

Abstract
A study of DXP synthase has revealed flexibility in the acceptor substrate binding pocket for non-polar substrates, and has uncovered new details of the catalytic mechanism to show that pyruvate can act as both donor and acceptor substrate.
Members of the isoprenoid class of natural products are all biosynthesized from the common precursors isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP). The mevalonate independent 2-C-methyl-D-erythritol 4-phosphate (MEP) pathway was identified as an alternative to the mammalian mevalonic acid (MVA) pathway for the production of these essential isoprenoid precursors.1 This pathway to isoprenoids is widespread in plants and human pathogens, including the majority of human bacterial pathogens2 and the malaria parasite Plasmodium falciparum,3 and is under intense investigation as a potential target for the development of new anti-infective agents and herbicides.4
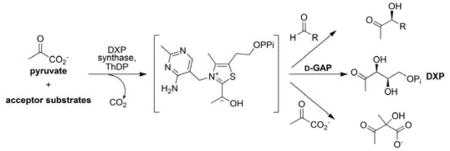
The first enzyme in the MEP pathway (Figure 1), 1-deoxy-D-xylulose 5-phosphate (DXP) synthase, represents a new class of thiamine-dependent enzymes that catalyzes the formation of DXP from pyruvate and D-glyceraldehyde 3-phosphate (D-GAP). This transformation is reminiscent of transketolases and decarboxylases, where a two-carbon intermediate bound to thiamine diphosphate (ThDP) is condensed with an acceptor substrate to generate a new carbon-carbon bond. Homologs of DXP synthase include transketolase and acetolactate synthase which are known to follow the classical ping-pong kinetic mechanism, where release of CO2 from the thiamine-bound intermediate precedes binding of the acceptor substrate. In contrast, DXP synthase follows an ordered kinetic mechanism,5 where binding of the second substrate, D-GAP, is required for release of CO2 and formation of a kinetically competent complex. Interestingly, structural analysis of DXP synthase indicates the active site is located between domains of the same monomer of the homodimer, in contrast to its homologs, where the active site is at the dimer interface.6 Taken together, these studies reveal distinctive characteristics of DXP synthase, placing this enzyme in the growing list of potential targets for anti-infective drug development.
Figure 1.

DXP synthase catalyzes the first step in the MEP pathway.
We have hypothesized that a study of DXP synthase substrate specificity will disclose important substrate binding determinants to help define the substrate binding pockets, and reveal previously undiscovered catalytic activities of this novel enzyme class. Existing assays used to study DXP synthase substrate specificity involved laborious TLC resolution of radioactive products7 or insensitive UV- or refractive index-based detection of unnatural products.8 These studies suggested the enzyme processes a small subset of aldose and aldose phosphate acceptor substrates, closely resembling the natural acceptor substrate. While the scope of substrates that could be tested was limited by these assays, the results nevertheless suggested a potential utility of DXP synthase for generating 1-deoxysugars.
Reports describing the incorporation of fluorophores to enhance sensitivity of detection, use harsh and/or labor intensive derivatization protocols9 or do not resolve substrates from products.7 Reports are lacking on tolerance of this important enzyme class toward structurally diverse substrates. To more thoroughly explore the scope of DXP synthase catalytic activity, a sensitive and flexible assay is needed. Here, we report a versatile, robust HPLC-based assay for DXP synthase, and present results demonstrating its utility for probing new reactions catalyzed by DXP synthase from Escherichia coli. Notably, our results indicate that DXP synthase demonstrates a valuable capacity to process non-polar aliphatic aldehydes that are structurally distinct from the natural substrate. Additionally, our assay has uncovered new details of the catalytic mechanism of DXP synthase. Here, we provide the first direct evidence to support the hypothesis formulated by Eubanks and Poulter5 that a second molecule of pyruvate can bind to DXP synthase. We further demonstrate that DXP synthase, in fact, utilizes pyruvate as the second substrate in the generation of acetolactate.
For sensitive detection of DXP synthase activity in this HPLC-based discontinuous assay, we have exploited the reactivity of the carbonyl groups in both substrates and product toward 2,4-dinitrophenylhydrazine (2,4-DNP) to give the corresponding hydrazones (Figure 2). Derivatization reactions were performed by treating quenched enzymatic reaction mixtures with excess 2,4-DNP in sulfuric acid at room temperature. Hydrazone formation was found to be complete within 2–3 min (Figure S2b). Acid-catalyzed hydrolysis of the phosphoryl moieties in D-GAP and DXP was noted in derivatization mixtures after ~30 min. Thus, derivatization reactions were adjusted to pH 5–7 after 5 min to prevent degradation. Hydrazone mixtures were then easily resolved and detected using HPLC equipped with a UV photodiode array detector. For rapid sample analysis, we have employed the use of a short Rocket® column such that HPLC analysis is complete within 10 min for each sample.
