

Abstract
Investigations detailed herein demonstrate the ability of chiral bidentate N-heterocyclic carbenes to promote directly – without the need for a Cu salt – site- and enantioselective C–C bond forming reactions. Within this context, catalytic allylic alkylations of various allylic phosphates with dialkylzinc and trialkylaluminum reagents, performed with chiral bidentate imidazolinium salts and in the absence of a Cu salt, are described. The Cu-free transformations deliver products bearing tertiary or (all-carbon) quaternary stereogenic centers with exceptional site- (>98:<2 SN2′:SN2) and high enantioselectivity [up to 97.5:2.5 enantiomer ratio (er)]. A chiral Zn-based N-heterocyclic carbene (NHC) complex, which serves as the catalyst for the enantioselective allylic alkylation reactions, is isolated and fully characterized. Spectroscopic and X-ray crystallographic studies indicate that the sulfonate group within the stereogenic-at-Zn bidentate complexes coordinates syn to the proximal phenyl substituent of the NHC backbone (vs the initially expected anti). Investigations regarding a related chiral NHC–Al based complex reveal similar structural attributes. The studies outlined provide a plausible rationale regarding the mechanism of Cu-free allylic alkylation reactions that involve dialkylzinc or trialkylaluminum reagents as well as the previously reported alkylmagnesium halides. Based on the research disclosed, it is proposed that bidentate metal-carbene complexes can serve as effective bifunctional catalysts, a critical attribute that differentiates such NHC-based complexes from the corresponding monodentate variants.
Introduction
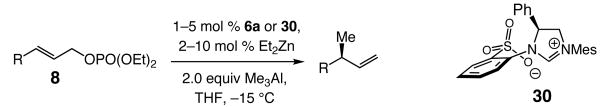
The research described herein sheds light on mechanistic details of C–C bond forming reactions which are promoted by chiral bidentate N-heterocyclic carbene (NHC) complexes and involve alkylmetal reagents (Grignard, dialkylzinc and trialkylaluminum reagents).1 Significant insight has been obtained through detailed examination of Cu-free2,3,4 catalytic allylic alkylations, transformations which give rise to tertiary or (all-carbon) quaternary stereogenic centers with high site- (>98:<2 SN2′:SN2) and enantioselectivities [up to 97.5:2.5 enantiomer ratio (er)]. The present studies have led to the synthesis, isolation and characterization of stereogenic-at-Zn and Al-based NHC complexes,5 allowing us to identify a critical, and somewhat unexpected, stereochemical feature of these complexes. We demonstrate that the high reactivity and selectivity levels observed in C–C bond forming processes promoted through the use of bidentate NHC ligands6,7 might be largely due to the ability of the corresponding metal-based complexes to serve as bifunctional catalysts.8
Discovery and development of chiral catalysts for enantioselective addition of readily available nucleophiles to easily accessible electrophilic substrates remains an important objective in modern chemical synthesis.9 One area of interest relates to catalytic enantioselective allylic alkylations of allylic halides or phosphates with organometallic nucleophiles, such as Mg-, Zn- or Al-based reagents.10 Such transformations often require the presence of an additional Cu-based salt. Thus, it is often a chiral Cu catalyst, generated in situ, which promotes C–C bond formation (see top pathway in Scheme 1). The higher activity of chiral Cu-based complexes, versus when only metal salts are used or in reactions where only an organometallic reagent is present (C–M in Scheme 1), is due to the activation provided by one or more Lewis basic ligands11 that are bound to the transition metal (i and ii, Scheme 1). The donor ligands (LB in Scheme 1) raise the nucleophilicity of the Cu-bound carbon (ii), while enhancing the π-Lewis acidicity of the transition metal (Lewis base activation of a Lewis acid). The above factors collectively translate into a highly effective alkylating agent (cf. conversion of iii to iv and the alkylation product in Scheme 1).
Scheme 1.

Lewis Base Activation in Cu-Based and Cu-Free Pathways to Catalytic Allylic Alkylation
An alternative strategy, also illustrated in Scheme 1 (bottom pathway), involves direct activation of the organometallic reagent by a nucleophilic Lewis base (v, Scheme 1). That is, association of the Lewis basic catalyst (LB) with the Mg-, Zn- or Al-based alkylating agent (C–M) might lead to its activation in the same manner as described above for the Cu-based system, resulting in alkylation of the substrate via vi (Scheme 1) and precluding the need for a Cu salt or a Cu-based complex. Association of the leaving group (LG), typically a Lewis basic entity, with the Lewis acidic metal center should might facilitate the alkylation process (see vi).
We recently demonstrated the validity of the latter approach through development of a class of reactions that involves γ-chloro-α,β-unsaturated esters as substrates, a chiral NHC as the catalyst and alkylmagnesium halide reagents.3 We have illustrated that, in the absence of a Cu salt, NHC–Mg-catalyzed reactions proceed to afford products bearing an all-carbon quaternary stereogenic center12 with moderate to high site- (78:22–93:7 SN2′:SN2) and enantioselectivity (81.5:18.5–99:1 er); an example is illustrated in eq 1. In comparison, as depicted in eq 2, in the presence of a Cu salt (CuCl2•2H2O), catalyst turnover frequency is higher (0.5 mol % catalyst in 0.5 h vs 5 mol % in 24 h) and the transformation proceeds with similar enantioselectivity. The desired chiral products, however, are usually obtained in lower yields, partly due to inferior site-selectivity, as demonstrated by the representative processes shown (eq 1–2). Cu-free catalytic
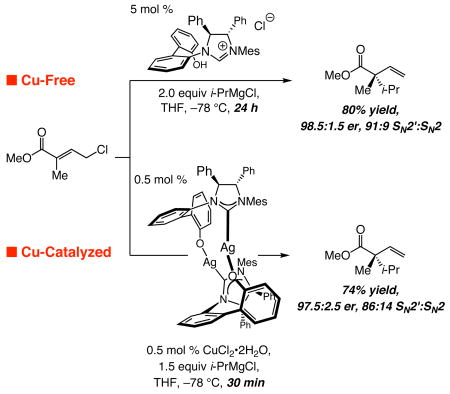 |
(1,2) |
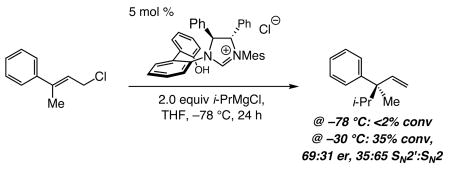 |
(3) |
enantioselective allylic alkylation (EAA) reactions, unlike those performed in the presence of a Cu salt,6 are inefficient and proceed with little or no site- or enantioselectivity with substrates that lack a carboxylic ester group; the example depicted in eq 3 is representative.
