

Abstract
Background
Cardiovascular disease is the leading cause of death for both men and women in the United States and the world. There is a profound pattern in the time of day at which the death occurs; it is in the morning, when the endothelium is most vulnerable and blood pressure surges, that stroke and heart attack most frequently happen. Though the molecular components of circadian rhythms rhythmically oscillate in blood vessels, evidence of a direct function for the ‘circadian clock’ in the progression to vascular disease is lacking.
Methods and Results
In the current study, we have found increased pathological remodeling and vascular injury in mice with aberrant circadian rhythms, Bmal1 (Bmal1-KO) and Clock (Clockmut). In addition, naïve aortae from Bmal1-KO and Clock mutant mice exhibit endothelial dysfunction. Akt and subsequent nitric oxide signalling—a pathway critical to vascular function—was significantly attenuated in arteries from Bmal1-KO mice.
Conclusions
Our data reveals a new role for the circadian clock during chronic vascular responses which may be of significance in the progression of vascular disease.
Keywords: circadian rhythm, endothelium, remodeling, thrombus, vasculature
The cardiovascular system behaves rhythmically over the course of a day,1-3 coordinating tissue perfusion in accordance to oscillating metabolic and functional demands. These oscillations, which occur in blood vessels as variations in contractility and blood pressure, follow a distinctive temporal pattern—a circadian rhythm. Aberrations to circadian rhythm meet with pathological consequences. Shift work provokes a 40% increase in the risk of cardiovascular disease4 while disturbance of daily blood pressure rhythms elevates the incidence of vascular disease.5, 6 In addition, the onset of acute vascular events such as myocardial infarction7 and stroke8 also exhibit circadian variation. However, direct evidence to implicate the molecular components of circadian rhythms in the chronic progression of disease is lacking.
The molecular components that generate circadian rhythms—the circadian/biological clock—constitute a unique collaboration of genes and proteins that govern by virtue of transcriptional, translational, and post-translational mechanisms. The transcriptional driving force is comprised of the basic helix-loop-helix transcription factors, Bmal1 and Clock (or Npas2). Bmal1/Clock or Bmal1/Npas2 heterodimerize to transactivate Period (Per) and Cryptochrome (Cry), the braking force of the loop, which then restrain Bmal1/Clock/Npas2, and consequently, their own transcription. Further modification of the core complex, including phosphorylation and degradation, refines timing to the daily cycle.9 Indeed vascular cells10, 11 contain all necessary components of this unique molecular metronome. Though recent data has implicated the circadian clock in aspects of acute vascular function,12-14 there is no data directly implicating Bmal1 or Clock in the chronic process of vascular disease and even less-so is known regarding the downstream mechanisms involved.
Methods
Animals
All animal studies were performed according to protocols approved by the Medical College of Georgia Institutional Committee for Use and Care of Laboratory Animals. Bmal1-KO mice and were housed under standard 12 hour light/dark conditions (LD). Clockmut mice were either held in LD or held in constant dark conditions (DD) for 6 weeks (2 weeks acclimation, 4 weeks experimentation) as indicated. Bmal1-KO and littermate wild type mice were formerly produced by gene targeting in 129Sv/J embryonic stem cells15 which we backcrossed 3 times to C57BL/6J. Clock heterozygote mutant mice which served as parental strains for mutants and control wild-types in respective studies were purchased from Jackson laboratories (C57BL/6J-Clockm1Jt/J, stock number 002923) formerly generated by ENU mutagenesis in C57BL/6J mice causing an A to T transversion at the third base position of the 5′ splice donor site of intron 19 of Clock.16
Materials
P-eNOS (1177), PDK1 and Akt1 polyclonal antibodies were purchased from Cell Signaling, GAPDH monoclonal antibody from Ambion, P-Akt (Threonine-308), eNOS), and PAI-1 antibodies from BD Transduction Labs, PAR4 peptide from Advanced ChemTech and PPACK was purchased from Calbiochem.
Functional Studies in Isolated Aortic Arteries
7-10 week old (young) Bmal1-KO mice or age-matched littermate wild-type controls were anesthetized with ketamine/xylazine and subsequently exsanguinated. Residual blood was removed by perfusing physiologic saline by cardiac puncture. The thoracic aorta was carefully dissected free and excised from the aortic arch to the point of the diaphragm. To avoid damage to the endothelium, perivascular fat carefully dissected and the aorta was cut into rings (2 mm thickness) for placement into organ chambers containing Krebs buffer maintained under physiologic conditions. The composition of KHS (in mmol/L) was: NaCl 118.3, KCl 4.7, CaCl2 2.5, MgSO4·7H2O 1.2, KH2PO4 1.2, NaHCO3 25, dextrose 5.6, and equilibrated with 95%O2-5%CO2 to maintain pH of 7.4 at 37 °C. The rings were suspended by two tungsten wires (25 μm diameter) and mounted in a vessel myograph system (6 ml chamber size, Multi Myograph, Danish Myo Technology). Isometric tension was measured using a force transducer coupled to data acquisition system. A resting tension of 1.0 g was used throughout the experiments. After an equilibration period of 60 minutes (during which time KHS was changed every 10 minutes and the resting tension was re-adjusted), rings were precontracted with phenylephrine until a plateau was reached. Vessels were then washed with KHS and this was repeated at least three times in order to stabilize the tissue. Aortae were then precontracted with phenylephrine and concentration-dependent responses to the endothelium-dependent agonist, acetylcholine (Ach; 1 × 10-9-5 ×10-5 M) and the endothelium-independent NO-donor, sodium nitroprusside (SNP; 1 × 10-9-5 ×10-5 M.
Flow Dependent Vascular Remodeling and Femoral Artery Injury
The complete left common carotid (LC) artery ligation was performed as previously described.17 Briefly, the distal left common carotid artery (LC) artery and its bifurcation into the external and internal carotid were exposed using blunt dissection. 8-0 nylon sutures (USSC Sutures) were used to ligate the LC artery, just proximal to the external/internal carotid artery bifurcation. For intravascular wire injury, the left femoral artery of mice was repeatedly cannulated (5-7 times) by a straight wire (0.38 mm in diameter, No. C-SF-15-15, Cook, Inc.) as previously described.18 Left common carotid artery ligation and femoral artery injury were performed at times of day as indicated. Incisions were closed (5-0 suture) and mice were left to recover for 5 weeks or 1 week after which mice were euthanized at the same time of day at which the procedure was initiated.