Figure 2.

a) Derivatization of DXP synthase substrates and product with 2,4-DNP and b) HPLC timecourse analysis.
Confirmation of DXP formation was accomplished following treatment of DXP synthase enzymatic reaction mixtures with Antarctic phosphatase (AP) and comparison of the resulting product to authentic 1-deoxy-D-xylulose hydrazone (Figure S3). Optimization of the enzymatic reaction conditions was also possible using this assay because both substrates and products can be detected by HPLC. Importantly, we found that pyruvate and D,L-GAP are unstable in buffers often used in DXP synthase assays,7,10,11,12,13 including Tris and glycylglycine. Enzymatic reactions carried out at pH 8.0 in HEPES or phosphate buffer were found to be optimal, as substrates are stable under these conditions.
Further validation of this assay was accomplished through Michaelis-Menten kinetic analysis of DXP synthase in the presence of natural substrates. Kinetic parameters were measured for pyruvate and D-GAP (Figure S4; KmD-GAP = 226 ± 32 μM, Kmpyruvate = 1.1 ± 0.3 mM, kcat = 246 ± 13 min−1) and were found to be in close agreement with reported values for DXP synthase from Rhodobacter capsulatus.5
Previous studies have demonstrated that DXP synthase utilizes glyceraldehyde (GA) as acceptor substrate, indicating a tolerance for absence of the phosphoryl moiety.5,12,14,15 We have confirmed this result using our assay. However, little is known about the effects of removing substrate hydroxyl groups or lengthening the carbon chain. This work focuses on investigating enzyme flexibility toward aliphatic aldehydes as alternative acceptor substrates.
Initial experiments tested acetaldehyde (3) as substrate for DXP synthase. The enzyme-catalyzed formation of acetoin (15) was measured by HPLC detection of the corresponding hydrazone derivative (Figure 3). Acetoin formation was confirmed by comparing the product to authentic acetoin hydrazone and by mass analysis. An apparent kcat/Km of (7.0 ± 0.2) × 102 M−1·min−1 was measured for acetaldehyde that is comparable to glyceraldehyde (kcat/Km of (21 ± 0.3) × 102 M−1·min−1, Table 1), revealing relaxed specificity for simple, non-polar acceptor substrates.
Figure 3.

Analysis of acetaldehyde (3) as an alternative acceptor substrate. a) HPLC timecourse showing acetoin (15) formation. Acetolactate (16) forms in the presence and absence of 3. b) Proposed mechanisms to form 15 and 16.
Table 1.
Summary of DXP synthase substrate specificity toward aliphatic aldehydes.
| substrate | product | kcat/Km × 102 (M−1·min−1) | relative kcat/Km |
|---|---|---|---|
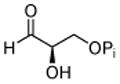 1: D-GAP |
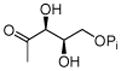 DXP |
8200 ± 200 | 390 |
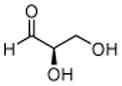 2: D-GA |
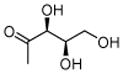 DX |
21 ± 3 | 1 |
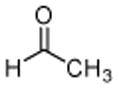 3: acetaldehyde |
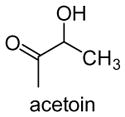 |
7.0 ± 0.2 | 0.33 |
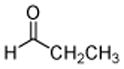 4: propanal |
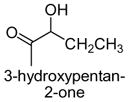 |
2.1 ± 0.4 | 0.10 |
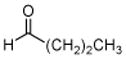 5: butanal |
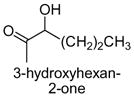 |
3.6 ± 0.2 | 0.17 |
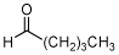 6: pentanal |
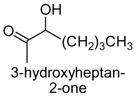 |
n.d. | -- |
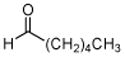 7: hexanal |
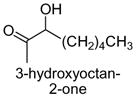 |
n.d. | -- |
n.d. = not determined
In addition, a second enzyme-dependent peak was observed (6.7 min, Figure 3a). Mass analysis of the hydrazone product was consistent with the hydrazone of acetolactate (16). In principle, acetolactate could be formed in a reaction of the thiamine-bound intermediate with a second molecule of pyruvate, similar to the reaction catalyzed by acetolactate synthase16 (Figure 3b). The acetolactate hydrazone was observed at 6.7 min, as well as a second peak corresponding to the acetoin hydrazone. These results are consistent with a mechanism involving reaction of the thiamine-bound intermediate with a second molecule of pyruvate to form acetolactate, which then undergoes decarboxylation to form acetoin (Figure 3b). In a previous kinetic study of DXP synthase,5 the rate of CO2 release was found to be significantly slower in the absence of D-GAP, providing evidence for an ordered kinetic mechanism. However, in the presence of high pyruvate concentrations, slow release of CO2 was observed. The authors speculated that a second molecule of pyruvate may bind to the active site to promote CO2 release from the thiamine-bound intermediate leading to a ThDP-acetaldehyde complex that could dissociate and allow additional binding of free ThDP.5 Our results provide the first direct evidence that pyruvate acts as acceptor substrate, to promote CO2 release and generate a new product, acetolactate.
