

Abstract
At excitatory synapses, decreases in cleft [Ca] arising from activity-dependent transmembrane Ca flux reduce the probability of subsequent transmitter release. Intense neural activity, induced by physiological and pathological stimuli, disturb the external microenvironment reducing extracellular [Ca] ([Ca]o) and thus may impair neurotransmission. Increases in [Ca]o activate the extracellular calcium sensing receptor (CaSR) which in turn inhibits nonselective cation channels at the majority of cortical nerve terminals. This pathway may modulate synaptic transmission by attenuating the impact of decreases in [Ca]o on synaptic transmission. Using patch-clamp recording from isolated cortical terminals, cortical neuronal pairs and isolated neuronal soma we examined the modulation of synaptic transmission by CaSR. EPSCs were increased on average by 88% in reduced affinity CaSR-mutant (CaSR−/−) neurons compared with wild-type. Variance-mean analysis indicates that the enhanced synaptic transmission was due largely to an increase in average probability of release (0.27 vs 0.46 for wild-type vs CaSR−/− pairs) with little change in quantal size (23 ± 4 pA vs 22 ± 4 pA) or number of release sites (11 vs 13). In addition, the CaSR agonist spermidine reduced synaptic transmission and increased paired-pulse depression at physiological [Ca]o. Spermidine did not affect quantal size, consistent with a presynaptic mechanism of action, nor did it affect voltage-activated Ca channel currents. In summary, reduced CaSR function enhanced synaptic transmission and CaSR stimulation had the opposite effect. Thus CaSR provides a mechanism that may compensate for the fall in release probability that accompanies decreases in [Ca]o.
Keywords: calcium, GPCR, depression, synaptic plasticity, synaptic transmission, synaptic vesicle release, synaptosome, extracellular, receptor
Introduction
Both the moderate decreases in extracellular [Ca] ([Ca]o) that occur during normal synaptic activity (Nicholson et al., 1978) and the substantial decreases in [Ca]o resulting from focal brain trauma or ischemia (Li et al., 1995; Nilsson et al., 1996) indicate that [Ca]o is dynamic. Additional supporting evidence comes from mathematical models (Smith, 1992; Vassilev et al., 1997; Egelman and Montague, 1998, 1999; Rusakov, 2001), voltage-activated Ca channel (VACC) current measurements in nerve terminals (Borst and Sakmann, 1999a; Stanley, 2000), and cleft [Ca] measurements with fluorescence probes (Rusakov and Fine, 2003).
A decrease in [Ca]o is predicted to sharply inhibit synaptic transmission (Dodge and Rahamimoff, 1967; Heidelberger et al., 1994; Schneggenburger and Neher, 2000). Thus, a physiological decrease in [Ca]o after exocytosis should impair subsequent transmitter release. However, recordings of sustained high-frequency activity indicate that this is not always the case (Taschenberger and von Gersdorff, 2000). Therefore, it has been proposed that a fall in cleft [Ca] may provide a retrograde signal to the nerve terminal to compensate for decreases in Ca entry (Vassilev et al., 1997; Rusakov and Fine, 2003). The components of such a signaling pathway were recently identified in neocortical nerve terminals as (1) a nonselective cation channel (NSCC) and (2) the extracellular calcium-sensing receptor (CaSR).
In neocortical nerve terminals increases in [Ca]o activate a receptor that blocks the activation of voltage-dependent NSCCs. Conversely, decreases in [Ca]o lead to activation of NSCCs (Smith et al., 2004). This [Ca]o-dependent NSCC current could modulate the probability of transmitter release by modifying the terminal resting membrane potential or action potential shape.
CaSR, a G-protein-coupled receptor, is activated by increases in [Ca]o, has a vital role in the regulation of serum [Ca] (Brown et al., 1993), and is expressed in the brain (Ruat et al., 1995; Rogers et al., 1997). We recently identified CaSR as a terminal [Ca]o sensor; increases in [Ca]o stimulate CaSR and reduce NSCC activity (Chen et al., 2006). Thus, a pathway including CaSR and NSCC could represent the retrograde signal communicating [Ca]o to the presynaptic terminal.
We hypothesized that if this pathway compensates for decreases in cleft [Ca], then activation of CaSR would reduce synaptic transmission. Conversely, if this pathway contributes to the steep [Ca]o sensitivity of exocytosis (Dodge and Rahamimoff, 1967; Heidelberger et al., 1994; Schneggenburger and Neher, 2000), then activation of CaSR should increase release probability.
To test whether CaSR modulates synaptic transmission, we examined the impact of a reduced-function CaSR mutation (CaSR−/−) and CaSR agonists on neurotransmission in mouse cortical neurons. Here, we show that CaSR−/− expression results in increased EPSCs, whereas CaSR agonists reduce basal synaptic transmission, indicating that CaSR activity modulates transmission. Therefore, the CaSR–NSCC pathway in nerve terminals may serve to obviate the reduction in synaptic transmission that would accompany physiological and pathological reductions in [Ca]o.
Materials and Methods
Synaptosome preparation.
Cortices were isolated from 6- to 8-week-old mouse brain and synaptosomes prepared and resuspended in Tyrode as described previously (Bergsman and Tsien, 2000). Synaptosomes were plated onto cover glasses and stored for 0.5–6 h at 4°C before use.
Neuronal culture preparation.
Neocortical cell cultures were prepared (Mermelstein et al., 2000) from postnatal day 1 (P1) to P2 wild-type or CaSR−/− mice (Ho et al., 1995). The cells were grown in 1 ml of MEM (Minimum Essential Medium; Invitrogen) with 28 mm glucose, 2.4 mm NaHCO3, 0.0013 mm transferrin, 2 mm glutamine, 0.0042 mm insulin, and 5% FBS, pH 7.35. Superior cervical ganglion (SCG) neurons were acutely dissociated from 14- to 21-d-old mice (Chen et al., 2007). Neurons were plated onto coverslips and maintained in a humidified atmosphere containing 5% CO2 in air at 37°C. Neurons were used 6–30 h after plating.
Genotyping CaSR mutant mice.
Heterozygotes for the CaSR mutation were mated to produce CaSR−/− as this mutation is lethal at P3–P30. DNA from mouse tail was released by treatment with 50 mm NaOH at 95°C for 15 min, followed by the addition of 1m Tris, pH 8.0, and 10 mm EDTA. PCR was then performed using DNA solution and three primers: CaSR 5′: TCTCTTCTCTTTAGGTCCTGAAAGA, CaSR 3′: TCATTGATGAACAGTCTTTCTCCCT, and r-neo2: TCTTGATTCCCACTTTGTCCTTGTA. The samples were run on a 1% agarose gel and identified as CaSR+/+, CaSR +/−, or CaSR−/−.
Electrophysiology.