Based on the above findings, we designed experiments that offer insight regarding the structural attributes of non-Cu-based chiral NHC–metal complexes and their ability to promote EAA; we aimed to elucidate on the mechanism by which such C–C bond forming processes are promoted. Our decision to focus initially on organozinc reagents (vs the originally examined alkylmagnesium halides) was partly due to higher stability of NHC–Zn complexes. Our efforts to secure and analyze rigorously an NHC–Mg complex,13 such as that derived from a chiral bidentate imidazolinium salt, proved unsuccessful. We therefore turned our attention to the corresponding Zn-based chiral carbene complexes, since we surmised that an NHC complex derived from a late transition metal would be less elusive. In addition, investigation of Cu-free EAA reactions with organozinc and organoaluminum reagents would allow us to determine whether such protocols are effective with a wider range of substrates (vs alkylmagnesium halides mentioned above).
Results and Discussion
1. Cu-Free, NHC-Catalyzed Enantioselective Allylic Alkylation (EAA) Reactions with Dialkylzinc Reagents
i. Evaluation of NHCs and other (non-carbene) Lewis bases
We began by examining the ability of an in situ-generated NHC–Zn complex to catalyze allylic alkylations. We probed the effectiveness of complexes derived from achiral and chiral imidazolinium salts 1–6 as well as Ag-based NHC complex 7 to promote the reaction with allylic phosphate 8a and Et2Zn (Table 1; see below for reactions with the corresponding chlorides). Transformations were carried out at 0 °C in THF and were allowed to proceed for 24 hours. As indicated in entry 1 of Table 1, there is no reaction between 8a and Et2Zn under such conditions in the absence of a catalyst. In the presence of 5.0 mol % achiral imidazolinium salt 1, which is likely deprotonated by the dialkylzinc reagent to afford the NHC–Zn complex, there is 20% conversion to 9, which is generated with complete site-selectivity (>98:<2 SN2′:SN2). When chiral bidentate NHC 2 is used, the amount of the desired product is nearly doubled (38% conv), a strong preference for SN2′ mode of addition persists, and 9 is formed in 86:14 er (entry 3, Table 1). Alkylation promoted by the carbene derived from 3a (entry 4) proceeds with high site-selectivity (>98:<2) and improved enantioselectivity (94:6 er), yet less efficiently than that catalyzed by 2 (18% conv vs 38% conv). Use of the methyl ether derived from imidazolinium salt 3 leads to complete erosion of catalytic activity, as shown in entry 5 of Table 1. Chiral NHC–Zn complexes from valinol-derived imidazolinium salt 4 as well as 5 are entirely ineffective (entries 6–7); this is either because deprotonation by the organometallic reagent and generation of the chiral Zn-based complex is inefficient, or, more likely, due to inability of the derived NHCs to provide the proper nucleophilic activation.
Table 1.
Allylic Alkylation Reactions with Et2Zn in the Presence of Various Nucleophilic Catalystsa
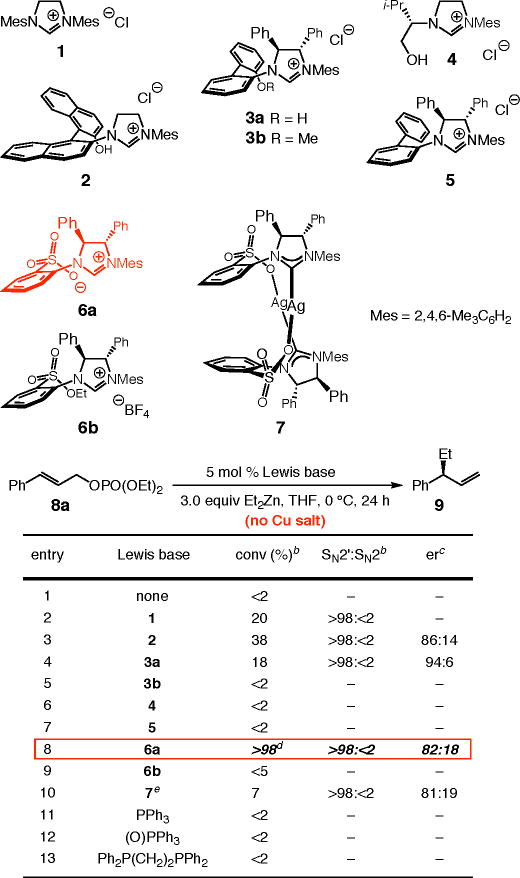 |
Reactions were performed under N2 atmosphere.
Conversion and site-selectivity were determined by analysis of 400 MHz 1H NMR spectra of product mixtures prior to purification.
Enantiomer ratios were determined by GLC analysis; see the Supporting Information for details.
Product isolated in 64% yield after purification.
2.5 mol % of dimer used.
The most active catalyst is the Zn-carbene derived from chiral imidazolinium sulfonate 6a: >98% conversion is observed and the desired product 9 is isolated in 64% yield with >98% site-selectivity and in 82:18 er (entry 8). As was the case with 3b in contrast to 3a (entries 4–5, Table 1), catalytic activity is lost with alkylsulfonate 6b (entry 9, Table 1). In spite of high site-selectivity and appreciable enantioselectivity, EAA with the derived Ag-complex 7 is inefficient (7% conv; entry 10). Spectroscopic studies (400 MHz 1H NMR) indicate a particularly sluggish Ag–Zn exchange as the likely source of the minimal catalytic activity. When reaction of a 10:1 mixture of Et2Zn and Ag-based complex 7 (d8-THF, 22 °C) is monitored, only 45–50% conversion to a new NHC complex is observed after 48 hours (the bidentate Zn complex; see below). When the process in entry 10 (Table 1) is allowed to proceed for 48 hours, conversion is increased to 38%, furnishing 9 with high site-selectivity (>98% SN2′) and in 78:22 er. It should be noted that the alkylation process in entry 10 of Table 1 is performed at 0 °C (vs 22 °C used for the spectroscopic analysis mentioned before).
The findings in entries 11–13 of Table 1 underline the unique ability of NHC-based complexes to serve as effective catalysts for EAA reactions with a dialkylzinc reagent. Attempts to promote alkylation through the use of triphenylphosphine (entry 11), the derived phosphine oxide (entry 12),8a,d,f or dppe (entry 13), resulted in quantitative recovery of the starting material (8a).