Histomorphometry
After 5 weeks of flow reduction induced by LC ligation, mice were anesthetized, exsanguinated, and perfused via the left ventricle with physiologic saline. In processing vascular tissues for western blotting, common carotid arteries or aortae were immediately dissected, flash frozen and stored at -80° C until further processing. In animal studies designed for histological/morphometric analysis of common carotid arteries, after saline infusion, mice were subsequently perfusion fixed with neutral buffered formalin. Both right and left common carotid arteries were carefully excised and post-fixed either overnight for morphometric studies or immediately embedded in frozen medium for cryotome processing. Morphometric analysis of carotid arteries was performed using video microscopy as described. Perimeter (p) of the vessel lumen was taken as the circumference (C) of a circle and lumen diameter (D) determined from the equation D=C/p, assuming that the vessel cross sections were circular in vivo. In order to determine thrombus area, the internal elastic lamina (IEL) and patent lumen were circumscribed to derive a radius (R) value from the formula R=2C/p, and then IEL area and luminal area (A) were calculated using the formula A=ΠR2. Thrombus area was derived from the difference of IEL area and lumen area.
Whole blood analysis of Platelet Activation
Blood was collected via cardiac puncture following carbon dioxide asphyxiation. Approximately 500-600 μl blood was drawn through a 27 gauge needle into a 1 ml plastic syringe with 70 μl 3.8% sodium citrate, 10 μM PPACK. Blood was immediately diluted 6-fold with a modified Tyrode's buffer (137 mM NaCl, 2.8 mM KCl, 1 mM MgCl2, 12 mM NaHCO3, 0.4 mM Na2HPO4, 5.5 mM glucose, 1 mM EDTA, 0.35% BSA, 10 mM Hepes, pH 7.4). P-selectin expression was monitored by flow cytometry (FACSCalibur, Becton Dickinson). To assess platelet activation, blood was stimulated with PAR4 and subsequently incubated with biotinylated rat anti-mouse CD62P antibody or an isotype-matched control antibody followed by phycoerythrin-conjugated streptavidin (immunoreagents from BD Pharmingen, San Diego, CA) to detect P-selectin by flow cytometry. Fluorescein isothiocyanate-conjugated rat anti-mouse CD41 antibody (BD Pharmingen, San Diego, CA) was used as a platelet identifier.
Statistical Analysis
The significance of differences were assessed by two-way analysis of variance (ANOVA) analysis to test for all sources of variation (genotype versus age or genotype versus light cycle conditions), and subsequent tests were made to assess between paired RC and LC lumen diameter from individual animals by paired Student's t-tests. Platelet responses and immunoblot densitometry were analyzed for significance by unpaired Student's t-tests. Error bars show the calculated standard error of the mean. Concentration response curves of vascular function were analyzed by repeated measures two-way and one-way analysis of variance (ANOVA) with a Bonferonni correction. Differences were considered significant when p<0.05.
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
Pathological vascular remodeling and enhanced vascular injury in mice with a dysfunctional circadian clock
Chronic blood flow alterations that mimic the human condition of vascular disease can be induced experimentally in vivo, by surgical ligation of the arterial circulation in animals. These ‘flow reduction models’ simulate the structural adaptation that occurs in obstructed arteries (vascular remodeling) during atherosclerosis and hypertension. We conducted studies to determine if disruption of the biological clock alters the response of blood vessels to a chronic reduction in blood flow. In young (6-10 weeks old) litter mate control wild-type mice (WT), reduction of blood flow in the left common carotid artery (LC) for a duration of five weeks induced wall thickening (Figure 1A) and inward remodeling (Figure 1B, C) of the LC as previously shown.19 In contrast, wall thickness of the remodelled LC of young Bmal1-KO mice was further increased (Figure 1A) while lumen was impaired in its ability to narrow (inward remodel) (Figure 1B, C), a response reminiscent of that in endothelial nitric oxide synthase (eNOS) knockout mice.20 The impairment in vascular remodeling in Bmal1-KO mice was striking; lumen diameters of left common carotid arteries (LC) in wild-type mice narrowed by 34±3.5% relative to right contralaterals (RC), while Bmal1-KO mice were only able to adapt by a 5.2±11% reduction. The aberrant remodeling in arteries of Bmal1-KO was accompanied by a substantial increase in collagen deposition in the medial layer as evidenced through trichrome staining (Figure 1B). Studies in aged Bmal1-KO revealed even more severe abnormalities. 25-30 week old Bmal1-KO mice exhibited a significant susceptibility to thrombosis (Figures. 1E, F) in the ligated vessel versus the non-ligated contralateral vessel and this response was absent in both the RC and ligated LC of wild-type mice. In addition, the thrombosis was accompanied by significant tissue remodeling around the site of thrombus formation (Figure 1E). In arterial regions that were thrombus-free, aged Bmal1-KO mice exhibited a paradoxical enlargement of lumen diameter after ligation relative to age-matched littermate controls (Figure 1D) reminiscent of stenosis induced dilatation.
Figure 1. Impaired remodeling and thrombosis in Bmal1-KO mice.
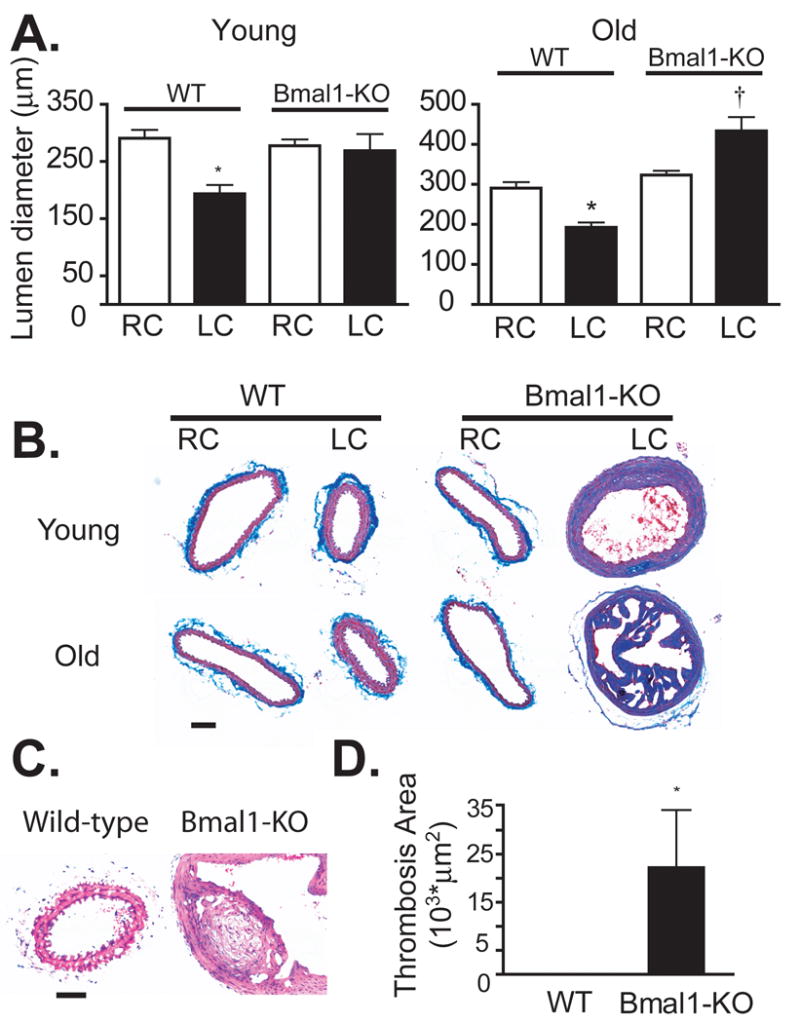
(A) Morphometric analysis of common carotid artery cross sections revealed typical vascular remodeling in male, wild-type (WT) mice after 5 weeks of arterial ligation, evident as wall thickening of the medial layer while thickening of the LC wall was exacerbated in age-matched Bmal1-KO mice (young). (B) Masson-trichrome staining showed a robust deposition of collagen staining (blue stain) in the media of Bmal1-KO mice relative to wild-type mice (bar=50 μm). (C) In contrast to a reduction in lumen diameter observed in WT mice, lumen diameter of male Bmal1-KO mice did not narrow (young, 7-10 weeks) or a even exhibited a paradoxical increase diameter (old, 25-30 weeks) (D) in regions that were free of thrombosis. (E) Hematoxylin and eosin staining of remodelled LC of old wild-type (left panel) reveals a normal, inward remodelled vessel with patent lumen, while Bmal1-KO (right panel) exhibits remodeling an enlarged lumen undergoing remodeling around the point of fibrosis (bar=50 μm). (N=9-10, *p<0.05, paired t-test RC versus LC; †p<0.05, two-sample t-test LC-WT versus LC-KO) (F) Thrombosis was increased in old Bmal1-KO mice (N=7-10/age group, (*p<0.05, WT versus KO).
