of transfected DNA. Approximately 18–24hr later, cells were washed in cold 1x PBS and lysed using either 25mM glycylglycine, 4mM EGTA, 15mM MgSO4, 1% Triton X-100 or 1x Passive Lysis Buffer (Promega). Luciferase assays were performed using the Dual Luciferase Assay® kit (Promega) or as previously described (11, 12). Activity of pCMV-renilla was used for normalization where indicated. Activity was measured using an LMaxII384 Luminometer (Molecular Devices). Values are presented as relative luciferase units of representative data from three or more independent experiments of triplicate samples.
siRNA transfections
For PTEN siRNA experiments, A498 cells were transiently transfected with 25nM of either negative control SMARTpool® siRNAs (Upstate, D-001206–13–05) or PTEN SMARTpool® siRNAs (Upstate, M-003023). These siRNAs were provided as a pool of four SMARTselected siRNA duplexes designed by Dharmacon and Upstate. Cells were plated in 6-well dishes at a density of 1.5×105 cells/well as described under ‘Transient transfections’ following the protocol for Effectene (Qiagen). The transfection reaction was the same whether siRNAs were transfected alone or siRNAs were co-transfected with either HRE-luc or 0.4MT-luc. Cells were left under transfection conditions for 24hrs, then washed twice with Hank’s Balanced Salt Solution (HBSS) and placed in fresh standard culturing media for an additional 48hrs. For YY1 siRNA experiments, pRc-9 cells were transiently transfected with 25nM of either non-targeting control siRNA (Santa Cruz Biotechnology, sc-37007) or YY1 siRNAs (Santa Cruz Biotechnology, sc-36863), which are a pool of three specific YY1 siRNA duplexes. For Akt siRNA experiments, 786-0 cells were transiently transfected with 50nM of either non-targeting control siRNA (Cell Signaling, 6201), SignalSilence Akt siRNA (Cell Signaling, 6211), or SignalSilence Akt2 siRNA (Cell Signaling, 6396). Cells were plated in 6-well dishes at a density of 1.5×105 cells/well as described under ‘Transient transfections’ and transfected according to the Lipofectamine 2000™ protocol for siRNA transfection (Invitrogen). Co-transfections of the pRc-9 or 786-0 cells with siRNAs and the HRE-luc or 0.4MT-luc were performed according to manufacturer’s instructions and as previously described (12). Cells were transfected for 24hrs before being washed once with cold 1x PBS and harvested for luciferase assays as described above or harvested for western blot analysis as described below. Values are presented as relative luciferase units of representative data from three or more independent experiments of triplicate samples.
Immunoblotting
Immunoblotting procedures were performed essentially as previously described (11, 12). Briefly, whole cell lysates were harvested from confluent 6-well plates by washing the cells twice with cold 1x PBS, adding 150μL of lysis buffer (50mM Tris-HCl, pH 6.8, 1% SDS, 1mM EDTA, pH 8.0, protease inhibitor cocktail) or SDS reducing buffer (60mM Tris-HCl, pH 6.8, 2% SDS, 14.4mM β-mercaptoethanol, 25% glycerol, 0.1% bromophenol blue), and boiling for 5 minutes. Proteins were resolved on 10% Tris/SDS-PAGE gels (Pierce) and electrotransferred to Immobilon-P PVDF membranes (Millipore). Membranes were blocked at room temperature for 1–2 hours with: 1) 3% milk in 1x PBS for PTEN blots; 2) 3% bovine serum albumin (BSA) in Tris- buffered saline 0.1% Tween-20 (TBST) for MT1-MMP blots; or 3) 5% milk in TBST for HIF-2α, VHL, YY1, P-Akt, total Akt, and actin blots. Primary antibodies were diluted in respective blocking buffer and incubated with the membranes overnight at 4°C with rocking, with the exception of P-Akt and total Akt antibodies, which were diluted in 5% BSA TBST, and MT1-MMP antibody, which was diluted in 1% BSA TBST. Appropriate secondary antibodies were diluted in blocking buffer and incubated with the membrane at room temperature for 1 hour. Proteins were visualized by Western Lightning Chemiluminescence Reagent (Perkin Elmer). Antibodies and dilutions are as follows: VHL, 1:100 (Neomarkers, Clone Ig33); HIF-2α, 1:1000 (Novus Biologicals, NB100–122); PTEN, 1:5000 (Upstate, siAb™ 07–016); P-Akt (ser473), 1:1000 (Cell Signaling, 9271); total Akt, 1:1000 (Cell Signaling, 9272); total Akt2, 1:1000 (Cell Signaling, 2962); YY1, 1:1000 (Active Motif, 39071); MT1-MMP, 1:2000 (R&D Systems, MAB918); actin, 1:10,000 (Oncogene, Ab-1); anti-mouse HRP conjugated antibody, 1:5000 (Santa Cruz Biotechnology); and anti-rabbit HRP conjugated antibody, 1:5000 (Amersham BioSciences).