ii. NHC–Zn-catalyzed reactions of allylic phosphates bearing disubstituted olefins
Cu–free EAA reactions, catalyzed by the NHC–Zn complex derived from imidazolinium sulfonate 6a, can be performed with a range of dialkylzinc reagents and allylic phosphates (Table 2). The achiral product, derived from SN2 mode of addition is not detected in any of the transformations depicted in Table 2. Effective substrates include those bearing a sterically demanding aryl unit (entry 2), an alkyl (entries 3–5) or a silyl (entries 6–7) substituent. The outcome of transformations represented in entry 8 of Table 1 and entries 1–7 in Table 2 demonstrate that Zn-catalyzed processes, although less efficient, proceed with similar enantioselectivity as the corresponding Cu-catalyzed variants. For example, EAA with (i-Pr)2Zn and 8c in the presence of 1.0 mol % of the Cu-based catalyst derived from treatment of Ag-based dimeric complex of imidazolinium salt 3a and CuCl2•2H2O proceeds to >98% conversion in 24 hours (−15 °C), delivering 14 (entry 5, Table 2) in 68% yield, >98:<2 SN2′:SN2 and 98.5:1.5 er6b (vs 81% conv, 64% yield, >98:<2 SN2′:SN2 and 96:4 er with 10 mol % NHC–Zn complex derived from 6a at 0 °C for 24 h). The Zn complex derived from 6a can be used to obtain the desired products in useful degrees of enantiomeric purity (89.5:10.5–96:4 er). Even in instances where use of a less reactive organozinc reagent and a sizeable alkene substituent requires EAA to be performed at ambient temperature to achieve complete conversion, transformations proceed with appreciable enantioselectivity (e.g., 93:7 er for alkylations of (PhMe2Si)-substituted alkene with n-Bu2Zn in entry 7, Table 2).
Table 2.
NHC–Zn-Catalyzed Enantioselective Allylic Alkylation Reactions of Disubstituted Olefins with Dialkylzinc Reagentsa
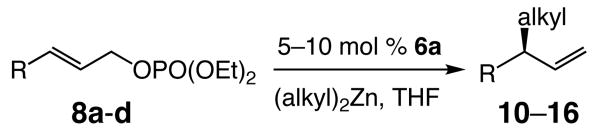 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | substrate (R) | (alkyl)2Zn | product | mol (%) 6a | temp (°C) | time (h) | conv (%);b yield (%)c | SN2′:SN2b | erd |
| 1 | 8a (Ph) | i-Pr2Zn | 10 | 5 | −15 | 60 | 67; 60 | >98:<2 | 90.5:9.5 |
| 2 | 8b (o-MeC6H4) | Et2Zn | 11 | 5 | −15 | 24 | >98; 62 | >98:<2 | 91.5:8.5 |
| 3 | 8c (Cy) | Me2Zn | 12 | 5 | −15 | 30 | 75; 54 | >98:<2 | 89.5:10.5 |
| 4 | 8c (Cy) | Et2Zn | 13 | 5 | −15 | 48 | 96; 79 | >98:<2 | 92.5:7.5 |
| 5 | 8c (Cy) | i-Pr2Zn | 14 | 10 | 0 | 24 | 81; 64 | >98:<2 | 96:4 |
| 6 | 8d (PhMe2Si) | Et2Zn | 15 | 5 | −15 | 48 | 86; 59 | >98:<2 | 94.5:4.5 |
| 7 | 8d (PhMe2Si) | n-Bu2Zn | 16 | 5 | 22 | 48 | 77; 59 | >98:<2 | 93:7 |
Reactions were performed under N2 atmosphere.
Conversion and site selectivity were determined by analysis of 400 MHz 1H NMR spectra of product mixtures prior to purification.
Yields of purified products.
Enantiomer ratios were determined by GLC analysis; see the Supporting Information for details.
iii. NHC–Zn-catalyzed reactions of allylic phosphates bearing trisubstituted olefins
Next, we sought to challenge the catalytic activity of the NHC–Zn complex in the context of reactions that involve trisubstituted olefins, and which generate all-carbon quaternary stereogenic centers.12 As the data illustrated in Table 3 show, the Cu-free catalytic conditions for EAA of dialkylzinc reagents to trisubstituted olefins of allylic phosphates give rise to formation of the desired products (18–24) in 52–91% yield and 94:6–97.5:2.5 er without any detectable amount of achiral SN2 product. Thus, Zn-catalyzed reactions of the more substituted olefins (Table 3) generally proceed with higher enantioselectivities than those of the corresponding disubstituted alkenes. It should also be noted that for reaction of sterically demanding (i-Pr)2Zn, reaction temperature must be raised to 0 °C; regardless, C–C bond formation proceeds without significant diminution in enantioselectivity.
Table 3.
NHC–Zn-catalyzed Enantioselective Allylic Alkylation Reactions of Trisubstituted Olefins with Dialkylzinc Reagentsa
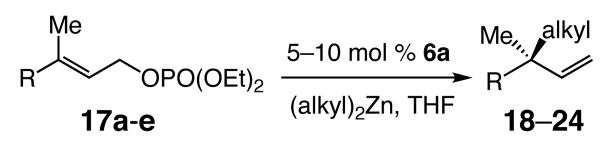 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | substrate (R) | (alkyl)2Zn | product | mol (%) 6a | temp (°C) | time (h) | conv (%);b yield (%)c | SN2′:SN2b | erd |
| 1 | 17a (Ph) | Et2Zn | 18 | 10 | −30 | 48 | 95; 91 | >98:<2 | 95.5:4.5 |
| 2 | 17a (Ph) | n-Bu2Zn | 19 | 10 | −30 | 72 | 72; 71 | >98:<2 | 95.5:4.5 |
| 3 | 17a (Ph) | i-Pr2Zn | 20 | 5 | 0 | 24 | 64; 52 | >98:<2 | 95.5:4.5 |
| 4 | 17b (p-CF3C6H4) | Et2Zn | 21 | 5 | −30 | 48 | >98; 68 | >98:<2 | 94.5:5.5 |
| 5 | 17c (p-NO2C6H4) | Et2Zn | 22 | 10 | −30 | 48 | >98; 82 | >98:<2 | 94.5:5.5 |
| 6 | 17d (Cy) | Et2Zn | 23 | 5 | −15 | 48 | 72; 58 | >98:<2 | 97.5:2.5 |
| 7 | 17e (CO2t-Bu) | n-Bu2Zn | 24 | 5 | −15 | 48 | >98; 85 | >98:<2 | 94:6 |
Reactions were performed under N2 atmosphere.
Conversion and site selectivity were determined by analysis of 400 MHz 1H NMR spectra of product mixtures prior to purification.
Yields of purified products.
Enantiomer ratios were determined by GLC analysis; see the Supporting Information for details.
2. Elucidation of the Structure of a Chiral Bidentate NHC–Zn(II) Complex
To establish the identity of the purported chiral NHC–Zn complex, we first set out to gain insight through spectroscopic studies. Thus, as illustrated in Scheme 2, subjection of 6a to 3.0 equivalents of Et2Zn at 22 °C for 24 hours leads to clean formation of a new complex with distinct signals indicative of a monomeric bidentate Zn-carbene (25). Representative key values are shown in Scheme 2. In the 1H NMR spectrum (400 MHz), the protons of the Et unit are represented by signals at δ +0.63 (methyl) and −0.63 ppm (methylene); the upfield shift of the signals of the Et group, particularly that of the methylene group of the EtZn(NHC) (from δ +0.02 ppm to −0.63 ppm), is congruent with the σ-donating effect of the N-heterocyclic carbene, causing an increase in the electron density of the bound carbon. The chemical shift value in the 100 MHz 13C NMR spectrum for the transition metal-bound carbene carbon (δ 204.0 ppm) is consistent with previously reported chemical shifts for monodentate NHC–Zn complexes.14
Scheme 2.
