

Abstract
Sensitivity to glucocorticoid (GC)-evoked apoptosis in lymphoid cell lines correlates closely with GC-mediated suppression of c-Myc expression. To establish a functional role for c-Myc in GC-mediated apoptosis, we have stably expressed MycER™, the human c-Myc protein fused to the modified ligand-binding domain of the murine estrogen receptor α, in GC-sensitive CEM-C7-14 cells. In CEM-C7-14 cells, MycER™ constitutively imparts c-Myc functions. Cells expressing MycER™ (C7-MycER™) exhibited a marked reduction in cell death after 72 h in 100 nM dexamethasone (Dex), with 10 – 20-fold more viable cells when compared to the parental CEM-C7-14 clone. General GC responsiveness was not compromised, as evidenced by Dex-mediated suppression of endogenous c-Myc and cyclin D3, and induction of c-Jun and the glucocorticoid receptor. MycER™ also blunted Dex-mediated upregulation of p27kip1 and suppression of the Myc target p53. In comparison to parental CEM-C7-14 cells, Dex-evoked DNA strand breaks were negligible and caspase activation was delayed, but the extent of G1 cell cycle arrest was similar in C7-MycER™ cells. Myc-ER™ did not result in permanent, complete resistance to GC however, and the GC-treated cells eventually died, indicative of redundant or interactive mechanisms in the GC-evoked lytic response of lymphoid cells. Our results emphasize the importance of c-Myc suppression in GC-evoked apoptosis of CEM-C7-14 cells.
Keywords: cell cycle, caspase 3, TUNEL, Max, p53, p27
Introduction
Glucocorticoids (GCs) kill immature thymocytes and sensitive malignant cells of lymphoid origin via classical apoptosis; consequently GCs have important roles in normal thymocyte selection and in the therapy of lymphoid malignancies (Gaynon and Carrel, 1999; Leung and Munck, 1975). GC-evoked lympholytic responses are mediated via the GC receptor (GR), and can be blocked by the GR antagoinst RU 38486 (Homo-Delarche, 1984; Thompson et al., 1995). Ligand-activated GR is known to function as a transcription factor for numerous genes; hence the current hypothesis for GC-evoked apoptosis favors receptor mediated alteration in expression of key vitality and/or death genes (Medh and Thompson, 2000; Montague and Cidlowski, 1995; Thompson, 1999). GR-mediated transcriptional activation (Dowd et al., 1991; Zhou and Thompson, 1996) and/or repression (Eastman-Reks and Vedeckis, 1986; Helmberg et al., 1995; Yuh and Thompson, 1989), as well as post-transcriptional regulation may play a role in GC-evoked apoptosis of thymocytes and leukemic cells.
The nuclear phosphoprotein c-Myc has been implicated as an important mediator of GC-evoked lympholytic responses. Cell cycle progression through the G1 - S boundary requires c-Myc, and deregulated expression or activation of c-Myc by chromosomal translocation or gene amplification is known to be a major causative factor for several human cancers, including lymphomas, lung carcinomas and neuroblastomas (Bishop, 1983; Spencer and Groudine, 1991). Both the c-Myc protein and c-myc mRNA are highly unstable, each with a half-life of approximately 30 min (Rabbitts et al., 1985), facilitating their tight regulation and reflecting c-Myc’s importance in controlling the proliferative state of the cell. In performance of many of its actions, c-Myc heterodimerizes with a homologous protein, Max, and the heterodimer activates transcription of genes containing the E-box consensus sequence CAC G/A TG (Blackwood and Eisenman, 1991). Max-independent actions of c-Myc may involve complex interactions with other coregulatory proteins (Peukert et al., 1997; Philipp et al., 1994; Sakamuro and Prendergast, 1999). Some of the growth promoting genes that are positively regulated by Myc-Max heterodimers include ornithine decarboxylase (odc; (Tobias et al., 1995). cdc25A, the cdk 2 and 4 activating cell cycle phosphatase gene (Galaktionov et al., 1996), the translation initiation factor eIF-4E (Jones et al., 1996) and the transcription factor p53 (Facchini and Penn, 1998; Reisman et al., 1993).
Several reports over the past few years have demonstrated the dual role of c-Myc in apoptosis (reviewed in (Thompson, 1998)). Depending on the cell type and conditions being studied, either deregulated overexpression or suppression of c-Myc can lead to apoptosis (Shi et al., 1992; Thompson, 1998). Based on reports where growth factor deprivation induced apoptosis is potentiated by enforced ectopic c-Myc expression (Askew et al., 1991; Evan et al., 1992), it has been proposed that c-Myc has a dual function of inducing cell proliferation as well as triggering apoptosis. In contrast to cell death provoked by overexpression of c-Myc, in several lymphoid cell systems, apoptosis is preceded by a severe down-regulation of c-myc message and c-Myc protein expression (Thompson, 1998). In normal mouse thymocytes, mouse S49 and human CEM and Jurkat cells. GC-evoked, GR-dependent apoptosis is associated with c-myc downregulation (Eastman-Reks and Vedeckis, 1986; Helmberg et al., 1995; Martins and Aguas, 1998; Yuh and Thompson, 1989). Only those CEM clones that apoptosed in response to GC exhibited such downregulation (Thulasi et al., 1993; Yuh and Thompson, 1989), further showing the relevance of this repression. GC resistance of one CEM clone can be overcome by simultaneous activation of the cAMP and GC pathways, with a concomitant restoration of c-Myc downregulation (Medh et al., 1998). In contrast to normal mouse thymocytes, those from non-obese diabetic (NOD) mice that are resistant to GC-induced apoptosis exhibit an increase in c-myc mRNA and c-Myc protein levels in response to GC treatment (Casteels et al., 1998), Transient expression of transfected c-myc in GC-sensitive CEM cells appeared to protect them against steroid-evoked death (Thulasi et al., 1993).
The above results have established a strong correlation between GC-evoked apoptosis and suppression of c-Myc in CEM leukemic lymphoblasts; however more direct evidence has not been forthcoming. Furthermore, a recent report suggests that GC-evoked apoptosis of a subclone of GC-sensitive CEM-C7 cells is not affected by expression of a tetracycline regulatable c-Myc construct (Loffler et al., 1999), contradicting several earlier observations. In murine P1798 cells, overexpression of c-Myc alone is not sufficient to protect from apoptosis, but coexpression of c-Myc and cyclin D3 complement each other and protect (Rhee et al., 1995). To better demonstrate the role of c-Myc in GC-evoked apoptosis of CEM cells, and to gain further insight into the mechanisms controlling apoptosis that are modulated by c-Myc, we have stably expressed a chimeric protein, MycER™, in CEM-C7 cells. We demonstrate here that MycER™ imparts c-Myc functionality and conveys significant protection from GC-evoked apoptosis, delaying cell death by 24–48 h. MycER™ expression reverses GC-mediated downregulation of the tumor suppressor protein p53, which is known to be regulated by c-Myc. Other independent GC-mediated events thought to play a role in apoptosis, including upregulation of c-Jun, repression of cyclin D3, and G1 growth arrest are not affected. Our data confirm the hypothesis that c-Myc suppression is important for GC-evoked apoptosis of CEM cells, and further assess the limits of c-Myc’s role in the process.
Results
We have been studying the mechanism of GC-evoked leukemic cell apoptosis using clones derived from the human acute lymphoblastic leukemic cell line CCRF-CEM. When GC-sensitive clonal CEM-C7 cells are treated with 100 nM dexamethasone (Dex), approximately 90% cells are killed by 72 h and by 96 h essentially all cells are dead (Medh et al., 1998). In the studies presented here, to ensure karyotypic and phenotypic purity, we have used a fresh subclone, CEM-C7-14, that replicates the GC response of the original CEM-C7 clone.