To determine if the effects of Bmal1 on vascular remodeling were related to the circadian rhythm function of the endogenous clock, we extended our studies to mice harbouring a mutation of the Clock gene (Clockmut). The advantage of this approach is that Clock mutant mice exhibit only a modest phase shift in circadian rhythm under standard light-dark conditions (LD) and become completely arrhythmic when housed under conditions of constant darkness (DD).16 Thus, by modifying circadian cues we can directly assess the importance of these transcription factors to the relationship between biological rhythms and vascular function. In DD conditions, the LC of Clockmut mice undergoing common carotid artery ligation exhibited pathological vascular responses which were manifest as an increase in wall thickness (Figure 2A) and an inability to inward remodel (Figure 2B) relative to WT mice. However, in LD, the wall thickening and inward remodeling induced by arterial ligation was not different between WT and Clockmut mice, providing strong evidence for a direct link between light cycle and circadian rhythm-dependent changes in vascular function.
Figure 2. Clock mutant mice exhibit pathological vascular responses.
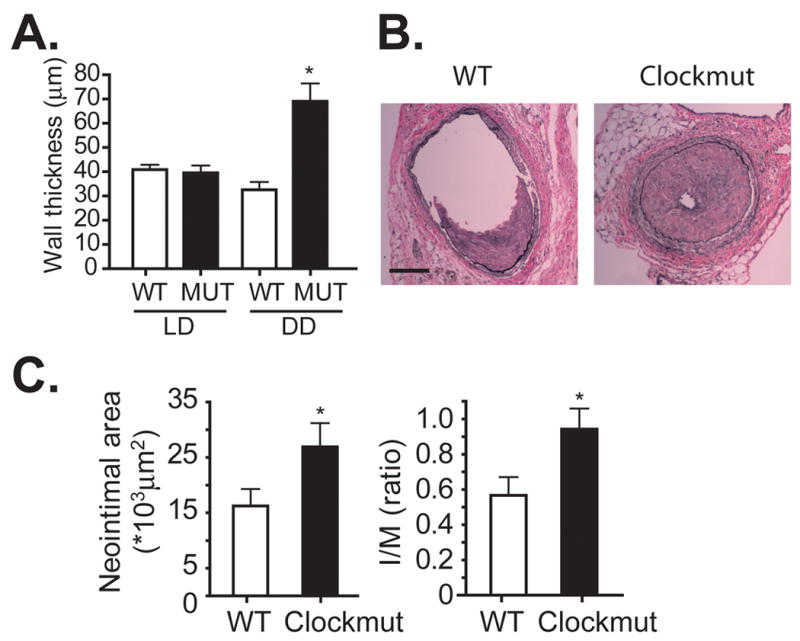
(A) After acclimation to either light/dark (LD) or constant dark conditions (DD), the left common carotid artery of wild-type (WT) and Clock mutant (MUT) mice (male, 15 to 20 weeks of age) was ligated for 5 weeks and remodelled LC thickness was measured, revealing a DD-dependent increase in wall thickness of LC in Clockmut mice (N=4, *p<0.05, Clockmut in DD versus WT in DD) and a DD-dependent impairment in inward remodeling (B). (C) Von Gieson staining of elastin fibers revealed enhanced intimal hyperplasia in the left femoral artery of male, Clockmut mice undergoing intraluminal wire injury reveals an enhanced injury relative to injured arteries of male, WT mice, that were housed in DD (bar=50 μm). (D). Morphometric analysis of femoral artery cross sections revealed an increase in neointimal area and intima to medial ratio indicative of an exacerbated response to wire injury. (N=7-10, *p<0.05).
We next sought to determine if the circadian clock might also influence other types of vascular injury such as intimal hyperplasia in response to intraluminal vascular injury. Five weeks after wire injury to the femoral artery, Clock mutant mice housed in DD exhibited a significant increase in arterial injury relative to wild-type mice, quantified as an increase in neointimal area and intima to medial ratio (Figure 2C, D). Though the extent of injury induced by arterial ligation (Supplemental Figure 1) and wire injury was not affected by gender (Supplemental Figure 2A, B) or time of the initial insult (Supplemental Figure 2C, 2D), wire injury in Bmal1-KO mice also induced a robust neointimal response relative to littermate wild-type control mice (Supplemental Figure 2B, 2D), providing further evidence for the importance of clock function in the chronic vascular response to injury.
Bmal1-KO mice exhibit intact coagulative responses
Thrombosis and vascular injury can occur due to a defect in the coagulation cascade localized to either the endothelium or blood platelets. To determine if the injury in Bmal1-KO mice was due to a defect in the coagulative properties of blood, we assessed the platelet response by FACS analysis in isolated whole blood of mice. Baseline levels of total platelets measured by the platelet marker CD41 were not different between wild-type and Bmal1-KO mice (Figure 3A). Stimulating platelets in isolated whole blood with the thrombin receptor agonist PAR-4 induced a robust and significant increase in platelet activation as measured by P-selectin (cd62) expression. However, the level of activation was not different between Bmal1-KO mice and wild-type mice (Figure 3B), indicating that an intrinsic defect in platelet activation could not account for the increased thrombosis apparent in Bmal1-KO mice.