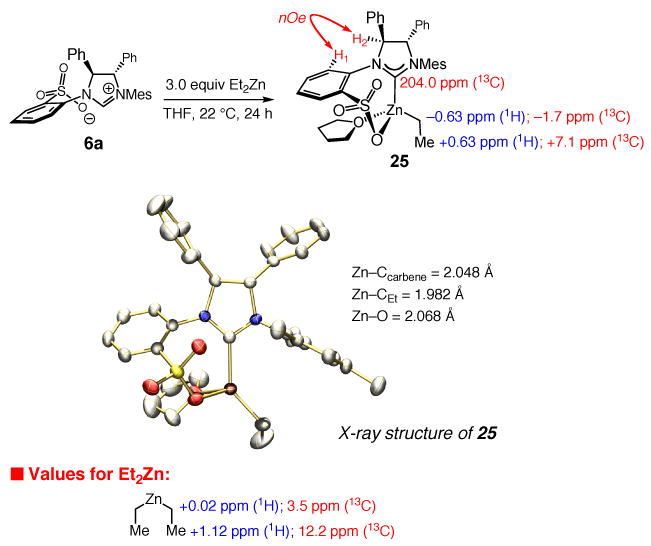
Synthesis and Structure of NHC–Zn(II) Complex 25a
aAll NMR data were measured in THF-d8. Mes = 2,4,6-Me3-C6H2.
The nOe studies resulted in an unexpected observation; as illustrated in Scheme 2, spectroscopic investigations led us to establish that the arylsulfonate coordinates to the Zn metal such that it is situated syn to the adjacent Ph moiety of the NHC. The results of the spectroscopic studies, as depicted in Scheme 2, are supported by X-ray crystallographic studies. The unexpected stereochemical mode of sulfonate coordination, involving a Zn–O bond that is not longer than expected,15 is in contrast to that observed in the dimeric Ag-based complex 7 (Table 1). 7d It appears that chelation of the sulfonate with the Zn center causes tilting of the N-aryl unit (Ccarbene–N–C–C dihedral angle = 49.5°) such that the ortho unit of the same substituent (H1 in 25, Scheme 2) and the backbone of the N-heterocyclic carbene (H2 in 25) are brought into close proximity. Such a degree of propinquity would be destabilizing if the coordinating group and the neighboring phenyl group of the NHC were to adopt an anti orientation (i.e., transpose H2 and Ph in 25). The previously observed anti relationship (established by X-ray)7d between the phenyl group of the NHC and the sulfonate in dimeric Ag complex 7 is likely because chelation involves a metal that is bound to the other sulfonate of the dimeric complex. Thus, the smaller Ccarbene–N–C–C dihedral angle in the dimeric structure (∼2° vs a more significant tilt of ∼50° in 25) causes the steric repulsion between the sulfonate and the phenyl unit to be the dominant factor, leading the two groups to be oriented anti to one another.16 It is for similar reasons that the sulfonate unit is anti to the NHC's phenyl substituent in imidazolinium salt 6a (X-ray).7d
3. A Catalytic Cycle for NHC–Zn-Catalyzed EAA Reactions
With a well-characterized enantiomerically and stereoisomerically pure chiral bidentate NHC–Zn complex in hand, we turned to examining the ability of 25 to serve as a catalyst for EAA reactions. Subjection of allylic phosphate 8a to one equivalent of 25 under conditions used for transformations in Table 1 (THF, 22 °C, 24 h), results in complete recovery of the starting material (Scheme 3). When 30.0 equivalents of Et2Zn are introduced, establishing a catalyst:dialkylzinc ratio congruous with the catalytic transformation (1:30–60), the allylic phosphate is completely consumed, affording 9 in 82:18 er and with >98% SN2′ selectivity.
Scheme 3.

Catalytic EAA with NHC–Zn Complex 25 and a Proposed Mechanism
The above findings indicate that the ability of the chiral NHC–Zn complex (25) to serve as an alkylation catalyst requires the presence of a dialkylzinc. It appears that, unlike the initially suggested scenario in Scheme 1 (see vi), the chiral complex does not directly alkylate the allylic phosphate; that is, the alkyl group of 25 is likely not transferred to the substrate. Alternatively, we put forward the mechanistic hypothesis presented in Scheme 3, involving the intermediacy of I. The alkylation process might therefore commence with replacement of the bound THF in 25 by a substrate molecule; the Lewis basic phosphate serves as the point of association with the Lewis acidic Zn center. Generation of the catalyst•substrate complex results in activation of the allylic phosphate towards reaction with the dialkylzinc reagent, which, in turn, is delivered by the protruding Lewis basic oxygen of sulfonate unit. The intermediacy of I provides an explanation for the high levels of SN2′ selectivities observed in all alkylation reactions.
The Lewis basicity of the sulfonate is likely enhanced due to strong electron donation from the NHC via the transition metal. The significance of a sulfonate oxygen that is properly situated to cause delivery of the dialkylzinc reagent, an attribute directly linked to the conformationally rigid structure of the bidentate NHC, is underlined by the ineffectiveness of the corresponding alkyl-sulfonate 6b (see Table 1). The inactivity of 6b is in agreement with the contention that it is likely the bidentate NHC complex that promotes alkylation (vs the dissociated zwitterionic variant). The above scenario is further supported by the fact that the corresponding allylic chlorides (vs phosphates) are ineffective as substrates in this class of Zn-catalyzed EAA reactions;17 the less Lewis basic halide likely does not chelate as effectively with the Zn center and the bound substrate is properly situated for directed alkylation. In cases where R1 (Scheme 3) is smaller in size than R2, such as the catalytic alkylations in Tables 2–3, high efficiency and enantioselectivity is observed. If R1 is more sterically demanding than R2, however, delivery of the dialkylzinc reagent becomes energetically unfavorable (steric repulsion with the coordinated alkylmetal reagent). Consistent with this proposal, when the Z olefin isomer corresponding to 17a is subjected to the same conditions as used in Table 3 (THF, −15 °C, 24 h), only 11% conversion is attained and rac-18 is isolated. The suggested mechanistic scheme also offers a plausible rationale regarding the unique efficiency of the sulfonate-containing NHC complexes in promoting EAA of allylic phosphates with dialkylzinc reagents: the sulfonate oxygen is properly oriented to coordinate and deliver the alkylzinc reagent to react with the alkene of the Lewis acid-activated substrate. In addition, the aforementioned chelation elevates the nucleophilicity of the bound dialkylzinc reagent. In brief, the bidentate chiral NHC complex likely serves as a bifunctional catalyst.8,18
The critical role that we put forward for the chiral catalyst's sulfonate, in particular the pseudo-equatorially disposed sulfonate oxygen, to alkylation efficiency and enantioselectivity is consistent with the inferior activity of the corresponding carbonyl-containing 27 (Scheme 4). That is, complex 27, which, based on spectroscopic studies (Scheme 4), likely bears a structure closely related to 25, does not contain an appropriately disposed Lewis basic oxygen to direct the addition of an alkylmetal reagent. Accordingly, in contrast to allylic alkylations performed in the presence of the NHC derived from sulfonate 6a, reactions involving carboxylate 26 are far less efficient and enantioselective; the example depicted in Scheme 4 is illustrative.