Whole-cell patch-clamp recordings from neocortical neurons and SCG neurons were performed with pipettes with resistances 3–7 MΩ and 1–3 MΩ, respectively. Synaptosome recordings used cell-attached configuration and pipette resistances 15–40 MΩ. The preparation was placed in a recording chamber on the stage of an inverted microscope. Test solutions were applied to the neurons at 1–3 ml/min from a nearby capillary tube and manifold. Membrane potentials were corrected for measured liquid junction potentials. In synaptosomal recordings the resting membrane potential was unknown and the described voltages are pipette potential multiplied by negative one. Synaptosome and cortical neuron recordings used a Tyrode bath solution [(in mm) 150 NaCl, 4 KCl, 1.1 CaCl2, 1.1 MgCl2 10 HEPES, and 10 glucose, at pH 7.35 with NaOH]. Bicuculline methiodide and spermidine were both obtained from Sigma-Aldrich. SCG recordings solutions were modified to isolate neuronal VACC currents (Chen et al., 2007). Tetrodotoxin (TTX, 1 μm; Alomone Labs) and TEACl (15 mm; NaCl decreased to 135 mm) were added to block Na and K currents respectively. Pipette solution was preparation dependent. In neocortical soma recordings, we used (in mm) 118 K-Gluconate, 14 creatine phosphate, 10 HEPES, 9 EGTA, 4 MgCl, 4 NaATP, 1 CaCl, and 0.3 NaGTP, pH 7.2 with KOH. In SCG recordings, we used (in mm) 120 N-methy-d-glucamine, 20 TEAOH, 11 EGTA, 10 HEPES, 10 MgCl2, 1 CaCl2, 4 NaATP, 0.3 NaGTP, 14 creatinine phosphate (pH 7.2 with methanesulfonic acid). Tyrode was used in synaptosome recordings and contained Mg and Ca at 2 mm. Voltage-clamp recordings were obtained using Pulse software with an Axopatch 200B (Axon Instruments) or EPC-9 (Heka Instruments) amplifier. Neurons that did not exhibit significant rundown of EPSC amplitude in a baseline period of ∼5 min were treated with 100 μm spermidine until a new stable baseline was achieved, after which time the agonist was washed off for ∼6 min. In SCG neurons, series resistance was compensated by 70–85%. Average EPSCs were obtained by measuring consecutive EPSCs (n = 16 ± 1) for each neuronal pair. To allow comparison between pairs of neurons data were pooled. Data were sorted by ascending mean EPSC amplitude for each genotype. The data sets were divided into groups, each containing the same number of measurements, based on the mean EPSC, and then the mean EPSC and EPSC variance plotted for each group.
Data analysis.
Data were analyzed using MiniAnalysis (Synaptosoft) and custom macros written in Igor Pro (Wavemetrics). Dose–response data (see Figs. 1, 4) were fit with:
where I represents current at concentration A of CaSR agonist, Imax is the maximum current, and n represents the Hill coefficient. Tests for paired or unpaired replicates were used as appropriate. Mean ± SEM values are reported in general. In the case of nonparametric data, the Mann–Whitney test was used and median values are reported. All tests were two-tailed and a p value <0.05 was considered significant. Graphpad Prism software was used to calculate p values.
Figure 1.
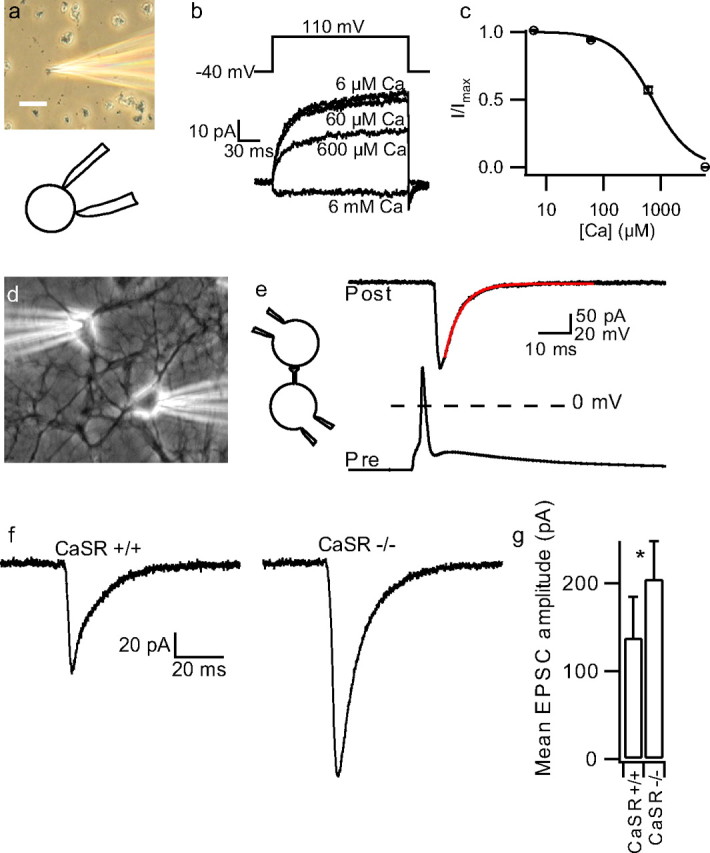
EPSC increased in CaSR−/− neurons. a, Photomicrograph and inset diagram of cell-attached recording from isolated synaptosome. Calibration bar is 20 μm in length. b, Currents activated by 150 mV step depolarizations in synaptosome recording at range of steady state bath [Ca] (6 μm–6 mm). c, Concentration-effect relationship for outward current and bath [Ca]. Average normalized data from 9 recordings made using Tyrode in pipette (2 mm Mg and Ca). Data from each recording were fit to Equation 1 (see Materials and Methods), where I was outward current at end of depolarizing step, Imax was I with 6 μm Ca in bath, and n was set to 1.3 (Smith et al., 2004). The curve was redrawn using the average value for IC50. d, Photomicrograph of whole-cell recording from two adjacent cortical neurons. e, Inset indicating paired recording and exemplar presynaptic action potential and postsynaptic current in neuron clamped at −70 mV. Broken line indicates 0 mV. f, Exemplars of EPSCs close to median values recorded from CaSR+/+ (n = 36) and −/− (n = 28) neuronal pairs. g, Histogram of mean ± SEM. EPSC for CaSR+/+ and −/− neuronal pairs. *p < 0.05.
Figure 4.

Low-dose spermidine does not inhibit VACC in SCG neurons. a, VACC currents activated by ramp depolarizations in an exemplar neuron were inhibited by spermidine (in mm). b, Diary plot of peak inward VACC current versus time and concentration of spermidine. c, Concentration-effect relationship for spermidine on peak VACC currents normalized against current in absence of polyamine. The curve was drawn using Equation 1, using the mean values and given IC50 of 9.5 ± 0.9 mm and n of 0.82 ± 0.06.
Results
Impaired CaSR facilitates synaptic transmission
To examine the role of the CaSR–NSCC signaling pathway in neurotransmission we used a CaSR mutant mouse (CaSR−/−) (Ho et al., 1995). CaSR−/− mice express a CaSR splice-variant which is activated by external Ca but has reduced affinity compared with wild-type (Oda et al., 2000). Cell-attached recordings from acutely dissociated neocortical terminals confirmed the [Ca]o-NSCC signaling pathway was present in mouse wild-type (CaSR+/+) terminals (Fig. 1a–c). Under conditions of low [Ca]o, depolarization activated outward currents over tens of milliseconds, with an IC50 for [Ca]o of 719 ± 45 μm (n = 9), moderately higher than reported in rat terminals (Smith et al., 2004). CaSR−/− mice usually died between 1 and 4 weeks of age precluding synaptosome recording. However, they were indistinguishable by physical examination at P1–P2 when cultures were prepared. To study the impact of CaSR on synaptic transmission, paired recordings were made from adjacent neurons after >14 d in culture (Fig. 1d,e). We injected depolarizing currents (1–3 ms, 100–500 pA) to evoke action potentials in the presynaptic neuron and isolated EPSCs in the postsynaptic neuron with the GABAA-antagonist bicuculline methiodide (20 μm) (Fig. 1e). Because the NSCC currents are inversely proportional to [Ca]o, we predicted that a lower affinity for Ca in CaSR−/− neuronal pairs would increase the NSCC current at basal [Ca]o and potentially modulate synaptic transmission. Consistent with our hypothesis, median average EPSC amplitude was almost twice as large in the CaSR−/− cultures compared with wild-type neurons (128 vs 68 pA; n = 28 and 36; p = 0.037), as illustrated in exemplar traces (Fig. 1f). The mean values were larger because of the positively skewed distribution of EPSC amplitude (Fig. 1g) (205 ± 43 pA vs 138 ± 47 pA, CaSR−/− and CaSR+/+ respectively). These data show that the monosynaptic EPSC amplitude was increased in neurons expressing the reduced Ca affinity mutant CaSR. Furthermore the data indicate that CaSR was not fully activated at basal [Ca] and [Mg] in CaSR−/− neurons.