The observation that acetoin is generated from pyruvate, in the absence of acetaldehyde, led us to seek further proof that acetaldehyde acts as acceptor substrate to generate acetoin independently. The rate of acetoin formation from pyruvate is 10-fold lower in the absence of acetaldehyde. Nevertheless, to rule out any uncertainty about the capacity of DXP synthase to use acetaldehyde as acceptor substrate to generate acetoin, the enzyme was incubated with pyruvate and d4-acetaldehyde. As expected, formation of the corresponding d4-acetoin hydrazone was observed (Table S1).
The results obtained in these preliminary experiments encouraged us to expand the scope of these studies to examine DXP synthase substrate promiscuity toward other aldehydes. Notably, DXP synthase exhibits similar reaction profiles in the presence of several alternative aliphatic aldehydes (results summarized in Table 1). Propanal (4), butanal (5), pentanal (6) and hexanal (7) are utilized by the enzyme (Figures S4–S8), indicating DXP synthase is considerably tolerant of long chain acceptor substrates. Specificity constants were measured for these substrates and compared to D-GAP (1), GA (2) and acetaldehyde (3) (Table 1). A near 400-fold decrease in kcat/Km results from the removal of the phosphoryl moiety from D-GAP, indicating the contribution of the phosphoryl moiety in substrate binding. However, removal of either hydroxyl group of GA or lengthening the carbon chain has only modest effects on kcat/Km values, demonstrating a flexibility in this binding pocket for lengthening the carbon chain and removal of hydroxyl moieties. This finding indicates that DXP synthase exhibits a relaxed specificity toward non-polar aldehyde substrates, rivaling that of TK,17 to generate the corresponding α-hydroxy ketones, an important compound class in the life-science industry.18,19
Not surprisingly, heptanal (8, Table S1) proved to be insoluble under these assay conditions. Branched aliphatic aldehydes are apparently not readily accepted by DXP synthase. Although minor product formation was observed in the presence of β-branched aldehyde 9 and cyclopropyl aldehyde 10 (Figures S9 and S10), more bulky α-branched aldehydes were not processed by the enzyme. Apparent binding of pentanal to protein was observed, indicating that accurate determination of specificity constants for longer chain aldehydes under these conditions will be difficult.
In summary, the lack of an appropriate assay combining the sensitivity and flexibility needed to study DXP synthase has imposed limitations in the exploration of substrate promiscuity. We have developed a robust HPLC-based assay that permits the detection of both substrates and product and provides the sensitivity required for detection of compounds that are not inherently chromophoric. Derivatization of carbonyl-containing substrates and products is complete within 2–3 min, and HPLC analysis for the reactions studied here could be accomplished within 10 min. We have used this assay to begin exploration of DXP synthase substrate promiscuity, and to resolve uncertainties about the catalytic mechanism.
Our findings reveal relaxed specificity of DXP synthase for non-polar aliphatic aldehydes, with the exception that bulky α-branched aldehydes are not well-tolerated by the enzyme. This relaxed specificity to produce α-hydroxy ketones points to DXP synthase as a potential biocatalyst for generating this important compound class, and newly reveals flexibility in the acceptor substrate binding pocket for non-polar substrates. The perplexing observation that CO2 release takes place in the absence of D-GAP, albeit at significantly reduced rates,5 was resolved during the course of our investigation. The assay reported here enabled the detection of enzyme-catalyzed acetolactate formation, providing confirmation that pyruvate acts as the second substrate to promote CO2 release in the absence of D-GAP. Our findings raise new questions about opportunities to target this unique enzyme, or to exploit it as a biocatalyst. Studies are underway to expand the scope of this reaction.