Scheme 4.

Synthesis and Catalytic Activity of NHC–Zn(II) Complex 27a
aAll NMR data were measured in THF-d8.
The above mechanistic paradigm offers a rationale for the varied and substantially lower efficiency with which bidentate NHC–Zn complexes derived from imidazolinium salts 2, 3a, and 4 promote Cu-free alkylations (see Table 1). In such complexes, it is likely the oxygen atom of the bridging aryloxy group that assists the addition of the dialkylzinc reagent. The attendant ineffectiveness of non-sulfonate complexes as catalysts might be partly due to the relative position of the Lewis basic directing group vis-à-vis the electrophilic π cloud of the bound substrate. The different degrees of catalyst activity (see Table 1) observed with complexes derived from 2, 3a, and 4 supports the latter contention.
The lower effectiveness of the NHC derived from the sulfonate-containing imidazolinium salt 6a (vs 3a) in reactions with alkylmagnesium halides may now be explained as well.3 As shown in Scheme 5 (II and III), the catalyst•substrate ensemble, containing an γ-chloro-α,β-unsaturated ester and the NHC–Mg complex, does not allow for facile delivery of the alkylmagnesium halide to the electrophilic olefin site due to undesirable relative orientation of the sulfonate group and the bound substrate. The electrophilic alkene is less removed from the metal center (vs alkene of the allylic phosphate in I, Scheme 3) and effective alkylation demands a different positioning of the Lewis basic directing group. Examination of molecular models indicates that the sulfonate group is posed to promote more readily the SN2 mode of addition (via II, Scheme 5); such undesired byproducts are obtained in significant amounts when 6a is used to promote Cu-free EAA reactions with alkylmagnesium halides. In contrast, the Lewis basic oxygen in phenoxy-containing IV (Scheme 5), which is more proximal to the NHC–bound metal center, is better suited to direct the addition of the Grignard reagent to a γ-chloro-α,β-unsaturated ester. The present model offers a plausible rationale regarding the requirement for the presence of a substrate that bears an unsaturated carboxylic ester group for efficient Cu-free EAA reactions involving alkylmagnesium halides (see eq 3).
Scheme 5.

Effect of Structure on the Ability of Bidentate NHC Complexes to Serve as Catalysts
In general, effective bifunctional catalysis hinges on cooperation between the two reaction-facilitating effects, which, individually, would not necessarily represent a sufficient driving force to catalyze a particular transformation. Such a principle implies that use of a more highly Lewis acidic complex derived from an NHC bearing an electron withdrawing sulfonate (6a) must be matched with a less reactive alkylmetal reagent (i.e., dialkylzinc vs alkylmagnesium halide). Otherwise, Lewis base-directed alkylation might not be required for efficient reactivity, causing a diminution in stereoselectivity. Consistent with such a proposal, catalytic alkylations of allylic phosphates in the presence of 6a and i-PrMgCl proceed with little or no enantioselectivity. As an example, reaction of 17a (entry 1, Table 3) with 5 mol % 6a and i-PrMgCl (2.0 equiv) leads to 54% conversion (−50 °C, THF, 24 h) and formation of nearly racemic 20 (SN2′:SN2 = 57:43). The higher Lewis acidity of the NHC complex obtained from the sulfonate-bearing 6a (vs phenol-containing 3a previously used in EAA with Grignard reagents) and the more Lewis acidic Mg(II) [vs (Zn(II) in reactions of Tables 1–2] could be responsible for such low selectivity. That is, with the strongly Lewis acidic NHC–Mg complex derived from sulfonate 6a as well as the more nucleophilic Grignard reagent (vs a dialkylzinc) alkylation can occur without assistance by the resident Lewis basic sulfonate, leading to little or no stereochemical induction. Directed catalytic additions with alkylmagnesium halides thus require the less Lewis acidic NHC derived from phenol-based (vs sulfonate) 3a, as shown in the representative case in eq 1 (via IV in Scheme 5).
4. Synthesis and Characterization of a Chiral Bidentate NHC–Al (III) Complex and Enantioselective Allylic Alkylation (EAA) Reactions with Alkylaluminum Reagents
i. Cu-free NHC-catalyzed reactions with Me3Al
In addition to Cu-catalyzed EAA reactions involving Mg- and Zn-based alkylating agents, more recently, we have developed a number of efficient and highly enantioselective processes that proceed in the presence of alkyl- or vinylaluminum reagents.6d-e We therefore decided to establish whether, similar to Zn(II)-based carbenes, the corresponding chiral bidentate NHC–Al(III) complexes can be prepared, characterized and used as catalysts.
In the absence of a catalyst, alkylation of allylic phosphate 8a proceeds only to 5% conversion in the presence of Me3Al (entry 1, Table 4). With imidazolinium salt 6a (entry 2, Table 4), there is only 10% conversion under Cu-free conditions.19 Similar to studies involving the dialkylzinc reagent (Table 1), use of ethylsulfonate 6b or Ag-based complex 7 does not lead to product formation (entries 3–4, Table 4).
Table 4.
Initial Studies on Allylic Alkylations with Me3Al in the Presence of N-Heterocyclic Lewis Base Catalystsa
 | ||||
|---|---|---|---|---|
| entry | Lewis base; (mol %) | conv (%)b | SN2′:SN2b | erc |
| 1 | none | 5 | 62:38 | – |
| 2 | 6a; 5 | 10 | 80:20 | 66.5:33.5 |
| 3 | 6b; 5 | <2 | – | – |
| 4 | 7; 2.5 | <2 | – | – |
Reactions were performed under N2 atmosphere.
Conversion and site-selectivity were determined by analysis of 400 MHz 1H NMR spectra of product mixtures prior to purification.