Establishment of CEM-C7-14 cells stably expressing ectopic c-Myc
CEM-C7-14 cells were transfected with the construct pBpuroMycER™ which contains the human c-Myc protein fused to the modified ligand binding domain (LBD) of the murine estrogen receptor α (ERα) (Littlewood et al., 1995). This modified LBD contains a point mutation (G525R) that is theoretically modified to render it incapable of binding to estrogen, while retaining normal affinity for the synthetic ligand 4-hydroxy tamoxifen (4HT). After selection in puromycin, the cells were termed C7-MycER™. These were tested for expression of the fusion protein. Parallel immunoblots probed with the c-Myc monoclonal antibody Mycl-9E-10.2 and sc-544, a monoclonal antibody raised against a C-terminal portion of ERα, recognized this protein in mass cultures of C7-MycER™ cells (Figure 1a). Its larger size makes it clearly distinguishable from endogenous c-Myc. Several clones of C7-MycER™ cells expressing moderate levels of MycER™ were isolated (Figure 1b). After a 24 h treatment with Dex, there is a considerable reduction of endogenous c-Myc in both CEM-C7-14 and C7-MycER™ cells, but not of MycER™ (Figure 1c). The presence of MycER™ protein despite Dex treatment suggested that there might be sustained c-Myc activity in Dex-treated cells. In correlation with their lytic response, CEM-C7-14 cells treated for 24 h with Dex exhibit approximately 80% downregulation of c-Myc immunoreactive protein (Figure 1c).
Figure 1.
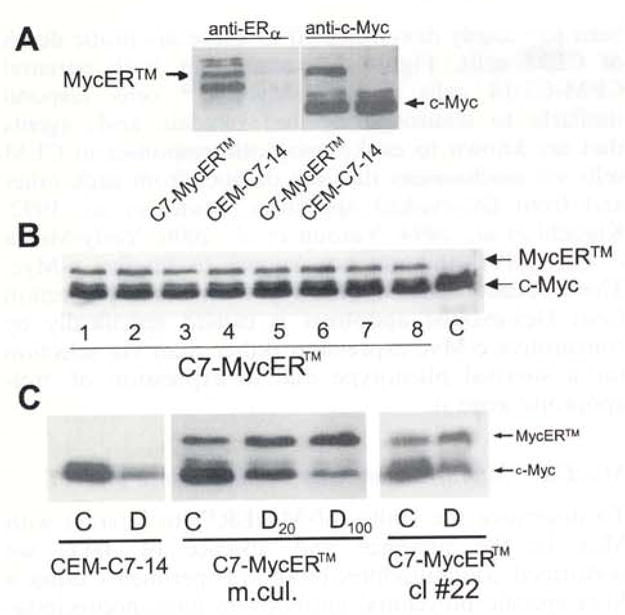
Expression of MycER™ in CEM-C7-14 cells. CEM-C7-14 cells were stably transfected with a plasmid pBpuroMycER™, selected for puromycin resistance and cloned in 0.5% soft agar. Individual colonies were isolated and amplified. Fifty microgram total cellular protein extracted from uncloned mass culture, individual clones or the parental untransfected CEM-C7-14 cells (C7) was resolved by SDS–PAGE, electroblotted, and the membranes were analysed by Western blotting. (a) Extracts of CEM-C7-14 cells and uncloned mass cultures of C7-MycER™ cells probed with an anti ERα antibody recognizing an epitope corresponding to the hinge region of ERα, or an anti-c-Myc monoclonal antibody Mycl-9E10.2) (b) MycER™ expression (antibody Mycl-9E10.2) in eight representative clones. (c) The parental CEM-C7-14 cells, uncloned C7-MycER™ (designated: m.cul.) and a representative clone (cl #22) of C7-MycER™ cells were treated with 20 nM Dex (D20), 100 nM Dex (D or D100) or ethanol vehicle (C) for 24 h. Whole cell extracts corresponding to 50 μg protein were subjected to SDS–PAGE and Western blotting using the anti-c-Myc antibody Mycl-9El0.2
MycER™ expression protects CEM-C7-14 cells from Dex-evoked death
Hormone dependence of proteins fused to nuclear receptor LBDs is attributed to simple steric hindrance (Picard et al., 1988), though outright structural proof of mechanism is not available yet. As to MycER, in the absence of hormone, the LBD is believed to mask the c-Myc leucine zipper domain, blocking interactions with Max. Various MycER chimeras have been shown to impart Myc-Max heterodimerization-dependent c-Myc functions to Rat-1a cells in an ER ligand dependent manner (Littlewood et al., 1995). However, there are instances where MycER has been shown to be constitutively active (Blyth et al., 2000; Vaillant et al., 1999), or to impart some Myc functions in an ER-ligand independent fashion (Philipp et al., 1994), suggesting that ER-ligand dependence may be restricted to certain model systems or specific Myc functions. We tested uncloned mass cultures and various C7-MycER™ clones for their sensitivity to Dex-evoked cell death in the presence or absence of 4HT, over a period of 4 days. Both the mass culture and all of the clones tested were 10- to 20-fold more resistant than the parental CEM-C7-14 clone after 3 days in 100 nM Dex in the absence of 4HT (Figure 2a). Up to 1 μM 4HT did not alter Dex-responsiveness in any of the clones tested (results from a representative clone, C7-MycER™ #22, are shown in Figure 2b). These data suggest that Myc mediated protection from GC-evoked death of C7-MycER™ cells may be imparted by MycER™ conferring a Max-independent function not requiring activation by 4HT, or that MycER™ is capable of forming Max-MycER™ complexes constitutively in an ER ligand-independent fashion in these C7-MycER™ cells. To rule out alterations in the MycER™ mRNA or DNA sequences in C7-MycER™ cells, cDNA isolated by RT – PCR and genomic DNA were subjected to PCR analysis and partial sequencing. The entire c-Myc coding sequence was found to be correct. The ER LBD was checked for integrity by PCR and did not indicate any large size reduction (data not shown). A third possibility is that transfection and selection of MycER™ expressing cells have inadvertently selected for a Dex-resistant phenotype. Though this seems unlikely to have occurred in the entire mass culture and every clone tested, we nonetheless carried out several tests of Dex-dependent GR function (see below). Another possibility for Dex resistance could be the inadvertent selection of an anti-apoptotic phenotype in the mass culture as well as clones. To rule out this possibility, we tested the ability of C7-MycER™ cells to respond to agents that have been previously demonstrated to cause apoptotic death of CEM cells. Figure 2c shows that both parental CEM-C7-14 cells and C7-MycER™ cells respond similarly to staurosporine and okadaic acid, agents that are known to evoke apoptotic responses in CEM cells via mechanisms that are distinct from each other and from GC-evoked apoptosis (Bruno et al., 1992; Kiguchi et al., 1994; Yatouji et al., 3000; Yerly-Motta et al., 1999), and are not known to involve c-Myc. These results confirm our hypothesis that protection from Dex-evoked apoptosis is caused specifically by constitutive c-Myc expression rather than via selection for a survival phenotype due to expression of anti-apoptotic gene(s).
Figure 2.