Real-time RT-PCR
Total cellular RNA was purified from confluent cultures assayed in triplicate using the RNeasy kit (Qiagen) with on-column DNase treatment. Reverse transcription (RT) was performed using either the Applied Biosystems Taqman Reverse Transcription Reagent Kit or the Moloney Murine Leukemia Virus Reverse Transcriptase Kit (Invitrogen) and following the manufacturers’ protocol and as described previously (11, 68). Real-time PCR reactions were performing using the Applied Biosystems Sybr Green master mix. Five hundred ng of input cDNA was used in each real-time PCR reaction. Each cDNA was assayed in duplicate using a MJ Research DNA Engine Opticon thermal cycler using the following parameters: 95°C 10min, 40 cycles of 95°C 15s, 60°C 1min, and a plate read. Primer sequences are as follows: PTEN: 5′-TGTTCAGTGGCGGAACTTGCAATC-3′ (sense) and 5′-TATCACCACACACAGGTAACGGCT-3′ (antisense); YY1: 5′-TGGAGGAATACCTGGCATTGACCT-3′ (sense) and 5′-TGGCCGAGTTATCCCTGAACATCT-3′ (antisense); actin: 5′-TGGCACCACACCTTCTACAAT-3′ (sense) and 5′-CACCGGAGTCCATCACGAT-3′ (antisense) Data are presented as relative expression as calculated by 2−ΔΔC(T) method (69).
Statistical analysis
Statistical significance was calculated using the student’s t-test available at http://www.physics.csbsju.edu/stats/t-test.html. Significance was assigned to p values <0.05.
Acknowledgments
We are grateful to the following for their generous gifts: Dr. William G. Kaelin, Jr. for use of the WT8 and pRc-9 cells and the VHL expression construct; Dr. Richard Bruick for the HRE-luc reporter and the HIF-2α expression construct; Dr. Jouko Lohi for the 0.4MT-luc reporter and the MTpc3SE expression construct; and Dr. Edward Seto for the pCEP4/YY1 construct. We thank Charles Coon for his technical assistance and valuable feedback. We also thank the members of the Brinckerhoff laboratory for critical reading of the manuscript. This work was supported by AR26599 and CA-77267 (C.E.B.) and by T32-AR-007576 (B.L.P.)
List of Abbreviations
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- HIF
hypoxia-inducible factor
- HRE
hypoxia-responsive element
- MT1-MMP
membrane type-1 matrix metalloproteinase
- PTEN
phosphatase and tensin homolog deleted on chromosome 10
- RCC
renal cell carcinoma
- TSA
trichostatin A
- VHL
von Hippel Lindau
- YY1
yin yang 1
Footnotes
Conflict of interest statement: The authors declare no financial conflicts of interest.