CaSR acts presynaptically
Mean EPSC amplitude varied widely for the 64 neuronal pairs examined (range: 13 pA to 1.73 nA) reflecting variation in release probability (p), number of transmitter release sites (N), or quantal size (q). We used variance–mean (V–M) analysis between neuronal pairs, coupled with study of spontaneous transmitter release, to determine whether changes in p, N, or q were responsible for the variation in EPSC amplitude for each genotype (Clements and Silver, 2000). Changes in p, N, or q alter the variance-mean relationship differently: the V–M relationship is parabolic for increases in p, V is proportional to M for increases in N, and V is proportional to M squared for increases in q (Foster and Regehr, 2004). The pooled V–M relationship was linear for lower EPSC values for both CaSR+/+ and CaSR−/− neuronal pairs (Fig. 2a,b) (r2 = 0.95 and 0.98, respectively) consistent with the EPSC increase arising from increases in N. At higher EPSC values for both genotypes the slope increased (Fig. 2a,b). This trend was opposite to the parabolic relationship expected if increases in p mediate the increases in mean EPSC, and was more consistent with that expected by increases in q. To measure q, we voltage-clamped cortical neurons at −70 mV and recorded miniature EPSCs (mEPSCs) in the presence of the Na channel blocker tetrodotoxin (TTX; 0.5 μm) and bicuculline (10 μm). Amplitudes of mEPSCs varied between 8 and 49 pA, which was insufficient to explain the >100-fold variation in average EPSC amplitude. Furthermore, we found no difference between CaSR+/+ and CaSR−/− mEPSC amplitudes (23 ± 4 pA, n = 10, and 22 ± 4 pA, n = 8, respectively; p = 0.69), confirming that increased quantal size did not underlie the increase in average EPSC amplitude in CaSR−/− neurons. The V–M relationship is linear when increases in N underlie changes in V and M (Foster and Regehr, 2004) as shown in the following equation:
Using the slopes of the best fit lines (Fig. 2a,b), mEPSC amplitudes for q, and Equation 2, we calculated pooled average release probability to be 0.27 for the CaSR+/+ pairs and 0.46 for the CaSR−/− pairs. Thus the majority of the CaSR genotype-dependent difference in EPSC amplitude may be attributed to an increase in release probability in CaSR−/− cultures.
Figure 2.
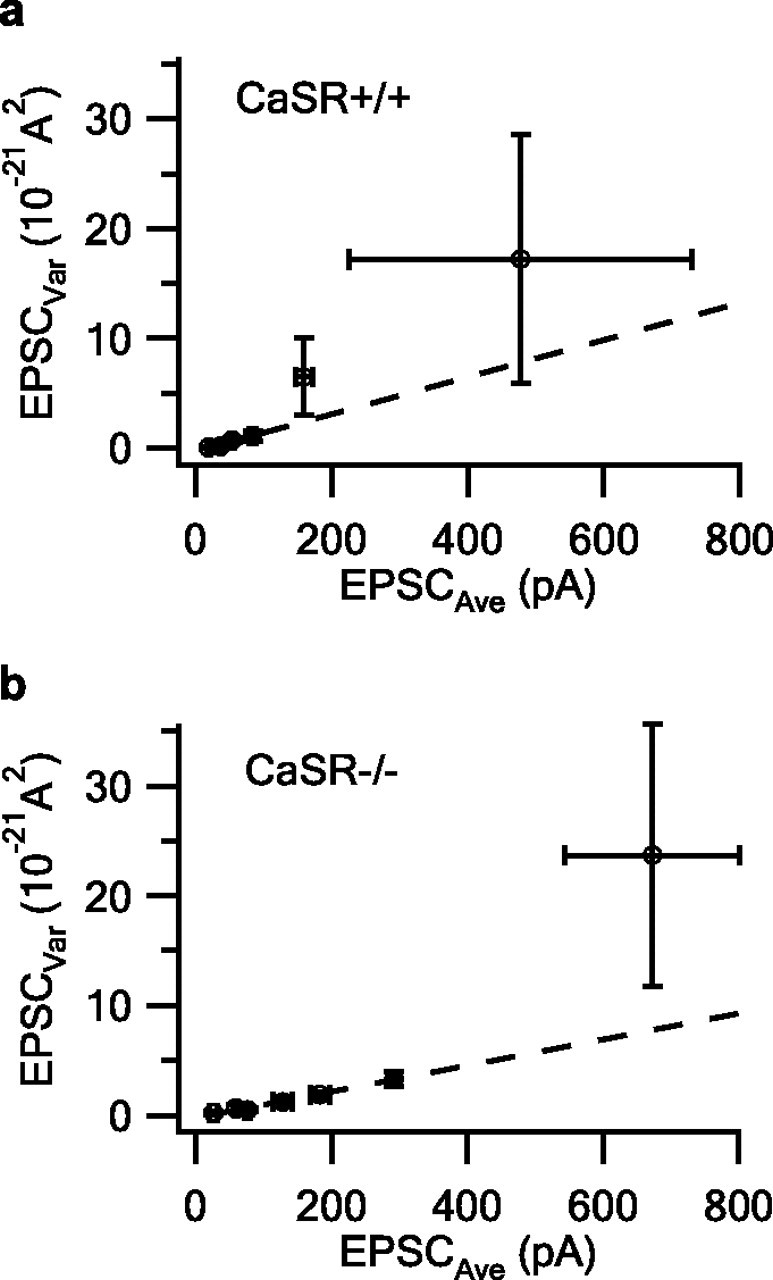
Pooled V–M for CaSR+/+ and −/− neuronal pairs. a, b, Plot of pooled average EPSC versus pooled average variance for CaSR+/+ (n = 36) and CaSR−/− (n = 28) neuronal pairs. Data are plotted as pooled mean ± SEM and is linear over lower range and described by lines with slopes of 16.9 pA and 11.9 pA for the CaSR+/+ and CaSR−/− neuronal pairs respectively.
Resting membrane potential (RMP) was measured in the cortical neurons and was relatively hyperpolarized in the CaSR−/− neurons (−78 ± 0.4 mV vs −80 ± 0.5 mV; p < 0.001; n = 269 and 205, respectively). NSCC activity in the CaSR−/− neuron was predicted to be increased at basal [Ca]o because of the lower affinity for Ca (Oda et al., 2000). In contrast to the observed hyperpolarization of RMP, increased NSCC current would depolarize the membrane potential. This dichotomy suggests a different mechanism for RMP regulation. Perhaps the genotype-dependent difference in membrane potential reflects a compensatory response comparable to the synaptic scaling observed after chronic antagonism of neurotransmitters (Turrigiano, 1999).