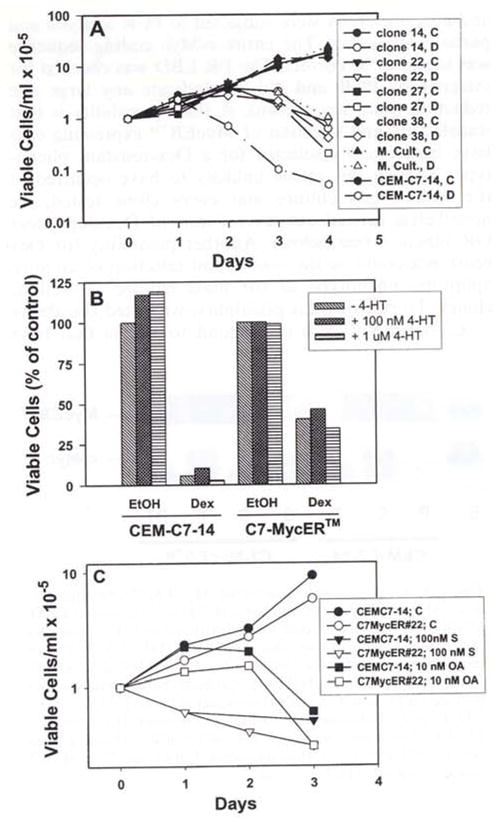
Protection of C7-MycER™ cells from Dex-evoked cell death, (a) CEM-C7-14 cells or the indicated individual C7-MycER™ clones or the mass culture (M. Cult.) were seeded at a density of 1×105 cells/ml and treated with ethanol (solid symbols) or 100 nM Dex (open symbols) for 96 h. Viable cells were counted at 24 h intervals by Trypan blue exclusion. Data plotted are averages of duplicate treatments. (b) CEM-C7-14 or C7-MycER™ clone #22 cells were treated in duplicate with ethanol or 100 nM Dex in the presence or absence of 100 nM or 1 μM 4HT. Trypan blue excluding cells were counted after 72 h. Data are plotted as a percentage of untreated time matched control for each cell line. (c) CEM-C7-14 or C7-MycER™#22 cells were treated for 72 h with either ethanol, 100 nM staurosporine or 10 nM okadaic acid, and viable cell counts were determined at 24 h intervals. Data plotted are averages of duplicate treatments from a representative experiment
MycER™ interacts with Max in the absence of 4HT
To determine the ability of MycER™ to interact with Max in the presence and absence of 4HT, we performed coimmunoprecipitation experiments using a Max specific polyclonal antibody in immumoprecipitation reactions, and the monoclonal anti-c-Myc antibody Mycl-9E-10.2 in Western blotting of the immunoprecipitated fractions. Figure 3 demonstrates that MycER™ coimmunoprecipitated with Max from C7-MycER™#22 cell lysates irrespective of whether the cells were treated with or without 4HT. There was possibly a small increase in extent of immunoprecipitated MycER™ after 4HT treatment, which may indicate a slightly greater efficiency of interaction between the leucine zipper domains of Max and MycER™ in the presence of 4HT. The data further confirms our hypothesis that MycER™ has been rendered constitutively active in our C7-MycER™ cells because of the inability of the unliganded ER LBD to significantly block MycER™-Max interactions. Treatment of cells with 100 nM Dex significantly reduces the amount of endogenous c-Myc coimmunoprecipitated with Max in both CEM-C7 and C7-MycER™#22 cells (Figure 3). Dex treated C7-MycER™#22 cells contain MycER™-Max heterodimers comparable in amounts to those in untreated cells. This is consistent with the protection against Dex-evoked apoptosis in this clone.
Figure 3.
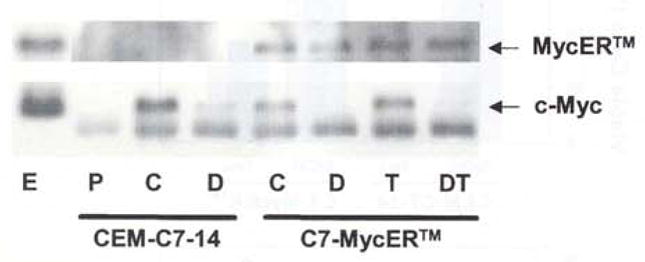
Coimmunoprecipitation of Myc-ER™ and endogenous c-Myc with anti-Max antibody. Whole cell extracts of CEM-C7-14 or C7-MycER™ cells treated with ethanol (C), 1 μM Dex (D), 250 nM 4HT (T) or Dex plus 4HT (DT) for 20 h were inmunoprecipitated with the polyclonal anti-Max antibody C-124. or non-specific antibody (P). Antibody bound proteins were purified on Protein A-Agarose beads and resolved by 10% SDS–PAGE and electroblotting. Whole cell extract (E) from C7-MycER™ cells was run in one lane as a control. The membrane was probed with the c-Myc monoclonal antibody Mycl-9E10.2, and developed by ECL reaction
Dex resistance ofC7-MycER™ cells is not caused by a general disruption of GC-responsiveness
We have confirmed that the GC response pathway is functional in C7-MycER™ cells and that they have not been inadvertently selected for a Dex resistant phenotype independently of MycER™ expression. First, as already noted, Dex evoked a downregulation of endogenous c-Myc (Figures 1c and 3), one demonstration that the GC pathway is functional in these cells. In tests of the induction of genes by Dex, we have confirmed by Western blotting that GR expression and induction is not compromised in C7-MycER™ cells (Figure 4a). GC-mediated downregulation of cyclin D3 has been demonstrated to be a Myc-independent event (Reisman and Thompson, 1995; Rhee et al., 1995). We demonstrate that the Dex-evoked downregulation of cyclin D3 in C7-MycER™#22 cells was comparable to that observed for CEM-C7-14 cells (Figure 4b). Expression and upregulation of c-Jun has been closely linked to GC-evoked apoptosis in CEM cells (Zhou and Thompson, 1996). Hence, we evaluated the ability of Dex to upregulate expression of the c-Jun immunoreactive protein in C7-MycER™#22 cells, when compared to parental CEM-C7-14 cells. Figure 4c demonstrates that C7-MycER™#22 cells exhibit c-Jun upregulation indistinguishable from that exhibited by CEM-C7-14 cells, further confirming that Dex responsiveness is not compromised in C7-MycER™ cells. These data provide further evidence that we have not selected for clones with alterations in the GR-response pathway following transfection of MycER™, and suggest that the observed resistance to Dex is attributable to the constitutive expression of ectopic MycER™.
Figure 4.
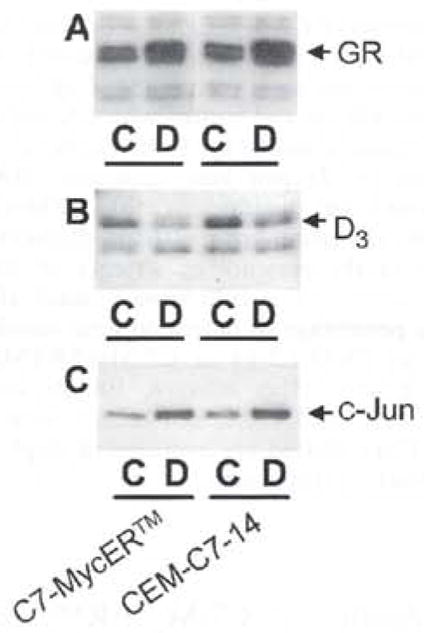
Regulation of GRα, c-Jun and cyclin D3 in CEM cells. Fifty μg protein from whole cell extracts of CEM-C7-14 and C7-MycER™#22 cells treated for 24 h with (D) or without (C) Dex (100 nM for a, 1 μM for b and c) were resolved by SDS–PAGE and transferred to PVDF membranes by electroblotting. Blots were probed with the polyclonal anti GRα antibody AhuGR150–175 (a), the anti-c-Jun polyclonal antibody (b), or the anti-cyclin D3 antibody (c)
MycER™ modulates expression of endogenous genes
To test whether the transactivation function of MycER™ in C7-MycER™ cells was dependent on 4HT, we evaluated the ability of MycER™ to overcome Dex-mediated repression of p53 in the presence and absence of 4HT. In CEM-C7-14 cells, Dex-evoked suppression of c-Myc results in the concomitant suppression of p53 protein presumably due to lack of c-Myc-mediated induction of p53 from E-box sequences within its promoter (Reisman et al., 1993). Although p53 has been shown to be nonfunctional in CEM cells (Cheng and Haas, 1990), its expression serves as a good marker for Myc functionality. As expected, in CEM-C7-14 cells, Dex evoked a reduction in p53 immunoreactive protein, and this result was not affected by 4HT treatment (Figure 5a). In C7-MycER™ mass culture and in C7-MycER™#22 cells, Dex did not evoke a significant suppression of p53, even in the absence of 4HT treatment, suggesting that the ectopic MycER™ is capable of constitutively maintaining p53 at near basal levels due to the Myc transactivation function of its Myc moiety. To normalize for loading discrepancies, blots were stripped and reprobed with anti-CREB (cyclic-AMP response element binding protein) antibody, since we have previously demonstrated that CREB expression is not altered after GC treatment. The cdk inhibitor p27kip1 is subject to c-Myc-mediated repression (Vlach et al., 1996), and Dex treatment of CEM-C7-14 cells causes an induction of p27kip1 immunoreactive protein following c-Myc suppression. We demonstrate that in C7-MycER™ mass culture and in C7-MycER™#22 cells Dex treatment does not induce p27kip1 expression to the same extent as it does in parental CEM-C7-14 cells (Figure 5b). Again, CREB was used as a normalizing control. These data suggest that MycER™ can mimic endogenous c-Myc in the repression of p27kip1.
Figure 5.
