

Abstract
Great strides have been made in the last 2 years in the field of frontotemporal lobar degeneration (FTLD), particularly with respect to the genetics and molecular biology of FTLD with ubiquitinated inclusions. It is now clear that most cases of familial FTLD with ubiquitinated inclusions have mutations in the progranulin gene, located on chromosome 17. It is also clear that most ubiquitinated inclusions in FTLD with ubiquitinated inclusions are composed primarily of TAR DNA-binding protein-43. Thus, FTLDs can be separated into 2 major groups (i.e. tauopathies and ubiquitinopathies), and most of the ubiquitinopathies can now be defined as TAR DNA-binding protein-43 proteinopathies. Many of the familial FTLDs are linked to chromosome 17, including both the familial tauopathies and the familial TAR DNA-binding protein-43 proteinopathies with progranulin mutations. This review highlights the neuropathologic features and the most important discoveries of the last 2 years and places these findings into the historical context of FTLD.
Keywords: Frontotemporal lobar degeneration, FTDP-17, FTLD-U, Progranulin, TAR-DNA binding protein-43, Tauopathy, Ubiquitinopathy
Historical: Pick Disease
More than 115 years ago, in 1892, Arnold Pick published an article describing 3 patients with clinical aphasia and pathologic circumscribed frontal and temporal atrophy (1). In 1911, Alois Alzheimer subsequently discovered argyrophilic “Pick bodies” in such cases (2), and then in 1922, Gans, one of Arnold Pick's students, coined the term “Pick disease” for frontal disorders with circumscribed atrophy and Pick bodies (3).
Frontal Lobe Dementia of the Non-Alzheimer Type: Pick Disease and Non-Pick Lobar Atrophy/Dementia Lacking Distinctive Histology
Twenty years have passed since Arne Brun defined “frontal lobe dementia of the non-Alzheimer type.” In his 1987 article, Brun et al (4) described the clinical presentation of frontal lobe dementia of the non-Alzheimer type as various combinations of alterations in behavior, personality, executive function, or language. He delineated 2 major underlying pathologic entities: the less common Pick disease and the more frequent non-Pick lobar atrophy, which has the same circumscribed frontal and temporal atrophy but lacks Pick bodies. At that point in the history of frontotemporal dementia, the neuropathology could be simply diagrammed (Fig. 1).
FIGURE 1.
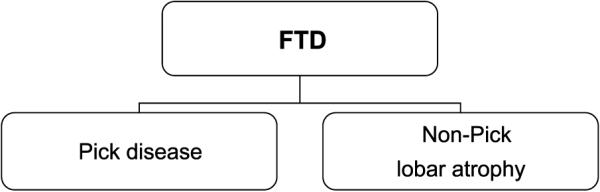
Frontotemporal dementia (FTD) pathologic subtypes, 1987.
In 1990, Knopman et al (5) described “dementia lacking distinctive histology (DLDH),” which in most cases includes the type of pathology found in “non-Pick lobar atrophy.” Dementia lacking distinctive histology has no immunohistochemically labeled inclusions, but does have some distinctive histologic findings, including circumscribed atrophy, variable caudate atrophy, and nigral pallor, both with corresponding neuronal loss and gliosis, and superficial microvacuolation and gliosis in frontal or temporal neocortex, or both. These features are also common to Pick disease (Fig. 2), but, whereas Pick disease has Pick bodies, DLDH has no pathologic inclusions (Fig. 3).
FIGURE 2.

Pick disease pathology. (A, B) Circumscribed frontal and temporal atrophy. (C) Pick bodies in dentate gyrus seen on hematoxylin and eosin (top; 40×) and with paired helical filament 1 immunohistochemistry (IHC) (bottom; 60×). (D) Pick bodies seen with paired helical filament 1 IHC (top; 60×) in frontal cortical layer II, which also shows microvacuolation and gliosis on hematoxylin and eosin (bottom; 10×).
FIGURE 3.