CaSR activation attenuates synaptic transmission
Alteration of external [Ca] or [Mg] will alter Ca entry through voltage-activated Ca channels (VACC) and impact exocytosis, complicating interpretation of CaSR modulation of synaptic transmission. We therefore tested whether mouse neocortical terminals were sensitive to spermidine, a CaSR agonist that blocks the activation of NSCC in rat terminals (Smith et al., 2004) but is less effective against VACC (Chen et al., 2007). In cell-attached recordings from mouse neocortical terminals, spermidine reversibly inhibited the outward [Ca]o-modulated NSCC currents activated by depolarizing steps (Fig. 3a,b). The block of the voltage-activated NSCC by external Ca (0.06, 0.6 and 6 mm; solid line) was dose-dependent. Spermidine application (10, 30, 100 and 300 μm; broken line) progressively blocked the NSCC current recorded with 60 μm external [Ca], achieved steady state within ∼10 s, and was rapidly reversible after removal from the bath (Fig. 3b). Spermidine had an IC50 of 155 ± 5 μm in the presence of 60 μm Ca (n = 10). We examined the effect of spermidine on EPSC amplitude at −70 mV in pairs of adjacent neurons (Fig. 3c,inset). The presynaptic neuron was current clamped and an action potential was triggered with a brief current injection every 10 s (Fig. 3c). Baseline current was adjusted before each cycle to set the RMP to −70 mV. EPSC amplitudes were highly variable within each recording (Fig. 3d,e; broken line denotes mean). The addition of spermidine reversibly decreased mean EPSC amplitude to 81 ± 4% of control (p = 0.002, n = 8) (Fig. 3e). Of these recordings, one neuronal pair did not respond to spermidine but the others showed an average decrease in EPSC amplitude of 22%. This spermidine-mediated inhibition indicates that the CaSR is not maximally activated by basal Ca and Mg levels and that the pathway may therefore be poised to facilitate synaptic transmission in the event of decreased cleft [Ca].
Figure 3.
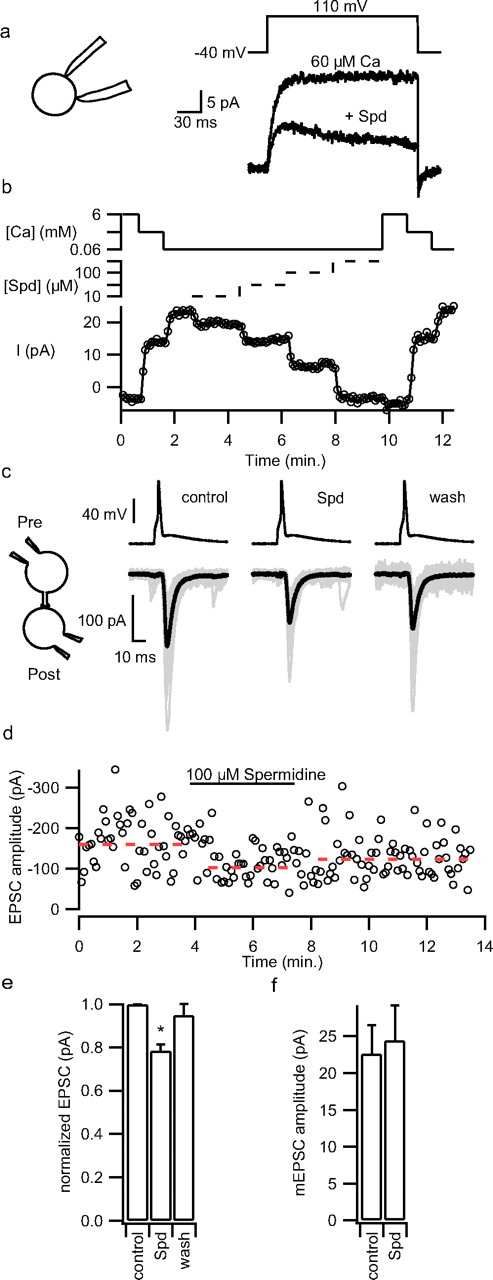
Spermidine reduces NSCC currents in synaptosomes and EPSC amplitude in cortical cultures. a, Outward currents activated with 60 μm bath Ca and 150 mV depolarizing step in a cell-attached synaptosome recording were decreased by bath application of spermidine (100 μm). Inset shows cell-attached configuration. b, Diary plot of NSCC amplitude versus time in same recording as a. Currents activated by 150 mV depolarization every 5 s were decreased by increases in bath [Ca] (60 μm–6 mm; upper unbroken trace) and bath spermidine concentration (10–300 μm; indicated by middle broken trace) reaching new steady states in ∼10 s. The Ca and spermidine axes are logarithmic and the absence of the broken trace indicates spermidine concentration is zero. c, Exemplar presynaptic action potentials and EPSCs before, during, and after spermidine application (100 μm) to paired recording from wild-type neurons. Average EPSCs in bold overlay the individual EPSCs (gray) recorded at steady state. d, Diary plot of EPSC amplitudes evoked by injection of depolarizing current into presynaptic neuron every 10 s. Broken line shows average EPSC values recorded during each phase of experiment. e, Histogram of mean effect of spermidine on EPSC amplitude (n = 8). f, Histogram of the effect of spermidine (100 μm) on mEPSC amplitude. *p < 0.05.
To check whether polyamines were acting presynaptically or postsynaptically we tested whether spermidine affected mEPSC size. The mEPSC amplitudes recorded in the presence of TTX (1–2 μm) were unaffected by spermidine, confirming a presynaptic site of action (Fig. 3f).
Because spermidine also inhibits N-type VACC currents by reducing channel permeation and shielding surface charge (Chen et al., 2007), we examined its potency as a general VACC antagonist. VACC currents were isolated in mouse superior cervical ganglion neurons which, like nerve terminals, contain N-, P/Q-, and R-type VACCs (Namkung et al., 1998). VACC currents activated by ramp depolarizations from −70 to 70 mV (1 mV/ms) in bathing solution with physiological divalent concentrations (1.1 mm [Ca] and [Mg]) were unaffected by spermidine at concentrations ≤300 μm (Fig. 4a,b). At higher concentrations spermidine inhibited VACC currents in a reversible, dose-dependent manner at rates faster than was resolved with the stimulation duty cycle (Fig. 4b). The average spermidine dose–response curve shows the peak inward VACC currents normalized against the currents recorded in the absence of the antagonists (n = 3) (Fig. 4c). The IC50 for spermidine was 9.5 ± 0.9 mm (n = 3). Application of Cd2+ (100 μm) blocked the vast majority of the inward current (95 ± 1%) and confirmed that the current was carried by VACCs (Fig. 4a,b). These results demonstrate that the decrease in EPSC amplitude observed in the presence of 100 μm spermidine cannot be accounted for by inhibition of VACCs, and thus is likely caused by activation of the CaSR–NSCC pathway.
CaSR activation enhances paired-pulse depression
Repetitive activity decreases cleft [Ca] and subsequent synaptic transmission (Rusakov and Fine, 2003). Subsequent EPSCs (P2) may thus be smaller than a primary EPSC (P1) in part because of the decrease in Ca electrochemical gradient. However, our data showing that [Ca]o decreases NSCC activity in nerve terminals, coupled with the above results from CaSR −/− neurons, strongly suggest that reduced CaSR signaling facilitates synaptic transmission. We tested whether CaSR activation was different for P1 and P2 by applying spermidine (100 μm), which should have its greatest effect when CaSR activation by cleft Ca is at its minimum. Paired stimulation of the presynaptic neuron (100 ms interstimulus interval) resulted in modest paired pulse depression (PPD) (Fig. 5a,b) (P2/P1 = 0.95 ± 0.04; n = 10). Application of spermidine resulted in enhanced PPD in 9 of 10 recordings, and average normalized PPR was reduced to 0.89 ± 0.04 (n = 10, p = 0.029). This is consistent with the CaSR–NSCC pathway playing a role in the facilitation of transmission during short-term plasticity.