Supplementary Material
Acknowledgments
We acknowledge Marc Rosen (Johns Hopkins University School of Medicine) for his assistance with mass spectrometry, and Jason Howard (Johns Hopkins University School of Medicine) for conducting initial derivatization experiments. We are grateful to Rahul Kohli and Philip Cole of Johns Hopkins University School of Medicine, and Richard Borch of Purdue University for helpful discussions and careful critiques of this manuscript. This work was supported by funding from the NIH (T32 GM007445) and Johns Hopkins Malaria Research Institute Pilot Grant for L.A.B.
Footnotes
Supporting Information Available: Assay protocols, unnatural product characterization, kinetic analyses.
References
- 1.Rohmer M, Knani M, Simonin P, Sutter B, Sahm H. Biochem J. 1993;295:517–524. doi: 10.1042/bj2950517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Testa C, Brown M. Curr Pharm Biotechnol. 2003;4:248–259. doi: 10.2174/1389201033489784. [DOI] [PubMed] [Google Scholar]
- 3.Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, Hintz M, Tübachova I, Eberl M, Zeidler J, Lichtenthaler H, Soldati D, Beck E. Science. 1999;285:1573–1576. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- 4.Eoh H, Brennan P, Crick D. Tuberculosis (Edinb) 2009;89:1–11. doi: 10.1016/j.tube.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eubanks L, Poulter C. Biochemistry. 2003;42:1140–1149. doi: 10.1021/bi0205303. [DOI] [PubMed] [Google Scholar]
- 6.Xiang S, Usunow G, Lange G, Busch M, Tong L. J Biol Chem. 2007;282:2676–2682. doi: 10.1074/jbc.M610235200. [DOI] [PubMed] [Google Scholar]
- 7.Querol J, Grosdemange-Billiard C, Rohmer M, Boronat A, Imperial S. Tetrahedron Lett. 2002;43:8265–8268. [Google Scholar]
- 8.Schürmann M, Schürmann M, Sprenger G. J Mol Catal B: Enzym. 2002;19–20:247–252. [Google Scholar]
- 9.Han Y, Sabbioni C, van der Heijden R, Verpoorte R. J Chromatogr, A. 2003;986:291–296. doi: 10.1016/s0021-9673(02)02016-2. [DOI] [PubMed] [Google Scholar]
- 10.Lee J, Oh D, Kim S. J Biotechnol. 2007;128:555–566. doi: 10.1016/j.jbiotec.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Lois L, Campos N, Putra S, Danielsen K, Rohmer M, Boronat A. Proc Natl Acad Sci USA. 1998;95:2105–2110. doi: 10.1073/pnas.95.5.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuzuyama T, Takagi M, Takahashi S, Seto H. J Bacteriol. 2000;182:891–897. doi: 10.1128/jb.182.4.891-897.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Altincicek B, Hintz M, Sanderbrand S, Wiesner J, Beck E, Jomaa H. FEMS Microbiol Lett. 2000;190:329–333. doi: 10.1111/j.1574-6968.2000.tb09307.x. [DOI] [PubMed] [Google Scholar]
- 14.Bailey A, Mahapatra S, Brennan P, Crick D. Glycobiology. 2002;12:813–820. doi: 10.1093/glycob/cwf100. [DOI] [PubMed] [Google Scholar]
- 15.Hahn F, Eubanks L, Testa C, Blagg B, Baker J, Poulter C. J Bacteriol. 2001;183:1–11. doi: 10.1128/JB.183.1.1-11.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crout D. The Chemistry of Branched Chain Amino Acid Biosynthesis: Stereochemical and Mechanistic Aspects. In: Barak A, Chipman J, editors. Biosynthesis of Branched Chain Amino Acids. VCH; Weinheim, Germany: 1990. pp. 199–242. [Google Scholar]
- 17.Hibbert E, Senussi T, Smith M, Costelloe S, Ward J, Hailes H, Dalby P. J Biotechnol. 2008;134:240–245. doi: 10.1016/j.jbiotec.2008.01.018. [DOI] [PubMed] [Google Scholar]
- 18.Müller M, Gocke D, Pohl M. FEBS J. 2009;276:2894–2904. doi: 10.1111/j.1742-4658.2009.07017.x. [DOI] [PubMed] [Google Scholar]
- 19.Marigo M, Jørgensen K. Chem Commun. 2006:2001–2011. doi: 10.1039/b517090g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.