References
- 1.Patel PH, Chadalavada RS, Chaganti RS, Motzer RJ. Targeting von Hippel-Lindau pathway in renal cell carcinoma. Clin Cancer Res. 2006;12:7215–20. doi: 10.1158/1078-0432.CCR-06-2254. [DOI] [PubMed] [Google Scholar]
- 2.Kaelin WG. The von Hippel-Lindau tumor suppressor protein and clear cell renal carcinoma. Clin Cancer Res. 2007;13:680s–4s. doi: 10.1158/1078-0432.CCR-06-1865. [DOI] [PubMed] [Google Scholar]
- 3.Motzer RJ, Russo P, Nanus DM, Berg WJ. Renal cell carcinoma. Curr Probl Cancer. 1997;21:185–232. doi: 10.1016/s0147-0272(97)80007-4. [DOI] [PubMed] [Google Scholar]
- 4.Zisman A, Pantuck AJ, Dorey F, Chao DH, Gitlitz BJ, Moldawer N, et al. Mathematical model to predict individual survival for patients with renal cell carcinoma. J Clin Oncol. 2002;20:1368–74. doi: 10.1200/JCO.2002.20.5.1368. [DOI] [PubMed] [Google Scholar]
- 5.Zisman A, Pantuck AJ, Wieder J, Chao DH, Dorey F, Said JW, et al. Risk group assessment and clinical outcome algorithm to predict the natural history of patients with surgically resected renal cell carcinoma. J Clin Oncol. 2002;20:4559–66. doi: 10.1200/JCO.2002.05.111. [DOI] [PubMed] [Google Scholar]
- 6.Atkins MB, Ernstoff MS, Figlin RA, Flaherty KT, George DJ, Kaelin WG, et al. Innovations and challenges in renal cell carcinoma: summary statement from the Second Cambridge Conference. Clin Cancer Res. 2007;13:667s–70s. doi: 10.1158/1078-0432.CCR-06-2231. [DOI] [PubMed] [Google Scholar]
- 7.Longo R, D’Andrea MR, Sarmiento R, Salerno F, Gasparini G. Integrated therapy of kidney cancer. Ann Oncol. 2007;18(Suppl 6):vi141–8. doi: 10.1093/annonc/mdm244. [DOI] [PubMed] [Google Scholar]
- 8.Iliopoulos O, Levy AP, Jiang C, Kaelin WG, Goldberg MA. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci U S A. 1996;93:10595–9. doi: 10.1073/pnas.93.20.10595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 10.Maynard MA, Ohh M. Von Hippel-Lindau tumor suppressor protein and hypoxia-inducible factor in kidney cancer. Am J Nephrol. 2004;24:1–13. doi: 10.1159/000075346. [DOI] [PubMed] [Google Scholar]
- 11.Petrella BL, Lohi J, Brinckerhoff CE. Identification of membrane type-1 matrix metalloproteinase as a target of hypoxia-inducible factor-2 alpha in von Hippel-Lindau renal cell carcinoma. Oncogene. 2005;24:1043–52. doi: 10.1038/sj.onc.1208305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petrella BL, Brinckerhoff CE. Tumor cell invasion of von Hippel Lindau renal cell carcinoma cells is mediated by membrane type-1 matrix metalloproteinase. Mol Cancer. 2006;5:66. doi: 10.1186/1476-4598-5-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 14.Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002;1:237–46. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 15.Kondo K, Kim WY, Lechpammer M, Kaelin WG. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003;1:E83. doi: 10.1371/journal.pbio.0000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, Klausner RD. The contribution of VHL substrate binding and HIF1-alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell. 2002;1:247–55. doi: 10.1016/s1535-6108(02)00044-2. [DOI] [PubMed] [Google Scholar]
- 17.Zimmer M, Doucette D, Siddiqui N, Iliopoulos O. Inhibition of hypoxia-inducible factor is sufficient for growth suppression of VHL−/− tumors. Mol Cancer Res. 2004;2:89–95. [PubMed] [Google Scholar]