Figure 3. Activation of platelets is normal in Bmal1-KO mice, but endothelial and liver PAI-1 is increased.
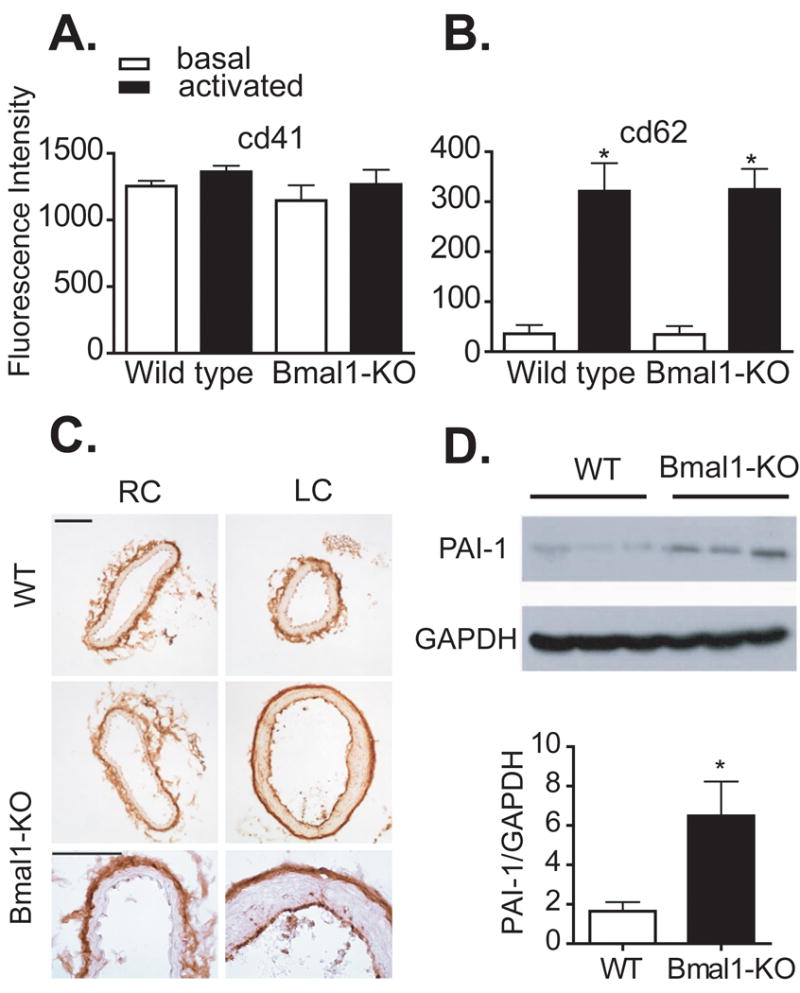
Platelet function was assessed in Bmal1-KO mice versus littermate control wild-type mice (male, 20-25 week old). Whole blood was isolated from mice and activated with PAR-4 to induce platelet aggregation, and analyzed for platelet markers by FACS analysis. There was no difference in total platelets (cd41) (A) or platelet activation (cd62) (B) in Bmal1-KO mice versus wild-type mice. (N=5, platelet aggregation studies *p<0.05, activated versus basal). (C) Protein expression of PAI-1 was increased in the endothelium (bar=30 μm) and livers (D) of 20-25 week old Bmal1-KO mice (liver immunoblot shows 3 wild type and 3 Bmal1-KO mice and right panel shows respective densitometry).
Endothelial dysfunction in Bmal1-KO mice and Clock mutant mice
Because the blood-borne response was intact, we next examined if a defect in endothelial function might contribute to the impairments in vascular remodeling and thrombosis observed in Bmal1-KO mice. PAI-1, a prothrombotic enzyme that prevents fibrinolysis, was upregulated in the endothelium of remodelled arteries (Figure 3C) and livers (Figure 3D) of Bmal1-KO mice. Indeed, endothelial dysfunction is known to promote pathological vascular remodeling and thrombosis. To directly assess endothelial function, we conducted organ bath studies using isolated aortic rings from Bmal1-KO mice. Aortae from Bmal1-KO mice, (Figure 4A, B) exhibited a severely impaired endothelium-dependent vasorelaxant response to acetylcholine relative to wild-type mice. Acetylcholine (5*10−6M) induced a robust 71±3.5% relaxation in wild type mice versus only 17±3.3% relaxation in Bmal1-KO mice. Endothelial dysfunction was also observed in aortae isolated from Clock mutant mice that were acclimated to conditions of constant darkness. Percent relaxation to Ach was only 56±11% in Clockmut mice versus 87±9.1 in WT mice also acclimated in DD. (Figure 4C). Importantly, the endothelial dysfunction in Clockmut mice was not apparent under light/dark conditions (Figure 4D) providing further evidence in support of a direct link between the biological clock and vascular integrity.
Figure 4. Endothelial dysfunction in mice with aberrant circadian rhythm.