Enantiomer ratios were determined by GLC analysis; see the Supporting Information for details.
ii. Synthesis of a chiral bidentate NHC–Al(III) complex through a ligand exchange process
Spectroscopic studies, similar to those performed with Et2Zn (see Scheme 2), indicate that treatment of 6a with five equivalents of Me3Al does not lead to NHC–Al complex formation after 24 hours (THF, 22 °C). Only when the THF solution is allowed to reflux for 100 hours, ∼20% of a new entity is generated [the desired NHC–Al(III) complex; see below]. Thus, somewhat surprisingly, in contrast to Et2Zn, Me3Al does not readily deprotonate the imidazolinium salt, generating the derived Al–carbene; similar observations are made with Et3Al. To access the desired NHC–Al(III) complex, we examined the possibility of a ligand exchange process between NHC–Zn(II) 25 and a trialkylaluminum reagent. As illustrated in Scheme 6, subjection of a THF-d8 solution of 25, generated from treatment of a sample of imidazolinium salt 6a and Et2Zn for 20 hours (22 °C), to three equivalents of Me3Al leads to formation of a new complex (55:45 25:29, same as that mentioned above) after 30 minutes based on analysis of the corresponding 400 MHz 1H NMR spectrum. Two new singlets appear at δ −1.40 and −1.74 ppm, which were assigned to the methyl groups of the Al-based complex 29; the 1H NMR signal for Me3Al appears at δ −0.92 ppm in THF-d8 (Scheme 6).20 When an additional 10 equivalents of Me3Al is introduced and the mixture is allowed to stir for an additional 30 minutes, Al-based 29 emerges as the major component (75% of the mixture) and the Zn carbene 25 constitutes 25% of the mixture (Scheme 6). Subsequent studies allowed us to secure a suitable crystal of the Al complex 29, the structure of which is illustrated in Scheme 6.21 Similar to Zn-based 25 (Scheme 2), the sulfonate tether in 29 is syn to the adjacent phenyl group of the NHC backbone.22
Scheme 6.
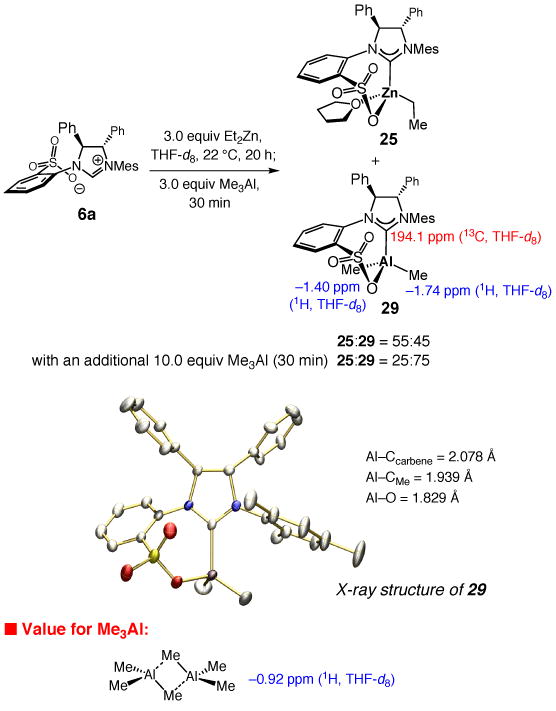
Synthesis and Structure of NHC–Al(III) Complex 29
iii. Cu-free catalytic reactions with Me3Al in the presence of NHC–Zn complex 25
In the presence of 1–5 mol % of chiral NHC–Zn complex 25, as illustrated in Table 5, enantioselective AA reactions with Me3Al proceed with high site- (up to >98:<2 SN2′:SN2) and appreciable enantioselectivity (79:21–87.5:12.5 er). The requisite NHC–Zn complex (25) is prepared in situ through reaction of imidazolinium salt 6a with Et2Zn; <2% products from addition of the ethyl group is observed in all cases shown in Table 5. Comparison of the data in Table 5 with those presented in Table 2, regarding reactions with dialkylzinc reagents, indicates that alkylation involving the more nucleophilic Me3Al requires shorter reaction time. The extent to which the transformations summarized in Table 5 involve catalysis by a Zn-based complex and the Al reagent via a complex such as I (Scheme 3), or the degree to which such processes are promoted by Al-based 29 is difficult to establish (due to rapid ligand exchange). It is likely, however, that the latter scenario is largely involved based on the Zn–Al exchange shown in Scheme 6. A final point regarding the AA process in Table 5 is that, as a comparison of the data in entries 1–2 and 4–5 indicate, in certain cases, the structurally modified NHC complex derived from 30 affords delivers higher efficiency and enantioselectivity.23
Table 5.
EAA Reactions of Disubstituted Allylic Phosphates with Me3Al in the Presence of NHC–Zn Complex 25a
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | substrate (R) | imidazolinium salt; mol % | mol (%) Et2Zn | product | time (h) | conv (%);b yield (%)c | SN2′:SN2b | erd |
| 1 | 8a (Ph) | 6a; 5 | 10 | 28 | 12 | >98; 88 | 98:2 | 79:21 |
| 2 | 8a (Ph) | 30; 1 | 2 | 28 | 24 | >98; 90 | 98:2 | 85.5:14.5 |
| 3 | 8b (o-MeC6H4) | 30; 5 | 10 | 31 | 24 | >98; 88 | 93:7 | 87:13 |
| 4 | 8e (o-NO2C6H4) | 6a; 5 | 10 | 32 | 24 | >98; 79 | 98:2 | 84.5:15.5 |
| 5 | 8e (o-NO2C6H4) | 30; 5 | 10 | 32 | 24 | 84; 81e | 98:2 | 85.5:14.5 |
| 6 | 8c (Cy) | 6a; 5 | 10 | 12 | 24 | >98; 83 | >98:<2 | 87.5:12.5 |
Reactions were performed under N2 atmosphere.
Conversion and site selectivity were determined by analysis of 400 MHz 1H NMR spectra of product mixtures prior to purification.
Yields of purified products.
Enantiomer ratios were determined by GLC analysis; see the Supporting Information for details.
After 48 h, >98% conv is observed (95% yield and identical selectivities as shown above).
Conclusions
We report a series of catalytic enantioselective allylic alkylations, including those that afford all-carbon quaternary stereogenic centers, which are promoted by a chiral bidentate NHC–Zn complex (25) in the absence of a Cu salt. Synthesis, isolation and characterization of the NHC–Zn complex reveals several important structural attributes of this emerging class of chiral Lewis basic ligands. The X-ray structure of stereogenic-at-Zn carbene 25 indicates that, unlike previously reported bidentate complexes containing a phenoxy ligation site, the sulfonate unit coordinates to the transition metal such that it is oriented syn to the proximal phenyl group of the N-heterocycle backbone. Similar studies regarding a derived Al-based chiral bidentate NHC complex (29) illustrate structural features similar to the Zn-based system.
The aforementioned stereochemical feature (sulfonate syn to the NHC phenyl group) is a unique attribute of the sulfonate-bearing class of chiral bidentate NHCs and will allow for a better appreciation of the reactivity modes and the selectivity levels observed in the reactions promoted by this class of metal complexes.6d-e,7f-g Within this context, we propose that a significant difference that accounts for the substantially higher degree of reactivity, site- and enantioselectivity exhibited by bidentate chiral NHC complexes might be due to the ability of such complexes to serve as bifunctional catalysts (Figure 1). Accordingly, the Lewis basic oxygen atoms associated with the second ligation site can deliver the Lewis acidic alkylmetal reagents to the bound substrate molecule, activated by coordination to the Lewis acidic Zn or Al center. Association of the strongly Lewis basic NHC enhances electron density at the heteroatomic sites, elevating their ability to associate with Lewis acidic dialkylzinc or trialkylaluminum reagents.