- 18.Kaelin WG, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Patel SA, Simon MC. Biology of hypoxia-inducible factor-2alpha in development and disease. Cell Death Differ. 2008;15:628–34. doi: 10.1038/cdd.2008.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005:re12. doi: 10.1126/stke.3062005re12. 2005. [DOI] [PubMed] [Google Scholar]
- 21.Hu CJ, Iyer S, Sataur A, Covello KL, Chodosh LA, Simon MC. Differential regulation of the transcriptional activities of hypoxia-inducible factor 1 alpha (HIF-1alpha) and HIF-2alpha in stem cells. Mol Cell Biol. 2006;26:3514–26. doi: 10.1128/MCB.26.9.3514-3526.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu CJ, Sataur A, Wang L, Chen H, Simon MC. The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol Biol Cell. 2007;18:4528–42. doi: 10.1091/mbc.E06-05-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lau KW, Tian YM, Raval RR, Ratcliffe PJ, Pugh CW. Target gene selectivity of hypoxia-inducible factor-alpha in renal cancer cells is conveyed by post-DNA-binding mechanisms. Br J Cancer. 2007;96:1284–92. doi: 10.1038/sj.bjc.6603675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alimov A, Li C, Gizatullin R, Fredriksson V, Sundelin B, Klein G, et al. Somatic mutation and homozygous deletion of PTEN/MMAC1 gene of 10q23 in renal cell carcinoma. Anticancer Res. 1999;19:3841–6. [PubMed] [Google Scholar]
- 25.Cairns P, Evron E, Okami K, Halachmi N, Esteller M, Herman JG, et al. Point mutation and homozygous deletion of PTEN/MMAC1 in primary bladder cancers. Oncogene. 1998;16:3215–8. doi: 10.1038/sj.onc.1201855. [DOI] [PubMed] [Google Scholar]
- 26.Kondo K, Yao M, Kobayashi K, Ota S, Yoshida M, Kaneko S, et al. PTEN/MMAC1/TEP1 mutations in human primary renal-cell carcinomas and renal carcinoma cell lines. Int J Cancer. 2001;91:219–24. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1034>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 27.Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1998;95:15587–91. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Velickovic M, Delahunt B, McIver B, Grebe SK. Intragenic PTEN/MMAC1 loss of heterozygosity in conventional (clear-cell) renal cell carcinoma is associated with poor patient prognosis. Mod Pathol. 2002;15:479–85. doi: 10.1038/modpathol.3880551. [DOI] [PubMed] [Google Scholar]
- 29.Land SC, Tee AR. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem. 2007;282:20534–43. doi: 10.1074/jbc.M611782200. [DOI] [PubMed] [Google Scholar]
- 30.Pore N, Jiang Z, Shu HK, Bernhard E, Kao GD, Maity A. Akt1 activation can augment hypoxia-inducible factor-1alpha expression by increasing protein translation through a mammalian target of rapamycin-independent pathway. Mol Cancer Res. 2006;4:471–9. doi: 10.1158/1541-7786.MCR-05-0234. [DOI] [PubMed] [Google Scholar]
- 31.Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–5. [PubMed] [Google Scholar]
- 32.Blancher C, Moore JW, Robertson N, Harris AL. Effects of ras and von Hippel-Lindau (VHL) gene mutations on hypoxia-inducible factor (HIF)-1alpha, HIF-2alpha, and vascular endothelial growth factor expression and their regulation by the phosphatidylinositol 3′-kinase/Akt signaling pathway. Cancer Res. 2001;61:7349–55. [PubMed] [Google Scholar]
- 33.Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12:122–7. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- 34.Toschi A, Lee E, Gadir N, Ohh M, Foster DA. Differential dependence of hypoxia-inducible factors 1 alpha and 2 alpha on mTORC1 and mTORC2. J Biol Chem. 2008;283:34495–9. doi: 10.1074/jbc.C800170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iliopoulos O, Kibel A, Gray S, Kaelin WG. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med. 1995;1:822–6. doi: 10.1038/nm0895-822. [DOI] [PubMed] [Google Scholar]