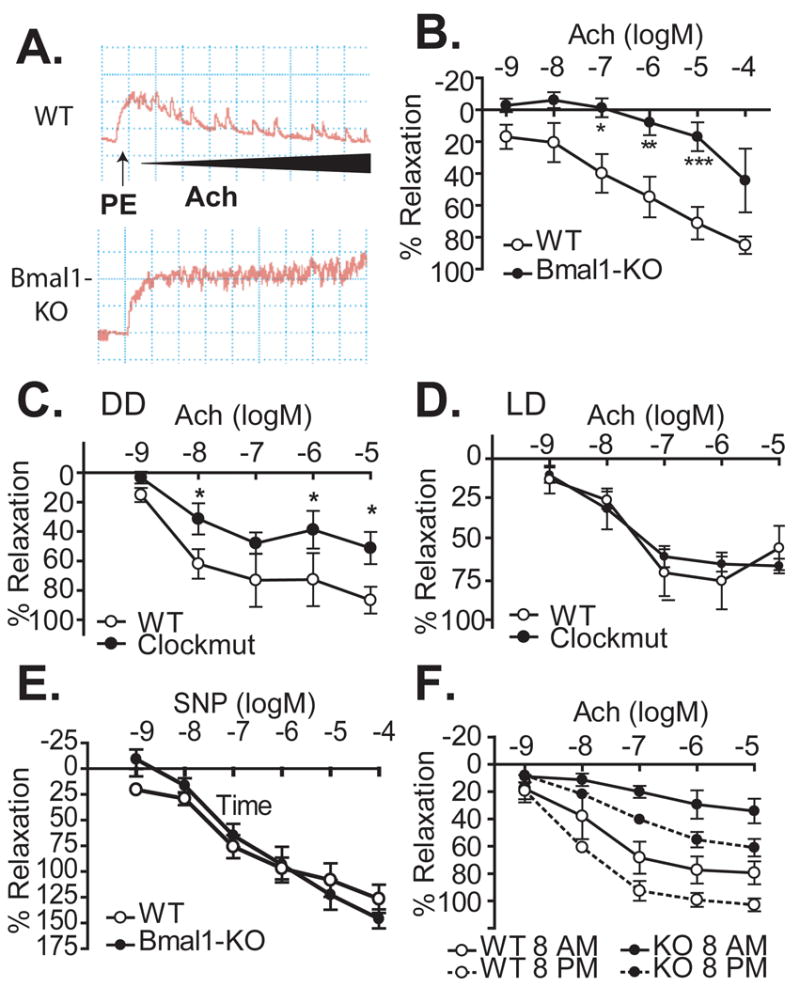
Isometric tension development was assessed in aortic rings prepared from Bmal1-KO mice (black squares) and litter-mate controls (white circles). After preconstriction with phenylephrine, concentration response to acetylcholine was measured. (A) Representative traces and summary data (B) of the acetylcholine response from wild-type and Bmal1-KO mice (10-15 week old, male) are shown. (C) Clock mutant mice were also assessed for their response to acetylcholine in dark-dark and light-dark conditions (D) (N=4 WT, N=6 Clockmut, *p<0.05 versus WT at corresponding concentration). (E) Relaxant responses to the endothelium-independent vasodilator SNP were not different between control and Bmal1-KO mice. (F) Ach response was assessed in WT and Bmal1-KO mice at 8 AM and 8 PM (N=5-7/group; *p<0.05, WT versus Bmal1-KO at respective time,†p<0.05 respective genotype at 8 AM versus 8PM)
Smooth muscle cell responses to nitric oxide as measured by relaxation to sodium nitroprusside (SNP) remained intact in Bmal1-KO mice (Figure 4E), providing further evidence of a direct impact of circadian rhythm on endothelial function. Finally, wild-type mice exhibited a variation in vascular function (measured by the response to Ach, Figure 4F), consistent with the temporal variation observed in prior studies.13 Interestingly, dysfunction in Bmal1-KO mice was further exacerbated at time points of reduced endothelial function in wild-type mice (Figure 4F).
Akt-eNOS signalling is blunted in remodelled arteries of Bmal1-KO mice
The serine-threonine kinase Akt1 protects cells from apoptosis,21 increases nitric oxide production via phosphorylation of eNOS,22, 23 and modulates vascular function.24 Previously, we assessed global expression patterns of circadian oscillating genes in the aorta and identified the serine/threonine kinase Akt1/PKB as a putative target of the circadian clock.11 Indeed, expression levels of Akt1 protein were significantly reduced in Bmal1-KO mice versus littermate control wild-type mice in left common carotid arteries undergoing 5 weeks of vascular remodeling (Figure 5A). In addition, the phosphorylated form of Akt at threonine 308, an index of Akt activation, was dramatically blunted in the remodelled arteries (Figure 5A, B). Because phosphorylation of Akt1 on threonine 308 is dependent on the upstream kinase, phosphoinositide dependent kinase 1 (PDK1),25, 26 we also assessed PDK1 expression. We found that protein expression of PDK1 was significantly decreased in Bmal1-KO mice relative to wild-type mice. In mice undergoing a shorter, 1-week duration ligation, we found that the contralateral control (unligated) RC of Bmal1-KO also exhibited substantially lower levels of P-Akt, Akt, and PDK (Figure 5C). Moreover, comparison of RC versus LC after 1 week of ligation revealed that P-Akt and t-Akt were robustly upregulated in remodelled LCs of WT mice, an effect that was still lower in Bmal1-KO mice, but upregulated nonetheless, which may reflect the impact of shear forces to regulate Akt.
Figure 5. Blunted Akt signaling in common carotid arteries of Bmal1 KO mice.
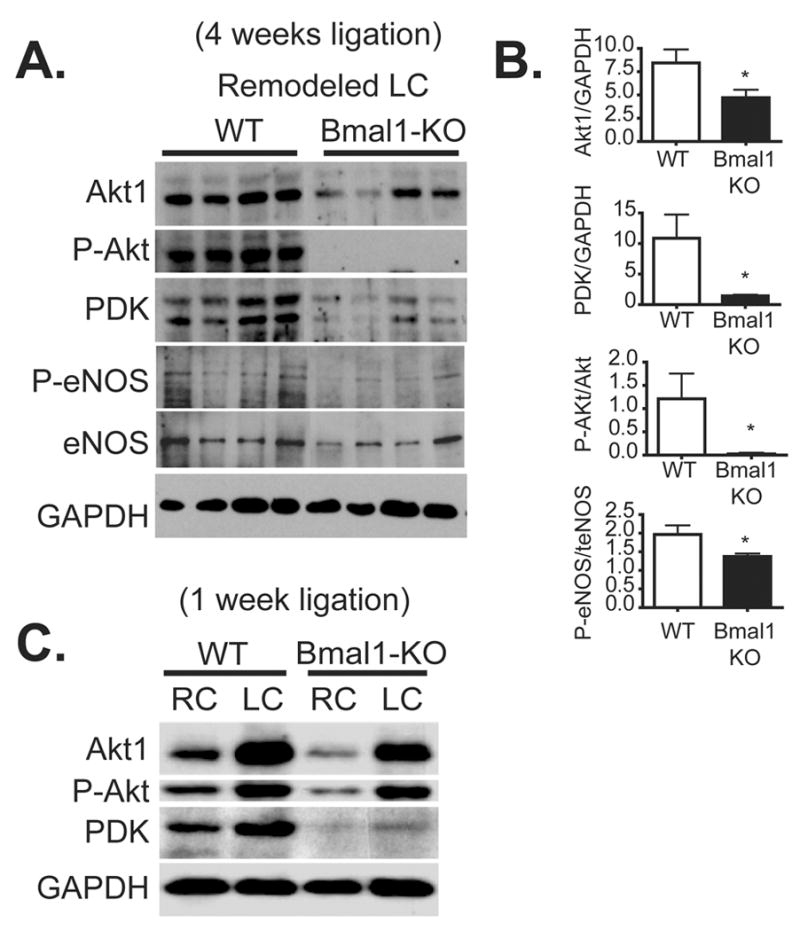
(A) After 5 weeks of complete carotid artery ligation, remodelled arteries (LC) were dissected and protein lysates were isolated and separated by SDS-PAGE electrophoresis from littermate control wild-type mice versus Bmal1-KO mice at a single time point (1 PM). Expression levels of total Akt, P-Akt1, and the phosphorylating kinase PDK were dramatically reduced in remodelled left common carotid arteries relative to wild-type mice. Remodelled arteries of Bmal1-KO mice also exhibited blunted phosphorylated levels of phosphorylated eNOS, though total eNOS on average did not change. (B) Changes were quantified by densitometry (*p<0.05 versus WT). (N=4 Wild-type, N=4 Bmal1-KO). (C) After 1 week of LC ligation, proteins were assessed in both RC and LC, revealing a attenuated Akt and P-Akt expression in the unligated RC of Bmal1-KO mice relative to WT mice.