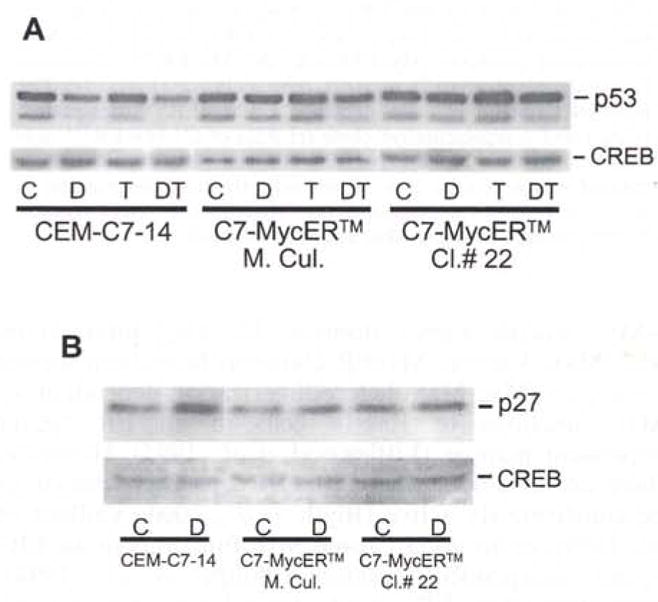
MycER™ modulates expression of endogenous c-Myc responsive genes, (a) CEM-C7-14, C7-MycER™ mass culture or C7-MycER™#22 cells were treated with ethanol (C), 100 nM Dex (D), 250 nM 4HT (T) or Dex plus 4HT (T) for 32 h. Whole cell extracts (50 μg protein) were resolved by 10% SDS–PAGE, transblotted on to PVDF membrane and probed with the monoclonal anti-p53 antibody. Membrane was stripped and reprobed with anti-CREB polyclonal antibody for normalization of loading, (b) CEM-C7-14, C7-MycER™ mass culture or C7-MycER™ #22 cells were treated with ethanol (C) or 1 μM Dex (D) for 24 h. Whole cell extracts (50 μg protein) were resolved by 10% SDS–PAGE and transblotted on to PVDF membrane and probed with a rabbit polyclonal antibody raised against p27kip1. Membrane was subsequently stripped and probed with a polyclonal antibody specific for CREB, to normalize for loading
MycER™ expression protects from Dex-evoked DNA strand breaks
We determined whether MycER™ mediated protection from Dex-evoked cell death was accompanied by a concomitant lack of certain biochemical changes associated with apoptosis. Flow cytometric TUNEL analysis of C7-MycER™#22 indicated that the percentage of cells with nicked DNA after 72 h of 100 nM Dex treatment was no more than 4%, comparable to that of untreated cells (Figure 6c,d). In contrast, parental CEM-C7-14 cells had approximately 40% cells with DNA nicks after 72 h of Dex treatment (Figure 6b). Figure 6e demonstrates a time dependent increase in TUNEL positive CEM-C7-14 cells upon Dex treatment, but no significant change in this classical component of Dex-evoked apoptosis in C7-MycER™ mass culture or C7-MycER™#22, further confirming our observation that expression of My-cER™ in CEM-C7-14 cells renders them GC-resistant.
Figure 6.
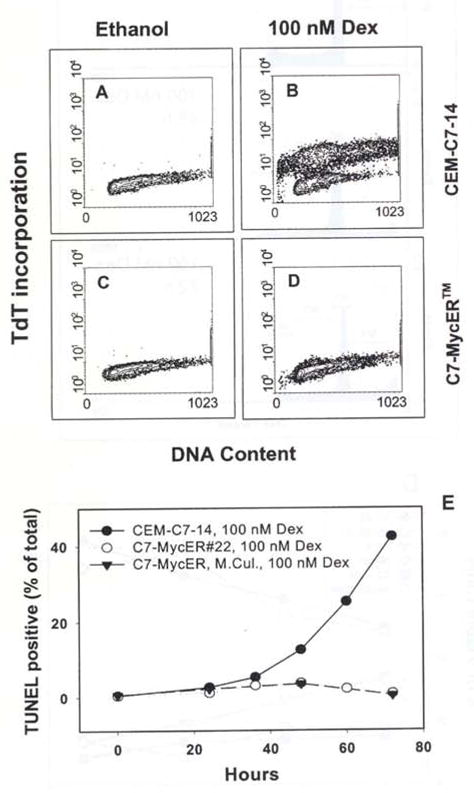
Lack of DNA strand breaks in Dex-treated C7-MycER™ cells, (a–d) CEM-C7-14 (a, b) or C7-MycER™ #22 (c, d) cells were treated with ethanol (a, c) or 100 nM Dex (b, d) for 72 h. Samples were harvested, and analysed for nicked DNA by TUNEL assay. Flow cytometry was carried out using a FACScan and Cell Quest 1.2 software from Becton Dickinson as described in Materials and methods, (e) Time course of DNA nicking. CEM-C7-14. C7-MycER™ mass culture and C7-MycER™ #22 cells were treated for 72 h with 100 nM Dex, and aliquots were harvested every 12–24 h for TUNEL analysis as described above
MycER™ expression prevents Dex-evoked caspase activation
There is increasing evidence that apoptosis triggered by diverse stimuli in a number of systems culminates in activation of a caspase cascade. In CEM-C7 cells, treatment with 100 nM Dex for 48 h results in a 6 – 7-fold increase in caspase 3 activity (Figure 7a). This is accompanied by cleavage of a carboxy terminal 85 kDa fragment from the intact 116 kDa caspase 3 substrate, poly ADP ribose polymerase (PARP; Figure 7b). C7-MyeER™#22 cells, when subjected to the same treatment, increased caspase 3 activity only slightly and showed minimal PARP cleavage up to 48 h (Figure 7a,b), suggesting that early caspase activation depends on c-Myc downregulation, and that this action can be prevented by ectopically expressed MycER™. By 72 h of Dex treatment, however, C7-MycER™ cells did exhibit detectable procaspase 3 cleavage (not shown) and PARP cleavage (Figure 7b).
Figure 7.
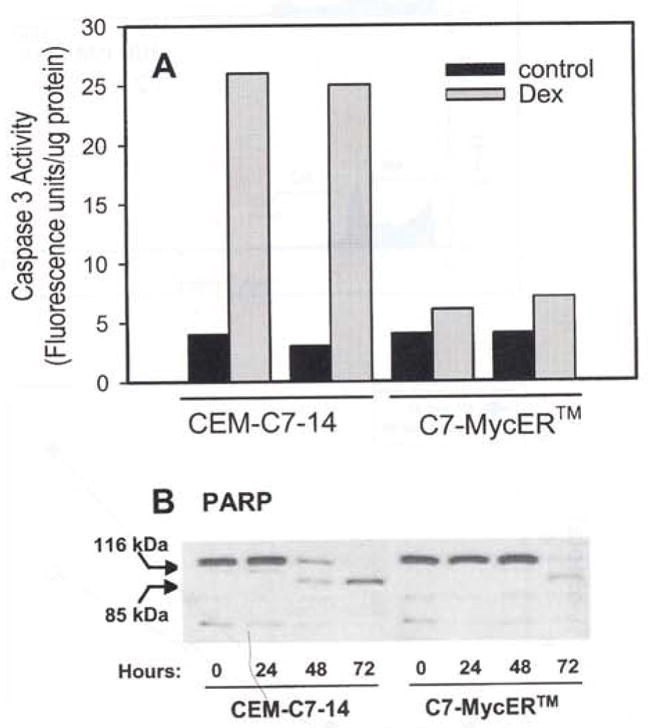
Caspase activation in Dex-evoked apoptosis. (a) Caspase 3 activity was measured using the substrate Z-DEVD-AFC in extracts of CEM-C7-14 and C7-MycER™ cells treated for 48 h with ethanol (solid bars) or 100 nM Dex (gray bars). Data are from a representative experiment, which consisted of two independent treatments; each is presented as a separate bar. Each bar represents an average of duplicate measurements. (b) CEM-C7-14 and C7-MycER™ cells were treated with 100 nM Dex for the indicated length of time, harvested, and whole cell extracts were prepared. Aliquots corresponding to 50 μg protein were resolved by SDS–PAGE, electroblotted, and the membrane was probed using an antibody (sc-7150, Santa Cruz Biotech.) that reacts with the uncleaved as well as cleaved PARP proteins
MycER™ expression does not prevent Dex-induced accumulation of cells in G1
Our data show that constitutive expression of MycER™ significantly protects cells from Dex-evoked death, delaying the outcome and preventing some well-documented concomitants, such as DNA nicking. Previous studies have demonstrated that Dex-evoked lysis of CEM cells is accompanied by growth arrest in the G0/G1 phase of the cell cycle (Harmon et al., 1979). To determine whether MycER™ was capable of overcoming Dex-evoked block in cell cycle progression, we evaluated the distribution of C7-MycER™#22 and parental CEM-C7-14 cells in individual phases of the cell cycle after various durations in Dex (Figure 8a,b). Flow cytometric analysis of the DNA content of propidium iodide stained cells again demonstrated less apoptosis in C7-MycER™#22 cells which had a significantly lower fraction of cells with a sub-G1 (apoptotic) DNA content after 72 h in 100 nM Dex when compared to CEM-C7-14 cells (Figure 8c). However, in both cell types, there was a similar increase in the percentage of viable cells in the G1 phase of the cells cycle up to 72 h after Dex treatment, with a parallel decrease in the percentage of cells in S phase (Figure 8d). Our data demonstrate that Dex-evoked growth arrest and apoptosis are separable functions, and while MycER™ is unable to prevent the former, it can delay the latter.