Figure 5.

Spermidine enhances paired pulse depression. a, Average EPSC evoked after presynaptic action potential triggering with a 100 ms interstimulus interval and duty cycle of 10 s before, during, and after spermidine bath application (100 μm). Spermidine reduced P1 by ∼20% but reduced P2 by more, and the effect was reversible. b, The histogram of P2/P1, normalized for the PPR before spermidine application, was reduced by bath spermidine (n = 10).
Impact of CaSR activation in neuronal networks
Frequently, excitation of a presynaptic neuron during a paired recording resulted in a brief burst of multiple action potentials (Fig. 6a), which could not be reduced in number by shortening the current injection. In contrast, VACC channel inhibition (100 μm Cd2+) blocked all but the first spike, consistent with one or more additional neurons being stimulated by the presynaptic neuron and also exciting the postsynaptic neuron (data not shown). Under these conditions, EPSCs were elicited in the postsynaptic neuron with each action potential (Fig. 6a, left panel). As above, activation of CaSR reduced synaptic transmission; spermidine (100 μm) reversibly reduced the number of action potentials and EPSCs elicited by the current injection (Fig. 6a). Thus, the impact of CaSR activation was enhanced in neuronal networks.
Figure 6.
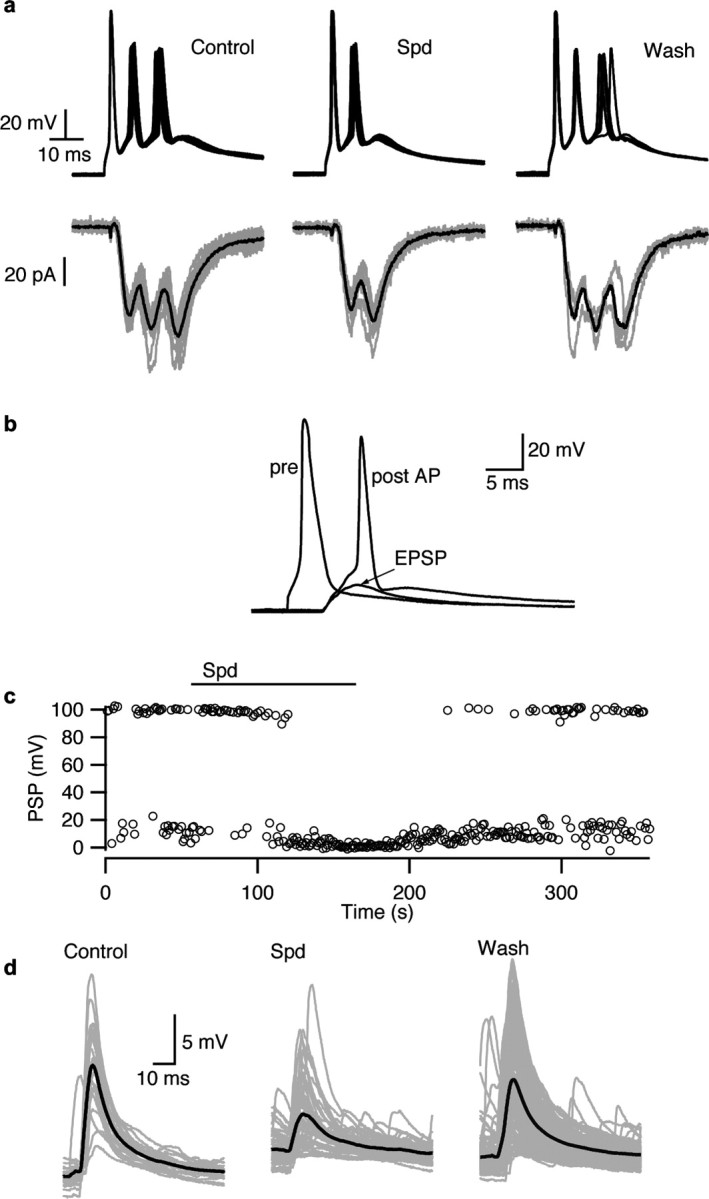
CaSR activation decreases synaptic transmission in polysynaptic networks and in current clamped neurons. a, Single brief stimulation of the presynaptic neuron every 10 s resulted in three action potentials and three EPSCs in the postsynaptic neuron under control conditions in these superimposed traces. The multiple firing presumably reflected synaptic feedback onto the neuron as it was prevented by VACC block. EPSC and action potential number were reversibly reduced by bath application of spermidine. b, Stimulation of the presynaptic neuron at 1 Hz resulted in a presynaptic action potential and EPSP or action potential. c, Spermidine (100 μm) reversibly decreased the probability of postsynaptic action potential firing from ∼0.5 to 0. d, Spermidine also reduced the amplitude of subthreshold EPSPs as shown in these superimposed traces (mean trace in bold).
The effects of the CaSR–NSCC pathway may be more evident when postsynaptic voltage is not clamped. In current-clamp experiments spermidine (100 μm) inhibited synaptic transmission between pairs of neurons. In one recording (Fig. 6b,c) the EPSP was close to action potential threshold and a postsynaptic action potential was elicited with 51% of presynaptic stimuli. Spermidine application reversibly blocked action potential initiation (Fig. 6c) because of the reduction in EPSP (Fig. 6d). This decrease in spike initiation and reduction in spike number in polysynaptic circuits point to an important role for CaSR signaling in modulating synaptic transmission.
Discussion
Decreases in brain [Ca]o resulting from excitatory synaptic transmission may inhibit subsequent transmission because of the exquisite sensitivity of transmitter release to [Ca]o (Dodge and Rahamimoff, 1967; Bollmann et al., 2000). Here, we demonstrate that a [Ca]o sensor, CaSR, modulates presynaptic NSCC activity and transmitter release in cortical neurons, and so may function to attenuate the impact of falls in [Ca]o after synaptic transmission. Based on our data we propose a model in which CaSR senses cleft [Ca] and signals to the NSCC (Fig. 7). A reduction in cleft [Ca] increases NSCC current that could influence action potential shape and thereby enhance neurotransmitter release.
Figure 7.

Diagram of proposed model for CaSR modulation of neurotransmitter release. a, Model depicts nerve terminal membrane containing CaSR (yellow oval) which is activated by extracellular Ca (red). A proportion of the NSCC are closed (gray) by the CaSR mediated signal (yellow circle). At physiological [Ca]o, CaSR is not fully activated and some active NSCC (yellow) facilitate vesicle fusion and transmitter release. b, At reduced [Ca]o, CaSR is no longer activated (gray) and in the absence of this signal NSCC may be activated (yellow) by depolarization. Because reduced [Ca]o will also decrease Ca entry through VACC and the probability of exocytosis, the increase in NSCC activity may partially compensate by facilitating vesicle fusion. c, At physiological [Ca]o reduced affinity CaSR−/− (gray hexagon) is less likely to be activated than CaSR+/+. Consequently, there is reduced signaling to NSCC, which remains active increasing the average probability of release. d, Application of spermidine (Spd; purple circles) and Ca to wild-type neurons fully activates CaSR reducing NSCC activity and transmitter release. Unlike Ca, spermidine does not enter the terminal and trigger exocytosis directly. Active molecules are colored yellow and inactive molecules are colored gray. The second messenger mediating signaling between NSCC and CaSR has not been identified and is denoted by a small intracellular circle.