As Akt exerts its regulatory role in vascular function, at least in part, by phosphorylating and subsequently activating eNOS, we further examined levels of eNOS in the remodelled arteries undergoing long-term, 4 week ligation.23 Indeed, expression levels of phosphorylated eNOS were significantly attenuated in Bmal1-KO mice relative to wild-type mice (Figure 5A, B), consistent with impaired endothelium-dependent vasomotion, pathological remodeling, and enhanced vascular injury.
Discussion
Seminal observations have led to the discovery of the circadian clock, and its importance to daily rhythms in central16, 27-30 and peripheral function.31-33 Central to activation of the circadian clock are the transcription factors Bmal1 and Clock. Bmal1 and Clock heterodimerize to bind E-box motifs within the promoter regions of downstream clock genes to induce the rhythmic changes in gene expression.34 On a functional level, disruption of Bmal127 or mutation of Clock16 in mice impairs circadian rhythms in daily activity. However, the biological significance of an aberrant endogenous clock in the long-term progression to vascular disease is unknown. Here, we demonstrate that Bmal1-KO and Clock mutant mice exhibit pathological responses to vascular injury which may in part stem from pronounced endothelial dysfunction in the vasculature. As Bmal1-KO mice age, the chronic response to arterial ligation became more severe, evident as a susceptibility to thrombosis, consistent with recent studies of photochemical injury in Bmal1-KO mice.35 Indeed, ageing itself is known to modify circadian clock function36. Other studies have also demonstrated that the ageing process is accelerated in Bmal1-KO mice, manifest as arthropathy15 and organ pathology37 by 35 to 40 weeks of age in contrast to wild-type mice of the same age.
Changes in other physiological and metabolic parameters in response to disruption of circadian rhythm do not appear to account for the changes in vascular remodeling observed in Bmal1-KO mice. Bmal1-KO mice did exhibit elevated levels of cholesterol and triglycerides relative to WT mice as previously demonstrated,38 however these differences were age-independent (Supplemental Table 1) and modest relative to mouse models of hypercholesterolemia. In ApoE-knockout mice, endothelial function is known to be preserved even in the face of striking 10 to 20 fold increases in cholesterol.39, 40 In addition, endothelial function is normal in atherosclerotic free regions of aorta, but impaired only at sites of plaque formation in cholesterol-fed apoE knockout mice.41 Thus, as it applies to our studies, it would be highly unlikely that the <1.5fold elevations in cholesterol in Bmal1-KO [which are atherosclerosis-free on a regular chow diet] would contribute to the endothelial dysfunction and impaired remodeling we observe.
In prior studies, we have shown that responses to insulin are improved in Bmal1-KO mice,38 which would not readily explain endothelial dysfunction, as insulin-resistance is intimately linked to compromised endothelial function. Finally, Bmal1-KO mice are not hypertensive, but actually hypotensive and lack the night-time spike in blood pressure.12
These findings are the first to demonstrate that the endogenous clock genes can orchestrate the progression to chronic vascular disease. In addition, the acute endothelial dysfunction we observed in Bmal1-KO and Clock mutant mice is also consistent with observations in mutant mice of another core clock component Per2.13 Further indication that the vascular effects of Bmal1 and Clock are dependent on overall clock function is that the development of vascular disease [as evidenced by wall thickening and impairment in inward remodeling] only occurred when Clockmut mice were acclimated to free running conditions (DD) versus normal rhythmicity (LD). Thus we have established a relationship between light-cycle and deterioration of vascular integrity which indicate that the vascular phenotype is not a mere consequence of Clock mutation or Bmal1 deficiency and secondary effects therein, but is dependent on the integrity of circadian rhythms.
Previously, we have demonstrated that eNOS knockout mice exhibit aberrant remodeling, a phenotype strikingly similar to that which we now demonstrate in Bmal1-KO mice. PDK and its downstream target Akt1, which plays a key regulatory step in controlling eNOS activity were attenuated in both naïve and remodelled common carotid arteries of Bmal1-KO mice. Moreover, 1-week of LC ligation revealed robust upregulation of P-Akt in remodeled LC of WT mice relative to RC of WT mice, similar to in vitro observations in sheared endothelial cells,42 hinting at a complex manner of Akt regulation involving both the circadian clock and shear stress. The reduction in Akt signalling and subsequent vascular pathology observed in Bmal1-KO mice is consistent with the established protective role for Akt and eNOS in the regulation of vascular function.24, 43, 44 In contrast to the blunting in Akt, PAI-1 which is prothrombotic, and antifibrinolytic was increased in Bmal1-KO mice. Indeed, there is ample evidence linking the regulation of PAI-1 to the molecular clock. PAI-1 oscillates with a circadian rhythm,45 and its expression is regulated by two clock components Bmal146 and Reverbα 47.48 Moreover, PAI-1 transgenic mice exhibit spontaneous age-dependent coronary thrombosis,49 a phenotype similar to the injury-induced thrombosis we observed in Bmal1-KO mice. Thus, biological oscillation driven by the molecular clock can influence events in an extended temporal process, which when perturbed, may precipitate a state of endothelial dysfunction, pathological vascular remodeling, and thrombosis that ultimately may forge a path to vascular disease.
Supplementary Material
Acknowledgments
We wish to thank Dr. Christopher A. Bradfield and Dr. Garret FitzGerald for graciously providing Bmal1 (MOP3) knockout mice. We also wish to thank Dr. Joseph Takahashi and Dr. Mario B. Marrero for insightful suggestions and Jina Kim, Monica Davis, James Mintz, and Pulkit Malik for their technical assistance.
Sources of Funding. This work was supported in part by grants from the National Institutes of Health (DK070658 and HL089576) and the American Heart Association (0665284B).
Footnotes
Disclosures. None.
Clinical Perspective
Vascular physiology exhibits a profound circadian rhythm, evident as 24-hour variations in blood pressure and vascular contractility. In addition, acute cardiovascular events also exhibit a distinct timing; heart attacks and strokes occur most frequently in the morning. However, evidence for the involvement of the transcription factors that generate circadian rhythms in the progression to vascular disease is lacking. This study demonstrates that the circadian transcription factors Bmal1 and Clock influence not only acute vascular responses, but also the long-term adaption of arteries as occurs during vascular remodeling and vascular injury. We found that mice with genetic disruption of the Bmal1 gene (Bmal1-KO) or mutation of the Clock gene (Clockmut) exhibited pathological vascular remodeling of the common carotid artery after vessel ligation and pronounced intimal hyperplasia of the femoral artery after intraluminal wire injury. Moreover, the extent of injury was conditioned by ageing, progressing to thrombosis in remodeled common carotid arteries of Bmal1-KO mice. This was further accompanied by an increase in the profibrinolytic and prothrombotic molecule plasminogen activation inhibitor (PAI-1), a known target of the circadian clock. Conversely, mechanisms protective to endothelial function were impaired, evident as attenuated vascular responses to acetylcholine and attenuation in protein expression of the Akt-eNOS pathway. These data provide direct evidence for the circadian clock in vascular remodeling and vascular injury that may ultimately be of significant importance in the progression to vascular disease.