Figure 8.
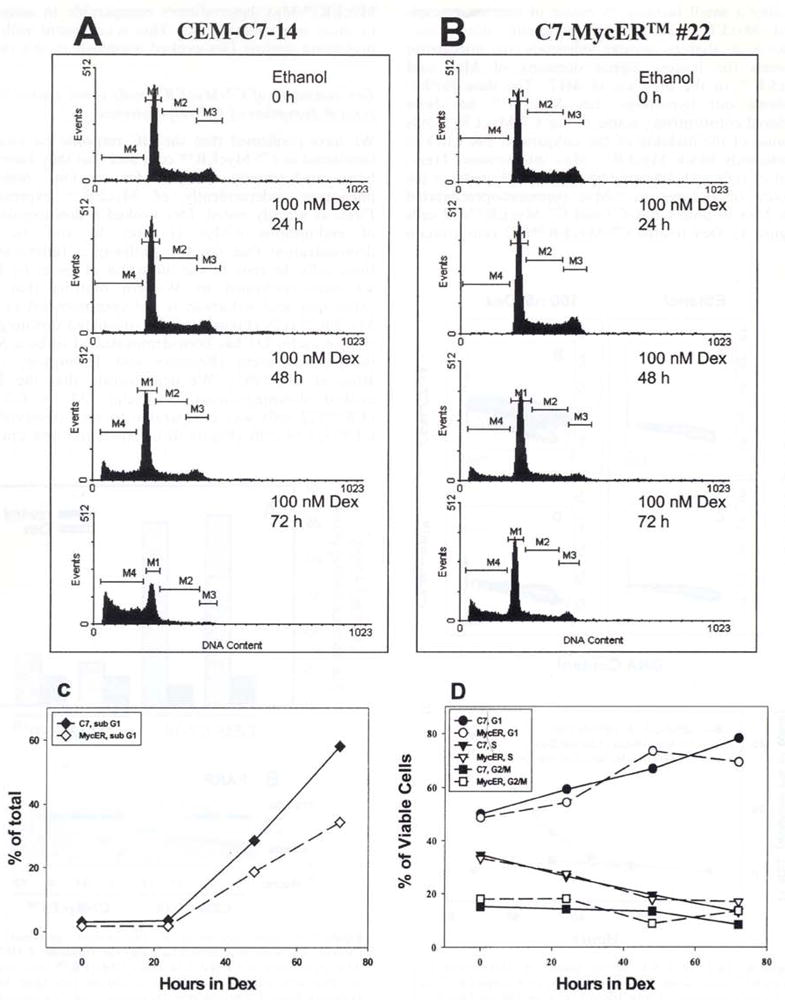
Flow cytometric analysis of Dex-mediated effects on cell cycle distribution. CEM-C7-14 and C7-MyeER™#22 cells were treated with ethanol vehicle or 100 nM Dex for the indicated times, harvested, and their DNA was stained with propidium iodide by sequential incubations in low salt and high salt solutions. The DNA content was measured flow cytometrically using the Cell Quest 1.2 software and a Becton Dickinson FACScan. (a) (CEM-C7-14) and (b) (C7-MycER™#22) show the distribution of 20000 singlet cells gated in individual windows based on their DNA content after treatment with 100 nM Dex for the indicated times. M1, M2, and M3 represent cells in G1, S and G2/M phases of the cell cycle. M4 corresponds to cells with a sub-G1 DNA content and are considered apoptotic. (c) depicts the time course of accumulation of cells with sub G1 DNA content in the presence of 100 nM Dex; data are plotted as a percentage of total cells analysed. In (d) Dex induced changes in distribution of non-apoptotic cells is measured as a function of the duration of Dex treatment. Cells in each phase of the cell cycle are represented as a percentage of viable cells (G1 + S + G2/M). Plots in a and b are from one representative of two independent experiments: data in c and d are averages of the two experiments
Discussion
The protooncogene product c-Myc is an important regulator of cell proliferation, oncogenic transformation and apoptosis. Various independent studies reveal two aspects of the relationship between the expression of c-Myc and apoptosis. Apoptosis associated with growth factor deprivation seems to be augmented by overexpression of c-Myc. In contrast, in T-lymphocytes and several growth factor independent leukemic cell lines, we and others have consistently demonstrated a close correlation between suppression of endogenous c-Myc levels and apoptosis (Helmberg et al., 1995; Medh et al., 1998; Rhee et al., 1995; Yuh and Thompson, 1989). Among the various clones of the human lymphoblastic leukemic cell line CEM, there is a strong correlation between susceptibility to drug-induced apoptosis and c-Myc suppression (Medh et al., 1998; Thulasi et al., 1993).
To further explore the importance of c-Myc reduction for Dex-evoked apoptosis in these lymphoid cells, we have now expressed a chimeric c-Myc protein, MycER™, in CEM-C7-14cells, In the transfected mass cultures and in all subclones tested, our data demonstrate significant protection from GC-induced apoptosis. Sustained constitutive expression of the c-Myc chimera prevented or delayed Dex-evoked events implicated in apoptosis, including altered expression of key genes, DNA strand breaks, caspase 3 activation and PARP cleavage. Though the cells, expressing a constitutively active c-Myc fusion protein, showed a dramatic delay in apoptosis, most eventually died. This delayed death occurred despite a lack of DNA nicking, and much lower caspase 3 activation and PARP cleavage. Hence we conclude that c-Myc suppression is important for classical Dex-evoked apoptosis, but that GCs also evoke other events which eventually kill the cells, albeit more slowly. Loffler et al., (1999) have recently reported that expression of a tetracycline regulatable c-Myc construct does not prevent GC-induced apoptosis of a subclone of CEM-C7 cells. Our data are in partial agreement with this study in that we do see eventual cell death in our C7-MycER™ cells. Unlike the results they reported however, we have observed a clear Myc-induced protection from Dex-evoked apoptosis for at least 72 h after steroid treatment. These seemingly contrasting findings may stem from different parental clones used, from variations in the level of expression and regulation of the transgene, or from distinct experimental conditions.
In considering the functions of c-Myc, one must take into account its multiple modes of action. Heterodimerization between Myc and Max is a prerequisite for c-Myc mediated transactivation from E-box sequences (Blackwood and Eisenman, 1991). The dependence on an ER ligand for gene activation by MycER hybrids has been proposed to be due to relief of steric hindrance for Max binding to the leucine zipper domain of the c-Myc molecule (Picard et al., 1988). The lack of 4HT-dependence for the c-Myc transactivation function in our C7-MycER™ cells may be due to an inadvertent mutation that has arisen in the ER-LBD, or more likely, to cellular conditions that cause relief from steric hindrance, e.g. inadequate expression of ER-LBD interacting factor(s). Whatever the specific cause, by employing coimmunoprecipitation methods, we have demonstrated that in CEM-C7-14 cells MycER™ interacts comparably with Max in the presence or absence of 4HT, explaining its constitutive transcriptional activating ability. Repressive functions of c-Myc may not require heterodimerization with Max and thus may not be affected by the ER LBD of MycER™.