CaSR function at the synapse
The steep dependence of release probability on [Ca]o (Dodge and Rahamimoff, 1967) lead to the hypothesis that even modest falls in cleft [Ca] will significantly impair synaptic transmission. Yet fast, sustained, synchronous synaptic transmission has been observed at rates of up to 800 Hz (Taschenberger and von Gersdorff, 2000), indicating that either falls in cleft [Ca] do not occur at all synapses or there are compensatory mechanisms to reduce the effect of decreases in cleft [Ca]. CaSR, a G-protein-coupled receptor, is activated by [Ca]o and triggers homeostatic pathways that defend serum Ca (Brown et al., 1993). CaSR is localized at central synapses (Ruat et al., 1995), where it detects falls in [Ca]o and indirectly activates a voltage-dependent NSCC (Smith et al., 2004; Chen et al., 2006). Our finding that CaSR−/− cultures had larger amplitude EPSCs at physiological [Ca] and [Mg] (Fig. 1) demonstrates that CaSR is positioned to compensate for decreases in [Ca]o. Furthermore our finding that the polyamine spermidine reduced EPSC size (Fig. 3) is important support for the proposal that CaSR is not saturated at physiological divalent concentrations and can respond to changes in [Ca]o under basal conditions. This effect was more pronounced in current-clamp recordings and when paired recordings included feedback pathways (Fig. 6). Spermidine did not alter mEPSC size, confirming a presynaptic site of action for this CaSR agonist.
As CaSR−/− mutants have reduced affinity for Ca (Oda et al., 2000), we propose NSCC currents are increased at basal [Ca] and [Mg], leading to increased action potential duration and prolongation of Ca entry through VACC (Fig. 7). The corresponding enhancement in synaptic transmission would counter the impact of the decreased Ca electrochemical gradient. Broadening of the action potential because of coincident activation of a NSCC is a well-recognized mechanism (Fatt and Katz, 1951). Furthermore, increased action potential width has been recorded during repetitive activity at central terminals (Borst and Sakmann, 1999b; Geiger and Jonas, 2000). As Ca entry occurs during the repolarization phase of the action potential, spike broadening is an effective method of increasing Ca entry and facilitating transmitter release.
Spermine and spermidine are other CaSR agonists that may be physiologically important (Masuko et al., 2003). CaSR is a potentially important site of action for endogenously released spermine because the ∼3 mm concentration estimated in synaptic vesicles (Masuko et al., 2003) is well above the concentration required to impact synaptic transmission (Fig. 3a) (DiScenna et al., 1994; Ferchmin et al., 1995).
CaSR and release probability
Increases in EPSC size may result from increases in N, p, or q, but an increase in spike broadening would solely manifest as an increase in p. The mEPSC amplitude was unchanged in our mutant cultures; this argues against an increase in quantal size underlying the larger EPSC. Our estimate of q from spontaneous mEPSC analysis is reasonable given the close matching of quantal size from V–M and miniature analysis (Meyer et al., 2001; Bailey et al., 2006). Moreover, V–M plots of the pooled data were linear over the lower part of the range consistent with the majority of the variation in M and V resulting from increases in release site number within each genotype. The average release probability for the pooled neuronal populations was 0.27 for the CaSR+/+ pairs and 0.46 for the CaSR−/− pairs (Fig. 2a,b). Given no genotype-dependent change in q, this 70% increase in p almost entirely accounts for the observed 88% increase in average EPSC size in mutant neurons. The observation that the V–M relationship becomes steeper at higher values (Fig. 2a,b) indicates that at higher EPSC amplitudes an increase in q may be contributing to the increasing variance, although the mechanism for this is unclear. Alternatively, the very high, nonlinear EPSC data may reflect a small group of neurons with different release properties. Our estimate of p in cortical neurons is close to values (0.34–0.51) reported for other cortical synapses (Lawrence et al., 2004).
Using the median EPSC amplitudes and the following equation:
we calculated that the average number of release sites were very similar for CaSR+/+ and CaSR−/− neuronal pairs (11 and 13 respectively). This contrasts with the reduced neurite length and branching observed in neurons after deletion or inhibition of CaSR (Vizard et al., 2008). Further studies are required to determine whether there is any relationship between CaSR regulation of synaptic transmission and neurite length.
The modest hyperpolarization in the CaSR−/− membrane potential was unexpected. Reduced NSCC activity could have hyperpolarized RMP but NSCC activity was expected to be increased in the CaSR−/− mutants, pointing to another cause. Manipulation of neuronal activity produces compensatory changes in quantal size and frequency (Turrigiano et al., 1998; Desai et al., 2002). It is possible that the change in membrane potential was a compensatory mechanism that countered the impact of doubling of EPSC size in the CaSR−/− mutants.
Together, the data support our hypothesis that decreased CaSR activation at nerve terminals increases release probability through its activation of the NSCC.
Ca in the cleft
Reductions in cleft [Ca]o are predicted to accompany brain activity resulting from presynaptic and postsynaptic Ca sinks, slow rates of replenishment of external Ca, small cleft volume, and relatively large intracellular volumes (Rusakov et al., 1998; Egelman and Montague, 1999; Kullmann et al., 1999). Data from Ca-sensitive electrodes, VACC currents and extracellular fluorescence probes show bulk and cleft [Ca]o is reduced by synaptic activity (Nicholson et al., 1978; Borst and Sakmann, 1999a; Rusakov and Fine, 2003). Moreover, this reduction can impair subsequent synaptic transmission (Rusakov and Fine, 2003). The majority of mouse nerve terminals (90%; n = 39) were sensitive to decreases in [Ca]o with a IC50 of 719 μm [higher than the 265 μm reported in rat terminals (Smith et al., 2004)] positioning them to respond to modest decreases in [Ca]o. At steady [Ca]o the NSCC currents are activated with a latency <1 ms after depolarization and may be activated by action potential waveforms (Smith et al., 2004), indicating one mechanism by which the CaSR–NSCC pathway could modulate synaptic transmission. But, can the CaSR respond to dynamic changes in cleft [Ca]? Our paired-pulse experiments indicate that spermidine accentuates paired-pulse depression. One interpretation of this is that decreases in [Ca]o were initiated by the first EPSC (Rusakov and Fine, 2003), reducing CaSR activation and enhancing the size of the second EPSC. Spermidine application should increase CaSR activation, blunting the compensation and increasing PPD. Contrary to this interpretation, decreases in [Ca]o affect NSCC in isolated terminals with a delay of ∼1 s (Smith et al., 2004) indicating this pathway may not respond rapidly enough to signal [Ca]o changes occurring in the 100 ms between EPSC1 and EPSC2. However, the response time of CaSR–NSCC signaling detected using the synaptosome-attached recording configuration may be underestimated if there are delays in penetration of the synaptic cleft by the applied solution (Smith et al., 2004, their Fig. 2I) or if high pipette [Ca] affects CaSR within the patch and attenuates the response to changes in bath solution. Further experiments are required to examine the dynamics of CaSR signaling and whether CaSR desensitization is physiologically important in the brain (Awumey et al., 2007). Downstream targets, other than NSCC, could also be affecting synaptic transmission by impacting vesicle pool size, intracellular [Ca] handling, VACC inactivation and glutamate receptor desensitization.