Figure 1.

Chiral Bidentate NHC–Metal Complex as a Bifunctional Catalyst.
The mechanistic proposals put forth herein account for the significant variations in the ability of bidentate chiral NHC complexes to promote different classes of allylic alkylations. The relative positioning of the Lewis basic heteroatom unit vis-à-vis the Lewis acidic metal is likely a critical determinant as to whether the alkylating agents can be effectively delivered to a particular set of electrophilic substrates upon binding to the chiral complex. Through such arguments, we provide a rationale regarding the effectiveness of two distinct classes of NHC-based chiral catalysts in promoting Cu-free EAA reactions with alkylmagnesium halides and dialkylzinc or trialkylaluminum reagents. Structural information, therefore, has led to elucidation of a plausible mechanism through which NHCs might activate alkylmetals and promote enantioselective C–C bond forming reactions. It should be noted that the catalytic pathways proposed herein are entirely different from those likely involved for the corresponding Cu-catalyzed processes, where it is likely the NHC–bound copper-alkyl that serves as the alkylating agent (vs the alkylzinc or alkylaluminum reagent, as described above). Nontheless, the observations detailed in this report have significant implications regarding the mechanism of NHC–Cu-catalyzed allylic alkylations6 as well as the related conjugate addition7b,e,f-g processes.
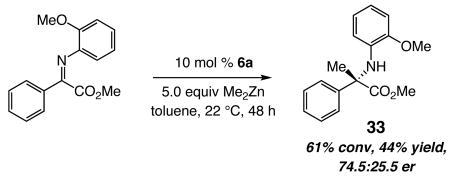
The insights gained through the studies detailed above will be crucial to our ability in utilizing various types of NHC-based chiral complexes towards future development of catalytic enantioselective methods. Preliminary evidence regarding the ability of chiral bidentate NHC–Zn complexes to activate common alkylmetal reagents directly towards enantioselective addition to other important classes of electrophilic substrates is provided in eq 4. Identification of new chiral complexes that readily and stereoselectively promote challenging C–C bond forming processes, such as that which affords α-quaternary amino ester 33, will be one of the objectives of future investigations in these laboratories.
 |
(4) |
Supplementary Material
Acknowledgments
The NSF (CHE-0715138) and the NIH (GM-47480) provided financial support. Y. L. is grateful for Schering-Plough (2006–7) and AstraZeneca (2007–8) graduate fellowships. We thank K-s. Lee for developing a synthesis of imidazolinium salt 26, and Dr. C. A. Baxter and Dr. K. Akiyama for preliminary experiments. Mass spectrometry facilities at Boston College are supported by the NSF (DBI-0619576).
Footnotes
Supporting Information Available. Experimental procedures and spectral data for substrates and products (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
Footnotes and References
- 1.For reviews on N-heterocyclic carbenes as ligands in metal-catalyzed processes, see: Kantchev EAB, O'Brien CJ, Organ MG. Angew Chem, Int Ed. 2007;46:2768–2813. doi: 10.1002/anie.200601663.Gade LH, Bellemin-Laponnaz S. Top Organomet Chem. 2007;21:117–157.Tekavec TN, Louie J. Top Organomet Chem. 2007;21:159–192.Glorius F, editor. N-Heterocyclic Carbenes in Transition Metal Catalysis. Springer-Verlag; Berlin, Heidelberg: 2007.
- 2.For Cu-free (non-enantioselective) conjugate addition and/or intramolecular allylic alkylation processes involving organozinc reagents, see: Komanduri V, Pedraza F, Krische MJ. Adv Synth Catal. 2008;350:1569–1576. doi: 10.1002/adsc.200800242.Kobayashi K, Ueno M, Naka H, Kondo Y. Chem Commun. 2008:3780–3782. doi: 10.1039/b807169a.
- 3.For a report regarding Cu-free enantioselective allylic alkylation reactions promoted by Mg-based chiral bidentate NHC complexes and involving alkylmagnesium halides, see: Lee Y, Hoveyda AH. J Am Chem Soc. 2006;128:15604–15605. doi: 10.1021/ja067456m.
- 4.For Cu-free catalytic enantioselective conjugate addition reactions involving dialkylzinc reagents, see: Bräse S, Höfener S. Angew Chem, Int Ed. 2005;44:7879–7881. doi: 10.1002/anie.200501732.
- 5.For overviews regarding stereogenic-at-metal complexes, see: Brunner H. Angew Chem, Int Ed. 1999;38:1194–1208. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1194::AID-ANIE1194>3.0.CO;2-X.Fontecave M, Hamelin O, Ménage S. Top Organomet Chem. 2005;15:271–288.For a recent example, see: Malcolmson SJ, Meek SJ, Sattely ES, Schrock RR, Hoveyda AH. Nature. 2008;456:933–937. doi: 10.1038/nature07594.
- 6.For applications of chiral bidentate NHC–Cu complexes to enantioselective allylic alkylation reactions, see: Larsen AO, Leu W, Nieto-Oberhuber C, Campbell JE, Hoveyda AH. J Am Chem Soc. 2004;126:11130–11131. doi: 10.1021/ja046245j.Van Veldhuizen JJ, Campbell JE, Giudici RE, Hoveyda AH. J Am Chem Soc. 2005;127:6877–6882. doi: 10.1021/ja050179j.Kacprzynski MA, May TL, Kazane SA, Hoveyda AH. Angew Chem, Int Ed. 2007;46:4554–4558. doi: 10.1002/anie.200700841.Gillingham DG, Hoveyda AH. Angew Chem, Int Ed. 2007;46:3860–3864. doi: 10.1002/anie.200700501.Lee Y, Akiyama K, Gillingham DG, Brown MK, Hoveyda AH. J Am Chem Soc. 2008;130:446–447. doi: 10.1021/ja0782192.
- 7.For applications of chiral bidentate NHC–Cu complexes to enantioselective conjugate addition reactions, see: Arnold PL, Rodden M, Davis KM, Scarisbrick AC, Blake AJ, Wilson C. Chem Commun. 2004:1612–1613. doi: 10.1039/b404614e.Lee Ks, Brown MK, Hird AW, Hoveyda AH. J Am Chem Soc. 2006;128:7182–7184. doi: 10.1021/ja062061o.Clavier H, Coutable L, Toupet L, Guillemin JC, Mauduit M. J Organomet Chem. 2005;690:5237–5254.Brown MK, May TL, Baxter CA, Hoveyda AH. Angew Chem, Int Ed. 2007;46:1097–1100. doi: 10.1002/anie.200604511.Martin D, Kehrli S, d'Augustin M, Clavier H, Mauduit M, Alexakis A. J Am Chem Soc. 2006;128:8416–8417. doi: 10.1021/ja0629920.May TL, Brown MK, Hoveyda AH. Angew Chem, Int Ed. 2008;47:7358–7362. doi: 10.1002/anie.200802910.Brown MK, Hoveyda AH. J Am Chem Soc. 2008;130:12904–12906. doi: 10.1021/ja8058414.