The stable C7-MycER™ transfectants serve as a very useful system with which to evaluate c-Myc’s role in GC-evoked lymphoid cell apoptosis. By examining c-Myc independent targets of GC action (Rhee et al., 1995; Zhou and Thompson, 1996), we have demonstrated that C7-MycER™ cells have not inadvertently lost all sensitivity to Dex, e.g. due to GR mutations. Thus, C7-MycER™ cells have responses comparable to those of the parental CEM-C7-14 cells for GR and c-Jun induction and for cyclin D3 and endogenous c-Myc downregulation (Figures 1 and 4). These data indicate that the basic GC-GR pathway is unaltered in C7-MycER™ cells. This knowledge allows the use of C7-MycER™ cells to evaluate in this system the dependence on c-Myc repression of various established correlates of the processes leading to cell death. One of these correlates is cell cycle arrest. We have previously shown that GC treatment leads to arrest in G0/G1 and that loss of clonogenicity correlates closely in time. Once the cells are arrested, removal of GC did not reverse the cell cycle block (Harmon et al., 1979). The c-Myc protein is an important regulator of the proliferative state of a cell by virtue of its ability to transcriptionally activate genes that are required for biosynthesis of essential components of cell proliferation and global transcriptional regulation (e.g. odc, cad, cdc25 and eIF-4E, (Galaktionov et al., 1996; Jones et al., 1996; Miltenberger et al., 1995) or repress genes such as the growth arrest and DNA damage inducible gene gadd45, and the growth arrest specific gene gas (Facchini and Penn, 1998). Changes in expression of these and other c-Myc-regulated genes could be responsible for the G1 arrest seen in CEM cells about 24 h after continual Dex treatment. However, restoring c-Myc function via expression of MycER™ did not protect cells from Dex-evoked G1 arrest (Figure 8). Therefore we conclude that the G1 arrest is not a consequence of c-Myc downregulation. Further, there is enhanced cell survival despite accumulating G1 arrest. Thus for the first time we have been able to separate Dex-evoked G0/G1 growth arrest of CEM cells from their Dex-evoked apoptosis. In P1798 cells grown in serum, these events are naturally separable (Wood and Thompson, 1984). suggesting that apoptosis of lymphoid cells is not a necessary immediate consequence of G0/G1 arrest by Dex. Our new data here with C7-MycER™ cells support this idea and give an opportunity to pursue the molecular systems that control and link two biological consequences of GC treatment.
One system that is critical for the control of the proliferative or apoptotic state of a cell is cyclin-cdk complexes (Afrakhte et al., 1998; Chiarugi et al., 1994). Binding of cdk inhibitors, including p27kip1 to cyclin D-cdk4 and cyclin E-cdk2 complexes causes their inactivation (Chiarugi et al., 1994; Perez-Roger et al., 1997). Expression of c-Myc and p27kip1 shows an inverse correlation, and the two proteins have been shown to antagonize each other’s actions (Muller et al., 1997; Vlach et al., 1996). For instance, c-Myc promotes p27kip1 phosphorylation and its subsequent degradation (Muller et al., 1997). In CEM-C7-14 cells Dex-evoked c-Myc suppression is associated with p27kip1 upregulation, and ectopic constitutive expression of c-Myc prevents p27kip1 levels from rising significantly above basal amounts in the presence of Dex (Figure 5b). We hypothesize that this may sustain cyclinE-cdk2 activity and hence contribute towards the protection of C7-MycER™ cells from Dex-evoked apoptosis, but it obviously does not explain the cell cycle arrest, the mechanism of which will require further study.
Several c-Myc independent actions of GCs are revealed in C7-MycER™ cells. These may contribute to the eventual, though much delayed, cell death caused by GCs in C7-MycER™ cells. Dex-evoked upregulation of c-Jun and downregulation of cyclin D3 are not altered by the ectopic MycER™ in C7-MycER™ cells. Sustained induction of c-Jun is important for the GC-evoked death of CEM cells and c-Jun elevation due to other stimuli is associated with apoptosis in several other systems (11). The downregulation of cyclin D3 may explain the cell cycle arrest.
These and other as yet unexplored GC-responsive genes may be important mediators of alternative pathways for apoptosis in CEM-C7-14 cells. Constitutive c-Myc expression facilitates a separation of c-Myc dependent from independent, slower modulators of GC-evoked apoptosis. Although detailed analyses of the c-Myc independent pathways for GC-evoked apoptosis are still ongoing, the data presented here already show that the delayed death of C7-MycER™#22 cells in Dex is significantly different from that of parental CEM-C7-14 cells. The fact that MycER™ delays but does not altogether prevent cell death argues against the inadvertent selection an anti-apoptotic factor expressing cell population. We have also demonstrated that susceptibility of C7-MycER™#22 cells to GC-independent apoptotic agents is not altered (Figure 2c). Our data suggest that either the functional level of c-Myc in MycER™#22 cells is insufficient for absolute protection or that c-Myc independent, slower lethal events, e.g. c-Jun induction, are simultaneously triggered by GC. This is analogous to the effect of ectopic expression of Bcl2 in several systems (Rosse et al., 1998; Wagner et al., 1993), or of inhibiting caspase action (Brunet et al., 1998).
In these studies, we have established that c-Myc suppression is crucial for an apoptosis pathway triggered by GCs, demonstrated the existence of parallel pathways, and most importantly, have shown for the first time that GC-evoked arrest of cells in G1/G0 is separable from GC-evoked apoptosis. C7-MycER™ cells thus give the opportunity to define the role of c-Myc reduction in Dex-evoked lymphoid cell apoptosis and understand the relationship between cell cycle arrest and apoptosis.
Materials and methods
Reagents
Dex, 4HT and other reagent grade chemicals were purchased from Sigma Chemical Co. (St. Louis. MO, USA). Staurosporine and okadaic acid were from Calbiochem (La Jolla. CA, USA). Reagents for SDS–PAGE as well as the Bradford protein assay reagent and the PVDP trans-blot transfer membrane were obtained from Bio-Rad (Richmond, CA, USA). The polyclonal antiserum AhuGR150–175, raised against a synthetic peptide corresponding to the amino acids 150 to 175 within the N-terminal domain of hGR (Srinivasan et al., 1994) was used for immunodetection of GR. The monoclonal c-Myc antibody Mycl-9E-10.2, raised against a synthetic peptide in the C-terminal region of c-Myc (Evan et al., 1985), was generated as a culture supernatant of the hybridoma cell line CRL1729 purchased from ATCC. The anti-p53 (sc-126) monoclonal antibody, and rabbit polyclonal antibodies against ERα (sc-544), Max (sc-765), PARP (sc-7150), p27kip1 (sc-528), CREB (sc-186) and c-Jun (sc-1694) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The polyclonal anti-cyclin D3 antibody was from Babco (Richmond, CA, USA). Horseradish peroxidase conjugated secondary antibodies were from Bio-Rad. Protein A-Agarose for immunuprecipitation reactions was from Gibco-BRL Life Technologies (Rockville, MD, USA).
Cell culture
Tissue culture media and components were purchased from Mediatech (Washington DC, USA). Fetal bovine serum (FBS) was from Atlanta Biologicals (Norcross, GA, USA). CEM-C7-14 cells used in this study are subcloned from the original GC-sensitive CEM-C7 clone derived from the parental line CCRF-CEM, obtained from a patient with acute lymphoblastic leukemia (Foley et al., l965). To obtain C7-MyeER™ cells, logarithmically growing CEM-C7-14 cells were washed in Ca++ and Mg++ free phosphate buffered, saline (PBS) and resuspended at a density of 1 × 106 cells/ml. Cells were electroporated at 960 μF and 300 V, with 20 μg of the construct pBpuroMycER™ (a gift from Dr T Littlewood, (Littlewood et al., 1995)), using a Bio-Rad Gene Pulser electroporator. Electroporated cells were allowed to recover for 24 h, then plated in medium containing puromycin (1.5 μg/ml) to select for cells expressing the transfected gene. The puromycin resistant population of cells expressing MycER™ was cloned in 0.5% soft agar to obtain multiple colonies that were individually tested for expression of MycER™. A representative clone, #22, was selected for detailed analyses, the results of which are presented here. All cells were cultured in RPMI 1640 medium supplemented with 5% FBS at 37°C in a humidified 5% CO2 incubator.