References
- 1.Hossmann V, Fitzgerald GA, Dollery CT. Circadian rhythm of baroreflex reactivity and adrenergic vascular response. Cardiovasc Res. 1980;14:125–129. doi: 10.1093/cvr/14.3.125. [DOI] [PubMed] [Google Scholar]
- 2.Millar-Craig MW, Bishop CN, Raftery EB. Circadian variation of blood-pressure. Lancet. 1978;1:795–797. doi: 10.1016/s0140-6736(78)92998-7. [DOI] [PubMed] [Google Scholar]
- 3.Panza JA, Epstein SE, Quyyumi AA. Circadian variation in vascular tone and its relation to alpha-sympathetic vasoconstrictor activity. N Engl J Med. 1991;325:986–990. doi: 10.1056/NEJM199110033251402. [DOI] [PubMed] [Google Scholar]
- 4.Boggild H, Knutsson A. Shift work, risk factors and cardiovascular disease. Scand J Work Environ Health. 1999;25:85–99. doi: 10.5271/sjweh.410. [DOI] [PubMed] [Google Scholar]
- 5.Pierdomenico SD, Lapenna D, Guglielmi MD, Costantini F, Romano F, Schiavone C, Cuccurullo F, Mezzetti A. Arterial disease in dipper and nondipper hypertensive patients. Am J Hypertens. 1997;10:511–518. doi: 10.1016/s0895-7061(96)00493-1. [DOI] [PubMed] [Google Scholar]
- 6.Rizzoni D, Porteri E, Guelfi D, Muiesan ML, Valentini U, Cimino A, Girelli A, Rodella L, Bianchi R, Sleiman I, Rosei EA. Structural alterations in subcutaneous small arteries of normotensive and hypertensive patients with non-insulin-dependent diabetes mellitus. Circulation. 2001;103:1238–1244. doi: 10.1161/01.cir.103.9.1238. [DOI] [PubMed] [Google Scholar]
- 7.Muller JE, Ludmer PL, Willich SN, Tofler GH, Aylmer G, Klangos I, Stone PH. Circadian variation in the frequency of sudden cardiac death. Circulation. 1987;75:131–138. doi: 10.1161/01.cir.75.1.131. [DOI] [PubMed] [Google Scholar]
- 8.Elliott WJ. Circadian variation in the timing of stroke onset: a meta-analysis. Stroke. 1998;29:992–996. doi: 10.1161/01.str.29.5.992. [DOI] [PubMed] [Google Scholar]
- 9.Ko CH, Takahashi JS. Molecular components of the mammalian circadian clock. Hum Mol Genet. 2006;15(Spec No 2):R271–277. doi: 10.1093/hmg/ddl207. [DOI] [PubMed] [Google Scholar]
- 10.Davidson AJ, London B, Block GD, Menaker M. Cardiovascular tissues contain independent circadian clocks. Clin Exp Hypertens. 2005;27:307–311. [PubMed] [Google Scholar]
- 11.Rudic RD, McNamara P, Reilly D, Grosser T, Curtis AM, Price TS, Panda S, Hogenesch JB, FitzGerald GA. Bioinformatic analysis of circadian gene oscillation in mouse aorta. Circulation. 2005;112:2716–2724. doi: 10.1161/CIRCULATIONAHA.105.568626. [DOI] [PubMed] [Google Scholar]
- 12.Curtis AM, Cheng Y, Kapoor S, Reilly D, Price TS, Fitzgerald GA. Circadian variation of blood pressure and the vascular response to asynchronous stress. Proc Natl Acad Sci U S A. 2007;104:3450–3455. doi: 10.1073/pnas.0611680104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Viswambharan H, Carvas JM, Antic V, Marecic A, Jud C, Zaugg CE, Ming XF, Montani JP, Albrecht U, Yang Z. Mutation of the Circadian Clock Gene Per2 Alters Vascular Endothelial Function. Circulation. 2007 doi: 10.1161/CIRCULATIONAHA.106.653303. [DOI] [PubMed] [Google Scholar]
- 14.Westgate EJ, Cheng Y, Reilly DF, Price TS, Walisser JA, Bradfield CA, Fitzgerald GA. Genetic Components of the Circadian Clock Regulate Thrombogenesis In Vivo. Circulation. 2008 doi: 10.1161/CIRCULATIONAHA.107.739227. [DOI] [PubMed] [Google Scholar]
- 15.Bunger MK, Walisser JA, Sullivan R, Manley PA, Moran SM, Kalscheur VL, Colman RJ, Bradfield CA. Progressive arthropathy in mice with a targeted disruption of the Mop3/Bmal-1 locus. Genesis. 2005;41:122–132. doi: 10.1002/gene.20102. [DOI] [PubMed] [Google Scholar]
- 16.Vitaterna MH, King DP, Chang AM, Kornhauser JM, Lowrey PL, McDonald JD, Dove WF, Pinto LH, Turek FW, Takahashi JS. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science. 1994;264:719–725. doi: 10.1126/science.8171325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudic RD, Brinster D, Cheng Y, Fries S, Song WL, Austin S, Coffman TM, FitzGerald GA. COX-2-derived prostacyclin modulates vascular remodeling. Circ Res. 2005;96:1240–1247. doi: 10.1161/01.RES.0000170888.11669.28. [DOI] [PubMed] [Google Scholar]
- 18.Yu J, Rudic RD, Sessa WC. Nitric oxide-releasing aspirin decreases vascular injury by reducing inflammation and promoting apoptosis. Lab Invest. 2002;82:825–832. doi: 10.1097/01.lab.0000018828.61722.bd. [DOI] [PubMed] [Google Scholar]
- 19.Rudic RD, Bucci M, Fulton D, Segal SS, Sessa WC. Temporal events underlying arterial remodeling after chronic flow reduction in mice: correlation of structural changes with a deficit in basal nitric oxide synthesis. Circ Res. 2000;86:1160–1166. doi: 10.1161/01.res.86.11.1160. [DOI] [PubMed] [Google Scholar]
- 20.Rudic RD, Shesely EG, Maeda N, Smithies O, Segal SS, Sessa WC. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J Clin Invest. 1998;101:731–736. doi: 10.1172/JCI1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 22.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 23.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo Z, Fujio Y, Kureishi Y, Rudic RD, Daumerie G, Fulton D, Sessa WC, Walsh K. Acute modulation of endothelial Akt/PKB activity alters nitric oxide-dependent vasomotor activity in vivo. J Clin Invest. 2000;106:493–499. doi: 10.1172/JCI9419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 26.Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, Coadwell J, Hawkins PT. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279:710–714. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- 27.Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–1017. doi: 10.1016/s0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, Schibler U. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110:251–260. doi: 10.1016/s0092-8674(02)00825-5. [DOI] [PubMed] [Google Scholar]