- 8.For a recent review on bifunctional enantioselective catalysis, see: Ma JA, Cahard D. Angew Chem, Int Ed. 2004;43:4566–4583. doi: 10.1002/anie.200300635.Nájera C, Sansano JM, Saá JM. Eur J Oreg Chem. 2009:2385–2400.For additional examples of bifunctional enantioselective catalysis, see: Kacprzynski MA, Hoveyda AH. J Am Chem Soc. 2004;126:10676–10681. doi: 10.1021/ja0478779.Kanai M, Kato N, Ichikawa E, Shibasaki M. Synlett. 2005:1491–1508.Carswell EL, Snapper ML, Hoveyda AH. Angew Chem, Int Ed. 2006;45:7230–7233. doi: 10.1002/anie.200603496.Paull DH, Abraham CJ, Scerba MT, Alden-Danforth E, Lectka T. Acc Chem Res. 2008;41:655–663. doi: 10.1021/ar700261a.Friel DK, Snapper ML, Hoveyda AH. J Am Chem Soc. 2008;130:9942–9951. doi: 10.1021/ja802935w.
- 9.Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis. Springer; Berlin, Germany: 1999. [Google Scholar]
- 10.For reviews on catalytic allylic alkylation reactions that involve “hard” alkyl- or arylmetal-based reagents, see: Hoveyda AH, Hird AW, Kacprzynski MA. Chem Commun. 2004:1779–1785. doi: 10.1039/b401123f.Yorimitsu H, Oshima K. Angew Chem, Int Ed. 2005;44:4435–4439. doi: 10.1002/anie.200500653.Falciola CA, Alexakis A. Eur J Org Chem. 2008:3765–3780.
- 11.For a review on Lewis base catalysis, see: Denmark SE, Beutner GL. Angew Chem, Int Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943.
- 12.Christophers J, Baro A, editors. Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis. Wiley–VCH; Wenheim: 2006. [Google Scholar]
- 13.For X-ray structures of monodentate NHC–Mg complexes, see: Schumann H, Gottfriedsen J, Glanz M, Dechert S, Demtschuk J. J Organomet Chem. 2001:617–618. 588–600.Arduengo AJ, III, Rasika Dias HVR, Davidson F, Harlow RL. J Organomet Chem. 1993;462:13–18.
- 14.For reports regarding X-ray structures of monodentate NHC–Zn(II) complexes, see: (a) Ref 13b. Wang D, Wurst K, Buchmeiser MR. J Organomet Chem. 2004;689:2123–2130.
- 15.A Zn–O bond length of 2.078 Å has been observed for Zn(OSO2CF3)4•THF complex (X-ray analysis; see: Amel'chenkova EV, Denisova TO, Nefedov SE. Zh Neorg Khim. 2006;51:1218–1263.
- 16.The Zn–C bond in Et2Zn has been calculated to be 1.950 Å; the corresponding values are increased to 1.982 Å in 25. However, due to large number of steric and electronic differences caused by NHC and its sulfonate tether, caution should be exercised as to whether comparison of these data shed any meaningful light on the effect of the bidentate carbene association with the alkylzinc reagent. For DFT calculations on lengths of bonds in dialkylzinc reagents, see: Markies PR, Schat G, Akkerman OS, Bickelhaupt F, Smeets WJJ, Spek AL. Organometallics. 1990;9:2243–2247.Haaland A, Green JC, McGrady GS, Downs AJ, Gullo E, Lyall MJ, Timberlake J, Tutukin AV, Volden HV, Østby KA. Dalton Trans. 2003:4356–4366.
- 17.For example, with the allyl chloride corresponding to 8a as the substrate, with 5 mol % 6a and three equivalents of Et2Zn at 0 °C, only 27% conversion to 9, which is formed in <55:45 er, is observed after 24 hours. At −15 °C, 20% conversion is achieved and 9 is isolated in ∼60:40 er.
- 18.For studies regarding enantioselective additions of dialkylzinc reagents promoted by a chiral bifunctional catalyst, see: Yamakawa M, Noyori R. J Am Chem Soc. 1995;117:6327–6335.Kitamura M, Okada S, Suga S, Noyori R. J Am Chem Soc. 1989;111:4028–4036.
- 19.Experiments with trialkylaluminum reagents are performed at −15 °C (vs 0 °C for dialkylzincs) since at elevated temperatures, alkylation products are formed through uncatalyzed processes.
- 20.The assignments of the chemical shifts for the two Al-bound methyls is based on the hypothesis that one alkyl unit resides directly below the π cloud of the mesityl group and should, due to anisotropic factors, be more effectively shielded (see X-ray of 29 in Scheme 6).
- 21.For a report regarding the X-ray structure of a monodentate NHC–AlMe3 complex, see: Li XW, Su J, Robinson GH. Chem Commun. 1996:2683–2684.For an X-ray structure of an NHC–AlH3, see: Arduengo AJ, III, Rasika Dias HV, Calabrese JC, Davidson F. J Am Chem Soc. 1992;114:9724–9725.
- 22.One feature of the Al-based complex 29 is the shortening of the C–Al bonds compared to those reported based on analysis of the X-ray crystal structure of Me3Al [1.94 Å vs 1.96–1.98 Å for non-bridging methyl (2.13 Å for the bridging methyl); Vranka RG, Amma EL. J Am Chem Soc. 1967;89:3121–3126.]. The cautionary note in ref 16 likely applies here as well. Nonetheless, it might be suggested that the shorter metal–methyl bond in 29 might be partly due to the strongly electron-withdrawing sulfonate ligand, which induces polarization at the metal center, in turn strengthening the Al–C bond through a strong electrostatic contribution. Similar Al–C bond shortening within a complex that bears an N,O-bidentate complex has been previously observed (by X-ray diffraction; Kai Y, Yasuoka N, Kasai N, Kakudo M. J Organomet Chem. 1971;32:165–179.). The relatively small degree of lengthening of the Zn–C bond in 25 (Scheme 2) is likely due to similar factors, since the aforementioned polarization effects are expected to be more dominant in the complex that involves the “harder” metal (Al). Such arguments, based on theoretical investigations, have been previously put forth to explain various structural attributes of bidentate methylzinc complexes ( Weston J. Organometallics. 2001;20:713–720. ).
- 23.For reports where the NHC–Cu complex derived from 30 and closely related bidentate chiral carbenes are superior to that obtained through the use of 6a, see: (a) Ref 6e. (b) Ref 7f-g.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.