Determination of sensitivity to Dex
Dex stock was prepared at a concentration of 2 mM in ethanol. Cells were treated with ethanol vehicle or Dex for various time intervals at a density of 1 × 105 cells/ml. Viable cells were counted by Trypan blue exclusion method using a hemacytometer.
SDS–PAGE and Western blotting
Cells plated at a density of 1–4 × 105 cells/ml were treated for the required time interval with ethanol alone or Dex (100 nM). For analysis of various proteins, approximately 8 × 106 cells were harvested, washed and lysed in buffer containing 50 mM Tris-HCl, pH 7.4, containing 150 mM NaCl, 1 mM EGTA, 1% NP-40, 20 μM leupeptin, 400 μM AEBSF (4-[2-aminoethyl] benzensulfonyl fluoride), 20 mM sodium molybdate, 5 mM sodium fluoride and 5% 2-mercaptoethanol. An equal volume of 2 × SDS–PAGE sample buffer (final composition: 120 mM Tris, 4% SDS, 20% glycerol, 5% 2-mercaptoethanol, 0.05% bromophenol blue, pH 6.8) was added, and samples were boiled for 3 min and centrifuged at 16 000 g for 20 min to obtain a whole cell extract as the supernatant fraction. Fifty microgram protein from each sample was electrophoresed on a 10% SDS polyacrylamide gel (Laemmli, 1970) using a mini-slab-gel electrophoresis apparatus from Bio-Rad and electroblotted on PVDF membrane. The transferred filters were blocked overnight at room temperature with 10% non-fat dry milk in PBS, followed by an overnight incubation in the appropriate primary antibody at 4°C. The filters were rinsed thoroughly before incubation for 2 h with horseradish peroxidase coupled secondary antibody. Bands were detected using the Enhanced Chemiluminescence kit (ECL Plus) from Amersham Pharmacia Biotech (Piscataway, NJ, USA).
Coimmunoprecipitation of Myc-Max complexes
CEM-C7-14 or C7-MycER™ treated with ethanol or Dex were extracted in lysis buffer (150 mM NaCl, 20 mM HEPES, pH 7.4, 1 mM EDTA, 0.5% NP-40, 0.5% sodium deoxycholate, 5 mM NaF, 20 μg/ml leupeptin, 400 μM mM AEBSF and 11 μg/ml soyabean trypsin inhibitor), centrifuged at 13 000 g for 10 min. An aliquot of the supernatant corresponding to 300 μg protein was incubated in 500 μl of RIPA buffer (phosphate buffered saline containing 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS) for 2h at 4°C with an anti-Max polyclonal antibody (sc-765, Santa Cruz Biotech), or nonspecific IgG. Each sample was subsequently added to a Protein A-Agarose pellet generated from 50 μl of a 50% suspension of beads, and incubated overnight on a rotary shaker at 4°C. Beads were washed four times with RIPA buffer and centrifuged at 500 g for 5 min to obtain a pellet which was resuspended in SDS–PAGE sample butter. Bound proteins were eluted by boiling for 3 min, and resolved by SDS – PAGE. Western blotting was performed using the anti-c-Myc monoclonal antibody Mycl-9E10.2.
Flow cytometric evaluation of cell cycle distribution and apoptosis
Cells seeded at a density of 1 × 105 cells/ml were treated with 100 nM Dex for 96 h. At 24 h intervals, three million cells were either directly lysed and stained sequentially in low and high salt solutions containing propidium iodide to measure DNA content, or subjected to terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-X-nick end labeling (TUNEL) reaction prior to staining. For TUNEL, cells were fixed in 4% paraformaldehyde and processed according to the protocol provided by the manufacturer (Boehringer Mannheim). Samples were analysed flow cytometrically using a FACScan, and the Cell quest 1.2 software (Becton Dickinson, Bedford, MA, USA).
Measurement of caspase 3 activity
Caspase 3 activity was measured as described by Sarin et al. (1996), with slight modifications. CEM-C7-14 cells treated with or without 100 nM Dex were harvested and 2.5 × 105 cells were lysed in buffer containing 50 mM HEPES, pH 7.5, 10% sucrose, 0.1% Triton X-100 and 10 μM dithiotreitol. One hundred microliters of the cell lysate was mixed with 50 μM of the caspase 3 specific fluorogenic substrate Z-DEVD-AFC (Z-Asp-Glu-Val-Asp-7-amido-4-trifluoromethyl-coumarin) (Enzyme Systems Products, Livermore, CA, USA) and incubated at room temperature for 1 h. The reaction was diluted to 1 ml with phosphate buffered saline and the fluorescence of AFC released was measured using Fluoro-Count (Packard, Downer’s Grove, IL, USA).
Acknowledgments
We thank Dr T Littlewood for providing us with the construct pBpuroMycER™, Dr Thomas Wood and the staff of the Recombinant DNA Laboratory of the Sealy Center for Molecular Sciences for help with sequence analysis of MycER cDNA isolated from C7-MycER™#22 cells, and Mr Mark Griffin for assistance with Flow Cytometric analyses. We also thank Ms Belly Johnson for cloning CEM-C7-14 cells and for her laboratory managerial skills. This work was supported by grants from the John Sealy Endowment Fund for Biomedical Research (to RD Medh), the Elsa U, Pardee Foundation (to RD Medh), and the National Cancer Institute (to EB Thompson, 2RO1 CA41047). This work was done in conjunction with the Walls Medical Research Foundation.
Abbreviations
- GCs
glucocorticoids
- GR
GC receptor
- cdks
cyclin dependent kinases
- Dex
dexamethasone
- ER
estrogen receptor
- ER™
G525R mutant of ER α
- LBD
ligand binding domain
- 4HT
4 hydroxy tamoxifen
- TUNEL
terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-X-nick end labeling
- PARP
poly ADP ribose polymerase
- FBS
fetal bovine serum
- PBS
phosphate buffered saline
- AEBSF
4-[2-aminoethyl]benzensulfonyl fluoride
- ECL
enhanced chemiluminescence
- AFC
7-amido-4-trifluoromethylcoumarin
References
- Afrakhte M, Heldin NE, Westermark B. Cell Growth Diff. 1998;9:983–988. [PubMed] [Google Scholar]
- Askew DS, Ashmun RA, Simmons BC, Cleveland JL. Oncogene. 1991;6:1915–1922. [PubMed] [Google Scholar]
- Bishop JM. Annu Rev Biochem. 1983;52:301–354. doi: 10.1146/annurev.bi.52.070183.001505. [DOI] [PubMed] [Google Scholar]
- Blackwood EM, Eisenman RN. Science. 1991;251:1211 – 1217. doi: 10.1126/science.2006410. [DOI] [PubMed] [Google Scholar]
- Blyth K, Stewart M, Bell M, James C, Evan G, Neil JC, Cameron ER. Oncogene. 2000;19:773–782. doi: 10.1038/sj.onc.1203321. [DOI] [PubMed] [Google Scholar]
- Brunet CL, Gunby RH, Benson RS, Hickman JA, Watson AJ, Brady G. Cell Death Diff. 1998;5:107–115. doi: 10.1038/sj.cdd.4400334. [DOI] [PubMed] [Google Scholar]
- Bruno S, Ardelt B, Skierski JS, Traganos F, Darzynkiewicz Z. Cancer Res. 1992;52:470 –473. [PubMed] [Google Scholar]
- Casteels KM, Gysemans CA, Waer M, Bouillon R, Laureys JM, Depovere J, Mathieu C. Diabetes. 1998;47:1033 – 1037. doi: 10.2337/diabetes.47.7.1033. [DOI] [PubMed] [Google Scholar]