Dementia lacking distinctive histology. Pathology (circumscribed frontal and temporal atrophy, as in Pick disease, is also present). (A) Caudate atrophy. (B) Pallor of the substantia nigra. (C) Superficial microvacuolation and gliosis, cortical layer II, frontal and temporal lobes (hematoxylin and eosin: 20×). (D) Neuronal loss and gliosis, caudate nucleus (hematoxylin and eosin: 40×). (E) Ubiquitin immunohistochemistry (IHC) of frontal lobe shows no inclusions (40×). (F) Ubiquitin IHC of dentate gyrus shows no inclusions (60×). (G) Neuronal loss and gliosis in substantia nigra (hematoxylin and eosin: 20×).
Tau Protein and the TAU gene (MAPT) in Frontotemporal Lobar Degeneration
In the 1990s, great strides were made in Alzheimer disease (AD) by studying of familial cases, identifying mutations and their effects, and looking for similar mechanisms to occur in sporadic AD (6-8). Specialists noted that up to 50% of clinically defined frontotemporal dementias (FTDs) were familial, and that many of them had insoluble tau deposits and were linked to chromosome 17. Some of these cases were described as familial multiple system tauopathy with dementia (9-11). This led to a consensus statement article designating such cases as “familial tauopathy with dementia linked to chromosome 17” (FTDP-17) (12). In 1998, Hutton et al (13) and Poorkaj et al (14) discovered that mutations in the MAPT gene were responsible for FTDP-17 cases with insoluble tau deposits. Like the mutations identified in familial AD, this exciting discovery allowed the development of research projects based on testable hypotheses for sporadic as well as familial tauopathies, with the goal of developing targeted drug therapy (15).
Currently, there are 64 MAPT mutations identified worldwide, 42 of which are pathogenic (16). MAPT missense mutations seem to result in partial loss of microtubule binding, whereas exon 10 splicing mutations seem to disrupt alternative splicing, thereby disturbing the normal 4R:3R tau isoform ratio (17-22). Both increase the tendency of tau protein to assemble into insoluble fibrils. These discoveries proved that tau dysfunction was sufficient to cause neurodegeneration and were significant because, whereas tau is also the major component of the neurofibrillary tangles of AD (23), no MAPT mutations were associated with AD. Tau was also found to be the major protein component of Pick bodies and the insoluble tau deposits typical of other sporadic degenerative disorders, including progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), amyotrophic lateral sclerosis (ALS)-Parkinson disease complex of Guam, and argyrophilic grain disease (24-27). Notably, many of these disorders present either as frontotemporal dementia/aphasia or as a movement disorder (parkinsonism or ALS), or both. The pathology of the FTDP-17s can be unique, or it can be similar to any of the sporadic tauopathies such as Pick disease, PSP, or CBD (Fig. 4). There remained a group of familial cases with clinical frontotemporal dementia that were linked to chromosome 17 but that had no mutations in MAPT (28-33).
FIGURE 4.

Familial tauopathy with dementia linked to chromosome 17 pathology. (A) Dentate gyrus with Pick-like bodies, L266V tau mutation (paired helical filament 1 immunohistochemistry [IHC]; 40×). (B) Progressive supranuclear palsy (PSP)-like pathology in frontal cortex, with neuronal PSP-type tangle (arrow) and tufted astrocyte (arrowhead; AT8 IHC; 60×). (C) Corticobasal degeneration-like cortical pathology with 2 large astrocytic plaques and abundant thread pathology (Gallyas stain; 20×). (D) Cortical gray-white junction with unique tau pathology consisting predominantly of globular white matter (oligodendroglial)-insoluble tau deposits (AT8 IHC; 20×).
Ubiquitinopathies
Meanwhile, progress was also being made with regard to frontotemporal lobar degeneration (FTLD) not associated with tau deposits. In 1991, Okamoto et al described ubiquitinated cytoplasmic and intranuclear inclusions in extramotor cortex in ALS (34). In 1992, Wightman et al (35) next found the same inclusions in hippocampus and neocortex in ALS patients with dementia. Two articles in 1995 and 1996 devised simple immunohistochemical means of pathologically subtyping frontotemporal dementia into major subtypes (Table 1) (36, 37), and in 2001, Woulfe et al (38) showed the same ubiquitinated inclusions in the hippocampus and neocortex of pathologically defined FTD cases without ALS. At the 2001 meeting of the American Association of Neuropathologists, Lipton et al (39) reported that FTLD with ubiquitinated inclusions (FTLD-U) was the single most common FTLD variant (Fig. 5). This and a similar article by Josephs et al (40, 41) were published in 2004. Most cases of DLDH, formerly thought to be the most common pathologic subtype, have now been shown to be FTLD-U, and DLDH is most likely rare to possibly nonexistent (40, 42-45).
TABLE 1.
Immunohistochemical Features of Neuronal Inclusions in Disorders Causing Frontotemporal Dementia
| Disease | Type of Neuronal Inclusion | Major Site | Tau Immunoreactivity | Ubiquitin Immunoreactivity |
|---|---|---|---|---|
| Pick disease | Pick body | Hippocampal and neocortical neurons | ++ | + |
| Corticobasal degeneration | Corticobasal inclusion | Layer II of neocortex, substantia nigra | ++ | - |
| Motor neuron disease-type dementia | Motor neuron disease-type inclusion | Layer II of neocortex, hippocampal dentate granule cells | - | ++ |
| Alzheimer disease | Neurofibrillary tangles | Hippocampal and neocortical neurons | ++ | + |
| Dementia of frontal type | None | - | - | - |
This concise table illustrates the simplicity with which tau and ubiquitin immunostains can distinguish pathologic subtypes of frontotemporal dementia based on positivity or negativity and type and distribution of inclusions. Reprinted with permission from Acta Neuropathol 1996;91:12734 (Fig. 1).
FIGURE 5.