Metabotropic glutamate receptors (mGluR), and the γ-aminobutyric acid B (GABAB) receptor (Brown et al., 1993; Kubo et al., 1998; Saunders et al., 1998; Wise et al., 1999) have also been shown to be modulated by [Ca]o. Recently, postsynaptic Group 1 mGluRs were shown to detect falls in [Ca]o and reduce postsynaptic AMPA receptor sensitivity (Hardingham et al., 2006). This pathway may operate in parallel with CaSR.
CaSR and disease
CaSR mutations cause diseases characterized by abnormal serum calcium levels (Brown and MacLeod, 2001; Gunn and Gaffney, 2004). In addition, CaSR mutations may be associated with seizures (Pearce et al., 1996; Kapoor et al., 2008). The profound decreases in [Ca]o that accompany many types of acute brain injury point to CaSR as a potential therapeutic target (Li et al., 1995; Nilsson et al., 1996). Our results show CaSR agonists reduce release of glutamate and so may protect against excitotoxicity.
In conclusion, we have shown that presynaptic CaSR senses changes in [Ca]o at the nerve terminal and modulates synaptic transmission. A decrease in [Ca]o activates a NSCC which can facilitate transmitter release and thus may provide a retrograde compensatory signal to the nerve terminal. This may be an important homeostatic pathway to sustain transmission when [Ca]o decreases during times of high activity.
Footnotes
This work was supported by Oregon Health & Science University–Howard Hughes Medical Institute Biomedical Support Program, National Institute of Neurological Disorders and Stroke (R01-NS 43444), and Oregon Health & Science University President's Fund. We thank Dr. Jeremy Bergsman for providing analysis software, Dr. Paul Mermelstein for advice with cell culture, Dr. Xiaohua Wang and Mr Nicholas Vyleta for comments on this manuscript, and Drs. J.G. Seidman and D. Connor for providing the CaSR mutant mice.
References
- Awumey EM, Howlett AC, Putney JW, Jr., Diz DI, Bukoski RD. Ca2+ mobilization through dorsal root ganglion Ca2+-sensing receptor stably expressed in HEK293 cells. Am J Physiol Cell Physiol. 2007;292:C1895–1905. doi: 10.1152/ajpcell.00404.2006. [DOI] [PubMed] [Google Scholar]
- Bailey TW, Jin YH, Doyle MW, Smith SM, Andresen MC. Vasopressin inhibits glutamate release via two distinct modes in the brainstem. J Neurosci. 2006;26:6131–6142. doi: 10.1523/JNEUROSCI.5176-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsman JB, Tsien RW. Syntaxin modulation of calcium channels in cortical synaptosomes as revealed by botulinum toxin C1. J Neurosci. 2000;20:4368–4378. doi: 10.1523/JNEUROSCI.20-12-04368.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollmann JH, Sakmann B, Borst JG. Calcium sensitivity of glutamate release in a calyx-type terminal. Science. 2000;289:953–957. doi: 10.1126/science.289.5481.953. [DOI] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Depletion of calcium in the synaptic cleft of a calyx-type synapse in the rat brainstem. J Physiol. 1999a;521:123–133. doi: 10.1111/j.1469-7793.1999.00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Effect of changes in action potential shape on calcium currents and transmitter release in a calyx-type synapse of the rat auditory brainstem. Philos Trans R Soc Lond B Biol Sci. 1999b;354:347–355. doi: 10.1098/rstb.1999.0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev. 2001;81:239–297. doi: 10.1152/physrev.2001.81.1.239. [DOI] [PubMed] [Google Scholar]
- Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC. Cloning and characterization of an extracellular Ca (2+)-sensing receptor from bovine parathyroid. Nature. 1993;366:575–580. doi: 10.1038/366575a0. [DOI] [PubMed] [Google Scholar]
- Chen W, Bergsman JB, Anderson E, Dauban P, Dodd RH, Ruat M, Smith SM. Decreases in extracellular Ca2+ activate a non-selective cation channel via the Ca2+ receptor in rat acutely-isolated neocortical nerve terminals. Soc Neurosci Abstr. 2006;32:235–20. [Google Scholar]
- Chen W, Harnett MT, Smith SM. Modulation of neuronal voltage-activated calcium and sodium channels by polyamines and pH. Channels. 2007;1:281–290. doi: 10.4161/chan.4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Silver RA. Unveiling synaptic plasticity: a new graphical and analytical approach. Trends Neurosci. 2000;23:105–113. doi: 10.1016/s0166-2236(99)01520-9. [DOI] [PubMed] [Google Scholar]
- Desai NS, Cudmore RH, Nelson SB, Turrigiano GG. Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci. 2002;5:783–789. doi: 10.1038/nn878. [DOI] [PubMed] [Google Scholar]
- DiScenna PG, Ferchmin PA, Eterovic VA, Teyler TJ. Spermine depresses NMDA, K/AMPA and GABAA-mediated synaptic transmission in the rat hippocampal slice preparation. Brain Res. 1994;647:353–356. doi: 10.1016/0006-8993(94)91335-8. [DOI] [PubMed] [Google Scholar]
- Dodge FA, Jr, Rahamimoff R. Co-operative action of calcium ions in transmitter release at the neuromuscular junction. J Physiol. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelman DM, Montague PR. Computational properties of peri-dendritic calcium fluctuations. J Neurosci. 1998;18:8580–8589. doi: 10.1523/JNEUROSCI.18-21-08580.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelman DM, Montague PR. Calcium dynamics in the extracellular space of mammalian neural tissue. Biophys J. 1999;76:1856–1867. doi: 10.1016/s0006-3495(99)77345-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatt P, Katz B. An analysis of the end-plate potential recorded with an intracellular electrode. J Physiol. 1951;115:320–370. doi: 10.1113/jphysiol.1951.sp004675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferchmin PA, Eterović VA, Rivera EM, Teyler TJ. Spermine increases paired-pulse facilitation in area CA1 of hippocampus in a calcium-dependent manner. Brain Res. 1995;689:189–196. doi: 10.1016/0006-8993(95)00568-b. [DOI] [PubMed] [Google Scholar]
- Foster KA, Regehr WG. Variance-mean analysis in the presence of a rapid antagonist indicates vesicle depletion underlies depression at the climbing fiber synapse. Neuron. 2004;43:119–131. doi: 10.1016/j.neuron.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Jonas P. Dynamic control of presynaptic Ca (2+) inflow by fast-inactivating K (+) channels in hippocampal mossy fiber boutons. Neuron. 2000;28:927–939. doi: 10.1016/s0896-6273(00)00164-1. [DOI] [PubMed] [Google Scholar]
- Gunn IR, Gaffney D. Clinical and laboratory features of calcium-sensing receptor disorders: a systematic review. Ann Clin Biochem. 2004;41:441–458. doi: 10.1258/0004563042466802. [DOI] [PubMed] [Google Scholar]
- Hardingham NR, Bannister NJ, Read JCA, Fox KD, Hardingham GE, Jack JJ. Extracellular Calcium Regulates Postsynaptic Efficacy through Group 1 Metabotropic Glutamate Receptors. J Neurosci. 2006;26:6337–6345. doi: 10.1523/JNEUROSCI.5128-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger R, Heinemann C, Neher E, Matthews G. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature. 1994;371:513–515. doi: 10.1038/371513a0. [DOI] [PubMed] [Google Scholar]