- Cheng J, Haas M. Mol Cell Biol. 1990;10:5502–5509. doi: 10.1128/mcb.10.10.5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi V, Magnelli L, Cinelli M, Basi G. Cell Mol Biol Res. 1994;40:603 – 612. [PubMed] [Google Scholar]
- Dowd DR, MacDonald PN, Komm BS, Haussler MR, Miesfeld R. J Biol Chem. 1991;266:18423–18426. [PubMed] [Google Scholar]
- Eastman-Reks SB, Vedeckis WV. Cancer Res. 1986;46:2457–2462. [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsay G, Bishop JM. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Facchini LM, Penn LZ. FASEB J. 1998;12:633–651. [PubMed] [Google Scholar]
- Foley GE, Lazarus H, Farber S, Uzman BG, Boone BA, McCarthy RE. Cancer. 1965;18:522–529. doi: 10.1002/1097-0142(196504)18:4<522::aid-cncr2820180418>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Galaktionov K, Chen X, Beach D. Nature. 1996;382:511 – 517. doi: 10.1038/382511a0. [DOI] [PubMed] [Google Scholar]
- Gaynon PS, Carrel AL. Adv Exper Med Biol. 1999;457:593–605. doi: 10.1007/978-1-4615-4811-9_66. [DOI] [PubMed] [Google Scholar]
- Harmon JM, Norman MR, Fowlkes BJ, Thompson EB. J Cell Physiol. 1979;98:267–278. doi: 10.1002/jcp.1040980203. [DOI] [PubMed] [Google Scholar]
- Helmberg A, Auphan N, Caelles C, Karin M. EMBO J. 1995;14:452–460. doi: 10.1002/j.1460-2075.1995.tb07021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homo-Delarche F. Cancer Res. 1984;44:431 – 437. [PubMed] [Google Scholar]
- Jones RM, Branda J, Johnston KA, Polymenis M, Gadd M, Rustgi A, Callanan L, Schmidt EV. Mol Cell Biol. 1996;16:4754 – 4764. doi: 10.1128/mcb.16.9.4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiguchi K, Glesne D, Chubb CH, Fujiki H, Huberman E. Cell Growth Diff. 1994;5:995–1004. [PubMed] [Google Scholar]
- Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Leung K, Munck A. Endocrinology. 1975;97:744–748. doi: 10.1210/endo-97-3-744. [DOI] [PubMed] [Google Scholar]
- Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffler M, Ausserlechner MJ, Tonko M, Hartmann BL, Bernhard D, Geley S, Helmberg A, Kofler R. Oncogene. 1999;18:4626–4631. doi: 10.1038/sj.onc.1202820. [DOI] [PubMed] [Google Scholar]
- Martins TC, Aguas AP. Immunology. 1998;95:377–382. doi: 10.1046/j.1365-2567.1998.00600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medh RD, Saeed MF, Johnson BH, Thompson EB. Cancer Res. 1998;58:3684–3693. [PubMed] [Google Scholar]
- Medh RD, Thompson EB. Cell Tissue Res. 2000;301:101 – 124. doi: 10.1007/s004419900159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miltenberger RJ, Sukow KA, Farnham PJ. Mol Cell Biol. 1995;15:2527 – 2535. doi: 10.1128/mcb.15.5.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montague JW, Cidlowski JA. Curr Topics Microbiol Immun. 1995;200:51–65. doi: 10.1007/978-3-642-79437-7_4. [DOI] [PubMed] [Google Scholar]
- Muller D, Bouchard C, Rudolph B, Steiner P, Stuckmann I, Saffrich R, Ansorge W, Huttner W, Eilers M. Oncogene. 1997;15:2561–2576. doi: 10.1038/sj.onc.1201440. [DOI] [PubMed] [Google Scholar]
- Perez-Roger I, Solomon DL, Sewing A, Land H. Oncogene. 1997;14:2373–2381. doi: 10.1038/sj.onc.1201197. [DOI] [PubMed] [Google Scholar]
- Peukert K, Staller P, Schneider A, Carmichael G, Hanel F, Eilers M. EMBO J. 1997;16:5672–5686. doi: 10.1093/emboj/16.18.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipp A, Schneider A, Vasrik I, Finke K, Xiong Y, Beach D, Alitalo K, Eilers M. Mol Cell Biol. 1994;14:4032 – 4043. doi: 10.1128/mcb.14.6.4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard D, Salser SJ, Yamamoto KR. Cell. 1988;54:1073–1080. doi: 10.1016/0092-8674(88)90122-5. [DOI] [PubMed] [Google Scholar]
- Rabbitts PH, Watson JV, Lamond A, Forster A, Stinson MA, Evan G, Fischer W, Atherton E, Sheppard R, Rabbitts TH. EMBO J. 1985;4:2009–2015. doi: 10.1002/j.1460-2075.1985.tb03885.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisman D, Elkind NB, Roy B, Beamon J, Rotter V. Cell Growth Diff. 1993;4:57–65. [PubMed] [Google Scholar]
- Reisman D, Thompson EA. Mol Endocrinol. 1995;9:1500–1509. doi: 10.1210/mend.9.11.8584027. [DOI] [PubMed] [Google Scholar]
- Rhee K, Bresnahan W, Hirai A, Hirai M, Thompson EA. Cancer Res. 1995;55:4188 – 4195. [PubMed] [Google Scholar]
- Rosse T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C. Nature. 1998;391:496–499. doi: 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- Sakamuro D, Prendergast GC. Oncogene. 1999;18:2942–2954. doi: 10.1038/sj.onc.1202725. [DOI] [PubMed] [Google Scholar]
- Sarin A, Wu ML, Henkart PA. J Exp Med. 1996;184:2445–2450. doi: 10.1084/jem.184.6.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Glynn JM, Guilbert LJ, Cotter TG, Bissonnette RP, Green DR. Science. 1992;257:212–214. doi: 10.1126/science.1378649. [DOI] [PubMed] [Google Scholar]
- Spencer CA, Groudine M. Adv, Cancer Res. 1991;56:1–48. doi: 10.1016/s0065-230x(08)60476-5. [DOI] [PubMed] [Google Scholar]
- Srinivasan G, Patel NT, Thompson EB. Mol Endocrinol. 1994;8:189–196. doi: 10.1210/mend.8.2.8170475. [DOI] [PubMed] [Google Scholar]
- Thompson EB. Annu Rev Physiol. 1998;60:575 – 600. doi: 10.1146/annurev.physiol.60.1.575. [DOI] [PubMed] [Google Scholar]
- Thompson EB. Trends in Endocrinol Metab. 1999;10:353–358. doi: 10.1016/s1043-2760(99)00187-3. [DOI] [PubMed] [Google Scholar]
- Thompson EB, Thulasi R, Saeed MF, Johnson BH. Ann N Y Acad Sci. 1995;761:261–275. doi: 10.1111/j.1749-6632.1995.tb31383.x. [DOI] [PubMed] [Google Scholar]
- Thulasi R, Harbour DV, Thompson EB. J Biol Chem. 1993;268:18306–18312. [PubMed] [Google Scholar]
- Tobias KE, Shor J, Kahana C. Oncogene. 1995;11:1721–1727. [PubMed] [Google Scholar]
- Vaillant F, Blyth K, Terry A, Bell M, Cameron ER, Neil J, Stewart M. Oncogene. 1999;18:7124–7134. doi: 10.1038/sj.onc.1203202. [DOI] [PubMed] [Google Scholar]
- Vlach J, Hennecke S, Alevizopoulos K, Conti D, Amati B. EMBO J. 1996;15:6595 – 6604. [PMC free article] [PubMed] [Google Scholar]
- Wagner AJ, Small MB, Hay N. Mol Cell Biol. 1993;13:2432–2440. doi: 10.1128/mcb.13.4.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood KM, Thompson EA., Jr Mol Cell Endocrinol. 1984;37:169 – 180. doi: 10.1016/0303-7207(84)90049-2. [DOI] [PubMed] [Google Scholar]
- Yatouji S, Liautaud-Roger F, Dufer J. Cell Prolif. 2000;33:51 – 62. doi: 10.1046/j.1365-2184.2000.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yerly-Motta V, Pavy JJ, Herve P. Biotechnic & Histochemistry. 1999;74:119 – 128. doi: 10.3109/10520299909047963. [DOI] [PubMed] [Google Scholar]
- Yuh YS, Thompson EB. J Biol Chem. 1989;264:10904 – 10910. [PubMed] [Google Scholar]
- Zhou F, Thompson EB. Mol Endocrinol. 1996;10:306–316. doi: 10.1210/mend.10.3.8833659. [DOI] [PubMed] [Google Scholar]