- 36.Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol. 2000;20:8969–82. doi: 10.1128/mcb.20.23.8969-8982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, Sellers WR. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1999;96:2110–5. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramaswamy S, Nakamura N, Sansal I, Bergeron L, Sellers WR. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell. 2002;2:81–91. doi: 10.1016/s1535-6108(02)00086-7. [DOI] [PubMed] [Google Scholar]
- 39.Gordon S, Akopyan G, Garban H, Bonavida B. Transcription factor YY1: structure, function, and therapeutic implications in cancer biology. Oncogene. 2006;25:1125–42. doi: 10.1038/sj.onc.1209080. [DOI] [PubMed] [Google Scholar]
- 40.Shi Y, Lee JS, Galvin KM. Everything you have ever wanted to know about Yin Yang 1. Biochim Biophys Acta. 1997;1332:F49–66. doi: 10.1016/s0304-419x(96)00044-3. [DOI] [PubMed] [Google Scholar]
- 41.Thomas MJ, Seto E. Unlocking the mechanisms of transcription factor YY1: are chromatin modifying enzymes the key? Gene. 1999;236:197–208. doi: 10.1016/s0378-1119(99)00261-9. [DOI] [PubMed] [Google Scholar]
- 42.Chen F, Kishida T, Duh FM, Renbaum P, Orcutt ML, Schmidt L, Zbar B. Suppression of growth of renal carcinoma cells by the von Hippel-Lindau tumor suppressor gene. Cancer Res. 1995;55:4804–7. [PubMed] [Google Scholar]
- 43.Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–86. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith K, Gunaratnam L, Morley M, Franovic A, Mekhail K, Lee S. Silencing of epidermal growth factor receptor suppresses hypoxia-inducible factor-2-driven VHL−/− renal cancer. Cancer Res. 2005;65:5221–30. doi: 10.1158/0008-5472.CAN-05-0169. [DOI] [PubMed] [Google Scholar]
- 45.Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11:72–82. doi: 10.1101/gad.11.1.72. [DOI] [PubMed] [Google Scholar]
- 46.O’Rourke JF, Tian YM, Ratcliffe PJ, Pugh CW. Oxygen-regulated and transactivating domains in endothelial PAS protein 1: comparison with hypoxia-inducible factor-1alpha. J Biol Chem. 1999;274:2060–71. doi: 10.1074/jbc.274.4.2060. [DOI] [PubMed] [Google Scholar]
- 47.Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000;19:4298–309. doi: 10.1093/emboj/19.16.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aprelikova O, Wood M, Tackett S, Chandramouli GV, Barrett JC. Role of ETS transcription factors in the hypoxia-inducible factor-2 target gene selection. Cancer Res. 2006;66:5641–7. doi: 10.1158/0008-5472.CAN-05-3345. [DOI] [PubMed] [Google Scholar]
- 49.Yao YL, Yang WM, Seto E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol Cell Biol. 2001;21:5979–91. doi: 10.1128/MCB.21.17.5979-5991.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310–8. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 51.Fath DM, Kong X, Liang D, Lin Z, Chou A, Jiang Y, et al. Histone deacetylase inhibitors repress the transactivation potential of hypoxia-inducible factors independently of direct acetylation of HIF-alpha. J Biol Chem. 2006;281:13612–9. doi: 10.1074/jbc.M600456200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kato H, Tamamizu-Kato S, Shibasaki F. Histone deacetylase 7 associates with hypoxia-inducible factor 1alpha and increases transcriptional activity. J Biol Chem. 2004;279:41966–74. doi: 10.1074/jbc.M406320200. [DOI] [PubMed] [Google Scholar]