- Ho C, Conner DA, Pollak MR, Ladd DJ, Kifor O, Warren HB, Brown EM, Seidman JG, Seidman CE. A mouse model of human familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism [see comments] Nat Genet. 1995;11:389–394. doi: 10.1038/ng1295-389. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Satishchandra P, Ratnapriya R, Reddy R, Kadandale J, Shankar SK, Anand A. An idiopathic epilepsy syndrome linked to 3q13.3-q21 and missense mutations in the extracellular calcium sensing receptor gene. Ann Neurol. 2008;64:158–167. doi: 10.1002/ana.21428. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Miyashita T, Murata Y. Structural basis for a Ca2+-sensing function of the metabotropic glutamate receptors. Science. 1998;279:1722–1725. doi: 10.1126/science.279.5357.1722. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Min MY, Asztely F, Rusakov DA. Extracellular glutamate diffusion determines the occupancy of glutamate receptors at CA1 synapses in the hippocampus. Philos Trans R Soc Lond B Biol Sci. 1999;354:395–402. doi: 10.1098/rstb.1999.0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JJ, Grinspan ZM, McBain CJ. Quantal transmission at mossy fibre targets in the CA3 region of the rat hippocampus. J Physiol (Lond) 2004;554:175–193. doi: 10.1113/jphysiol.2003.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li PA, Kristián T, Katsura K, Shamloo M, Siesjö BK. The influence of insulin-induced hypoglycemia on the calcium transients accompanying reversible forebrain ischemia in the rat. Exp Brain Res. 1995;105:363–369. doi: 10.1007/BF00233036. [DOI] [PubMed] [Google Scholar]
- Masuko T, Kusama-Eguchi K, Sakata K, Kusama T, Chaki S, Okuyama S, Williams K, Kashiwagi K, Igarashi K. Polyamine transport, accumulation, and release in brain. J Neurochem. 2003;84:610–617. doi: 10.1046/j.1471-4159.2003.01558.x. [DOI] [PubMed] [Google Scholar]
- Mermelstein PG, Bito H, Deisseroth K, Tsien RW. Critical dependence of cAMP response element-binding protein phosphorylation on L-type calcium channels supports a selective response to EPSPs in preference to action potentials. J Neurosci. 2000;20:266–273. doi: 10.1523/JNEUROSCI.20-01-00266.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AC, Neher E, Schneggenburger R. Estimation of quantal size and number of functional active zones at the calyx of Held synapse by nonstationary EPSC variance analysis. J Neurosci. 2001;21:7889–7900. doi: 10.1523/JNEUROSCI.21-20-07889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung Y, Smith SM, Lee SB, Skrypnyk NV, Kim HL, Chin H, Scheller RH, Tsien RW, Shin HS. Targeted disruption of the Ca2+ channel beta3 subunit reduces N- and L- type Ca2+ channel activity and alters the voltage-dependent activation of P/Q-type Ca2+ channels in neurons. Proc Natl Acad Sci U S A. 1998;95:12010–12015. doi: 10.1073/pnas.95.20.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, ten Bruggencate G, Stöckle H, Steinberg R. Calcium and potassium changes in extracellular microenvironment of cat cerebellar cortex. J Neurophysiol. 1978;41:1026–1039. doi: 10.1152/jn.1978.41.4.1026. [DOI] [PubMed] [Google Scholar]
- Nilsson P, Laursen H, Hillered L, Hansen AJ. Calcium movements in traumatic brain injury: the role of glutamate receptor-operated ion channels. J Cereb Blood Flow Metab. 1996;16:262–270. doi: 10.1097/00004647-199603000-00011. [DOI] [PubMed] [Google Scholar]
- Oda Y, Tu CL, Chang W, Crumrine D, Kömüves L, Mauro T, Elias PM, Bikle DD. The calcium sensing receptor and its alternatively spliced form in murine epidermal differentiation. J Biol Chem. 2000;275:1183–1190. doi: 10.1074/jbc.275.2.1183. [DOI] [PubMed] [Google Scholar]
- Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, Lewis-Barned N, McCredie D, Powell H, Kendall-Taylor P, Brown EM, Thakker RV. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor [see comments] N Engl J Med. 1996;335:1115–1122. doi: 10.1056/NEJM199610103351505. [DOI] [PubMed] [Google Scholar]
- Rogers KV, Dunn CK, Hebert SC, Brown EM. Localization of calcium receptor mRNA in the adult rat central nervous system by in situ hybridization. Brain Res. 1997;744:47–56. doi: 10.1016/s0006-8993(96)01070-0. [DOI] [PubMed] [Google Scholar]
- Ruat M, Molliver ME, Snowman AM, Snyder SH. Calcium sensing receptor: molecular cloning in rat and localization to nerve terminals. Proc Natl Acad Sci U S A. 1995;92:3161–3165. doi: 10.1073/pnas.92.8.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov DA. The role of perisynaptic glial sheaths in glutamate spillover and extracellular Ca (2+) depletion. Biophys J. 2001;81:1947–1959. doi: 10.1016/S0006-3495(01)75846-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov DA, Fine A. Extracellular Ca2+ depletion contributes to fast activity-dependent modulation of synaptic transmission in the brain. Neuron. 2003;37:287–297. doi: 10.1016/s0896-6273(03)00025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov DA, Harrison E, Stewart MG. Synapses in hippocampus occupy only 1–2% of cell membranes and are spaced less than half-micron apart: a quantitative ultrastructural analysis with discussion of physiological implications. Neuropharmacology. 1998;37:513–521. doi: 10.1016/s0028-3908(98)00023-9. [DOI] [PubMed] [Google Scholar]
- Saunders R, Nahorski SR, Challiss RA. A modulatory effect of extracellular Ca2+ on type 1alpha metabotropic glutamate receptor-mediated signalling. Neuropharmacology. 1998;37:273–276. doi: 10.1016/s0028-3908(98)00027-6. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse [see comments] Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- Smith SJ. Do astrocytes process neural information? Prog Brain Res. 1992;94:119–136. doi: 10.1016/s0079-6123(08)61744-6. [DOI] [PubMed] [Google Scholar]
- Smith SM, Bergsman JB, Harata NC, Scheller RH, Tsien RW. Recordings from single neocortical nerve terminals reveal a nonselective cation channel activated by decreases in extracellular calcium. Neuron. 2004;41:243–256. doi: 10.1016/s0896-6273(03)00837-7. [DOI] [PubMed] [Google Scholar]
- Stanley EF. Presynaptic calcium channels and the depletion of synaptic cleft calcium ions. J Neurophysiol. 2000;83:477–482. doi: 10.1152/jn.2000.83.1.477. [DOI] [PubMed] [Google Scholar]
- Taschenberger H, von Gersdorff H. Fine-tuning an auditory synapse for speed and fidelity: developmental changes in presynaptic waveform, EPSC kinetics, and synaptic plasticity. J Neurosci. 2000;20:9162–9173. doi: 10.1523/JNEUROSCI.20-24-09162.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci. 1999;22:221–227. doi: 10.1016/s0166-2236(98)01341-1. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Vassilev PM, Mitchel J, Vassilev M, Kanazirska M, Brown EM. Assessment of frequency-dependent alterations in the level of extracellular Ca2+ in the synaptic cleft. Biophys J. 1997;72:2103–2116. doi: 10.1016/S0006-3495(97)78853-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizard TN, O'Keeffe GW, Gutierrez H, Kos CH, Riccardi D, Davies AM. Regulation of axonal and dendritic growth by the extracellular calcium-sensing receptor. Nat Neurosci. 2008;11:285–291. doi: 10.1038/nn2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise A, Green A, Main MJ, Wilson R, Fraser N, Marshall FH. Calcium sensing properties of the GABA (B) receptor. Neuropharmacology. 1999;38:1647–1656. doi: 10.1016/s0028-3908(99)00119-7. [DOI] [PubMed] [Google Scholar]