- 29.van der Horst GT, Muijtjens M, Kobayashi K, Takano R, Kanno S, Takao M, de Wit J, Verkerk A, Eker AP, van Leenen D, Buijs R, Bootsma D, Hoeijmakers JH, Yasui A. Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature. 1999;398:627–630. doi: 10.1038/19323. [DOI] [PubMed] [Google Scholar]
- 30.Zheng B, Albrecht U, Kaasik K, Sage M, Lu W, Vaishnav S, Li Q, Sun ZS, Eichele G, Bradley A, Lee CC. Nonredundant roles of the mPer1 and mPer2 genes in the mammalian circadian clock. Cell. 2001;105:683–694. doi: 10.1016/s0092-8674(01)00380-4. [DOI] [PubMed] [Google Scholar]
- 31.Kornmann B, Schaad O, Bujard H, Takahashi JS, Schibler U. System-driven and oscillator-dependent circadian transcription in mice with a conditionally active liver clock. PLoS Biol. 2007;5:e34. doi: 10.1371/journal.pbio.0050034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagoshi E, Saini C, Bauer C, Laroche T, Naef F, Schibler U. Circadian gene expression in individual fibroblasts: cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell. 2004;119:693–705. doi: 10.1016/j.cell.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 33.Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, Menaker M, Takahashi JS. PERIOD2∷LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci U S A. 2004;101:5339–5346. doi: 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, Takahashi JS, Weitz CJ. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–1569. doi: 10.1126/science.280.5369.1564. [DOI] [PubMed] [Google Scholar]
- 35.Westgate EJ, Cheng Y, Reilly DF, Price TS, Walisser JA, Bradfield CA, FitzGerald GA. Genetic components of the circadian clock regulate thrombogenesis in vivo. Circulation. 2008;117:2087–2095. doi: 10.1161/CIRCULATIONAHA.107.739227. [DOI] [PubMed] [Google Scholar]
- 36.Van Reeth O, Zhang Y, Reddy A, Zee P, Turek FW. Aging alters the entraining effects of an activity-inducing stimulus on the circadian clock. Brain Res. 1993;607:286–292. doi: 10.1016/0006-8993(93)91518-w. [DOI] [PubMed] [Google Scholar]
- 37.Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006;20:1868–1873. doi: 10.1101/gad.1432206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rudic RD, McNamara P, Curtis AM, Boston RC, Panda S, Hogenesch JB, Fitzgerald GA. BMAL1 and CLOCK, Two Essential Components of the Circadian Clock, Are Involved in Glucose Homeostasis. PLoS Biol. 2004;2:e377. doi: 10.1371/journal.pbio.0020377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wikstrom L, Johansson C, Salto C, Barlow C, Campos Barros A, Baas F, Forrest D, Thoren P, Vennstrom B. Abnormal heart rate and body temperature in mice lacking thyroid hormone receptor alpha 1. Embo J. 1998;17:455–461. doi: 10.1093/emboj/17.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Villeneuve N, Fortuno A, Sauvage M, Fournier N, Breugnot C, Jacquemin C, Petit C, Gosgnach W, Carpentier N, Vanhoutte P, Vilaine JP. Persistence of the nitric oxide pathway in the aorta of hypercholesterolemic apolipoprotein-E-deficient mice. J Vasc Res. 2003;40:87–96. doi: 10.1159/000070705. [DOI] [PubMed] [Google Scholar]
- 41.Bonthu S, Heistad DD, Chappell DA, Lamping KG, Faraci FM. Atherosclerosis, vascular remodeling, and impairment of endothelium-dependent relaxation in genetically altered hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 1997;17:2333–2340. doi: 10.1161/01.atv.17.11.2333. [DOI] [PubMed] [Google Scholar]
- 42.Go YM, Boo YC, Park H, Maland MC, Patel R, Pritchard KA, Jr, Fujio Y, Walsh K, Darley-Usmar V, Jo H. Protein kinase B/Akt activates c-Jun NH(2)-terminal kinase by increasing NO production in response to shear stress. J Appl Physiol. 2001;91:1574–1581. doi: 10.1152/jappl.2001.91.4.1574. [DOI] [PubMed] [Google Scholar]
- 43.Ackah E, Yu J, Zoellner S, Iwakiri Y, Skurk C, Shibata R, Ouchi N, Easton RM, Galasso G, Birnbaum MJ, Walsh K, Sessa WC. Akt1/protein kinase Balpha is critical for ischemic and VEGF-mediated angiogenesis. J Clin Invest. 2005;115:2119–2127. doi: 10.1172/JCI24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phung TL, Ziv K, Dabydeen D, Eyiah-Mensah G, Riveros M, Perruzzi C, Sun J, Monahan-Earley RA, Shiojima I, Nagy JA, Lin MI, Walsh K, Dvorak AM, Briscoe DM, Neeman M, Sessa WC, Dvorak HF, Benjamin LE. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell. 2006;10:159–170. doi: 10.1016/j.ccr.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andreotti F, Kluft C. Circadian variation of fibrinolytic activity in blood. Chronobiol Int. 1991;8:336–351. doi: 10.3109/07420529109059170. [DOI] [PubMed] [Google Scholar]
- 46.Schoenhard JA, Smith LH, Painter CA, Eren M, Johnson CH, Vaughan DE. Regulation of the PAI-1 promoter by circadian clock components: differential activation by BMAL1 and BMAL2. J Mol Cell Cardiol. 2003;35:473–481. doi: 10.1016/s0022-2828(03)00051-8. [DOI] [PubMed] [Google Scholar]
- 47.Wang J, Yin L, Lazar MA. The orphan nuclear receptor Rev-erb alpha regulates circadian expression of plasminogen activator inhibitor type 1. J Biol Chem. 2006;281:33842–33848. doi: 10.1074/jbc.M607873200. [DOI] [PubMed] [Google Scholar]
- 48.Reidy MA, Irvin C, Lindner V. Migration of arterial wall cells. Expression of plasminogen activators and inhibitors in injured rat arteries. Circ Res. 1996;78:405–414. doi: 10.1161/01.res.78.3.405. [DOI] [PubMed] [Google Scholar]
- 49.Eren M, Painter CA, Atkinson JB, Declerck PJ, Vaughan DE. Age-dependent spontaneous coronary arterial thrombosis in transgenic mice that express a stable form of human plasminogen activator inhibitor-1. Circulation. 2002;106:491–496. doi: 10.1161/01.cir.0000023186.60090.fb. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.