- 53.Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol Cell Biol. 2006;26:2019–28. doi: 10.1128/MCB.26.6.2019-2028.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–24. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 55.Pantuck AJ, Seligson DB, Klatte T, Yu H, Leppert JT, Moore L, et al. Prognostic relevance of the mTOR pathway in renal cell carcinoma: implications for molecular patient selection for targeted therapy. Cancer. 2007;109:2257–67. doi: 10.1002/cncr.22677. [DOI] [PubMed] [Google Scholar]
- 56.He L, Fan C, Gillis A, Feng X, Sanatani M, Hotte S, et al. Co-existence of high levels of the PTEN protein with enhanced Akt activation in renal cell carcinoma. Biochim Biophys Acta. 2007;1772:1134–42. doi: 10.1016/j.bbadis.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 57.Hennenlotter J, Ohneseit PA, Simon P, Merseburger AS, Serth J, Kuehs U, et al. PTEN and p27Kip1 are not downregulated in the majority of renal cell carcinomas--implications for Akt activation. Oncol Rep. 2008;19:1141–7. [PubMed] [Google Scholar]
- 58.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–55. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 59.Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J Biol Chem. 2002;277:29936–44. doi: 10.1074/jbc.M204733200. [DOI] [PubMed] [Google Scholar]
- 61.Bracken CP, Whitelaw ML, Peet DJ. Activity of hypoxia-inducible factor 2alpha is regulated by association with the NF-kappaB essential modulator. J Biol Chem. 2005;280:14240–51. doi: 10.1074/jbc.M409987200. [DOI] [PubMed] [Google Scholar]
- 62.Maranchie JK, Zhan Y. Nox4 is critical for hypoxia-inducible factor 2-alpha transcriptional activity in von Hippel-Lindau-deficient renal cell carcinoma. Cancer Res. 2005;65:9190–3. doi: 10.1158/0008-5472.CAN-05-2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3:117–30. doi: 10.1016/s1535-6108(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 64.Li AG, Piluso LG, Cai X, Wei G, Sellers WR, Liu X. Mechanistic insights into maintenance of high p53 acetylation by PTEN. Mol Cell. 2006;23:575–87. doi: 10.1016/j.molcel.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 65.Li Y, Guessous F, Kwon S, Kumar M, Ibidapo O, Fuller L, et al. PTEN has tumor-promoting properties in the setting of gain-of-function p53 mutations. Cancer Res. 2008;68:1723–31. doi: 10.1158/0008-5472.CAN-07-1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lohi J, Lehti K, Valtanen H, Parks WC, Keski-Oja J. Structural analysis and promoter characterization of the human membrane-type matrix metalloproteinase-1 (MT1-MMP) gene. Gene. 2000;242:75–86. doi: 10.1016/s0378-1119(99)00549-1. [DOI] [PubMed] [Google Scholar]
- 67.Lohi J, Lehti K, Westermarck J, Kähäri VM, Keski-Oja J. Regulation of membrane-type matrix metalloproteinase-1 expression by growth factors and phorbol 12-myristate 13-acetate. Eur J Biochem. 1996;239:239–47. doi: 10.1111/j.1432-1033.1996.0239u.x. [DOI] [PubMed] [Google Scholar]
- 68.Burrage PS, Huntington JT, Sporn MB, Brinckerhoff CE. Regulation of matrix metalloproteinase gene expression by a retinoid X receptor-specific ligand. Arthritis Rheum. 2007;56:892–904. doi: 10.1002/art.22417. [DOI] [PubMed] [Google Scholar]
- 69.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 70.Osenkowski P, Toth M, Fridman R. Processing, shedding, and endocytosis of membrane type 1-matrix metalloproteinase (MT1-MMP) J Cell Physiol. 2004;200:2–10. doi: 10.1002/jcp.20064. [DOI] [PubMed] [Google Scholar]