Frontotemporal lobar degeneration with ubiquitinated inclusions pathology. (A) Superficial frontal cortex with neuronal cytoplasmic inclusions (arrow), neuronal intranuclear inclusions (NIIs; solid arrowhead), and dystrophic neurites (open arrowhead; 40×). (B) Dentate gyrus with neuronal cytoplasmic inclusions (60×). (C) Putamen neuron with NII (100×). (D) Dentate gyrus neuron with NII (60×). All are ubiquitin immunohistochemistry (Dako polyclonal, Carpinteria, CA).
The pathology of frontal lobe dementia of the non-Alzheimer type, now preferably termed “frontotemporal lobar degeneration,” could now be divided into 2 major categories: tauopathies and ubiquitinopathies (46, 47). The FTDP-17 cases without MAPT mutations also had ubiquitinated inclusions of the type seen in FTLD-U. The identity of the major protein component of the ubiquitinated inclusions remained elusive, as did the mutation or mutations in the FTDP-17 cases without MAPT mutations.
Non-MAPT FTDP-17 and Progranulin Mutations
Knowledge regarding FTLD-U “ballooned” beginning in the summer of 2006, with 2 papers published back-to-back in Nature in which familial FTLD-U cases linked to chromosome 17 were found to have mutations in the progranulin (PGRN) gene (48, 49). Work on PGRN advanced so quickly that, less than 2 years later, 98 PGRN mutations have been identified, 53 of which are known to be pathogenic (16). To date, most pathogenic mutations are nonsense mutations and produce a premature termination codon (“null” mutations) that results in haploinsufficiency (48, 49). However, a few missense mutations have been shown to be either pathogenic or major risk factors for FTLD-U; these likely result in low PGRN protein expression or secretion (50-54). Recently, a genomic deletion that included the entire PGRN locus was described in a Belgian FTD patient (55). Therefore, whereas mutations in MAPT seem to result in toxic gain of function, mutations in PGRN apparently result in loss of function (56).
PROGRANULIN was first described in relation to wound healing and tumorigenesis (57, 58). It is present in inactive, ramified microglia; in this form, it is anti-inflammatory and likely neurotrophic. Elastase cleaves PGRN into 7 proinflammatory granulin peptides that are present in ameboid microglia (59, 60). Serine leukocyte protease inhibitor (SLPI) protects PGRN from elastase cleavage (61). Because of the opposing effects of PGRN and GRN peptides, microglia can have both anti-inflammatory and proinflammatory effects and, therefore, may be involved in either a deficient or an overactive response to injury in the development of FTLD-U (59, 60). Alternatively, the loss of the neurotrophic support of PGRN may underlie FTLD-U, or there may be other, as yet unidentified, factors involved (59, 60). Progranulin in murine brain is located in microglia and in neurons of the superficial neocortex, hippocampal granular layer, and cerebellar Purkinje layer (62). Immunostains of human brain with antibodies to PGRN show similar positivity in a subset of cortical neurons and in activated microglia, including those surrounding senile plaques in AD, but no positivity of FTLD-U inclusions (48, 59, 63).
Progranulin mutations are found in many familial FTLD-U cases and in as many or more familial FTLD cases as are MAPT mutations (49, 64-66). Progranulin mutations are also found in apparently sporadic FTLD-U cases (64, 65). There is much heterogeneity in the clinical presentation even within the same family. Clinical presentation is usually early onset (fifth or sixth decade), but may also be late-onset, behavioral variant FTD or aphasia with or without parkinsonism (66-81). Clinical presentation may also be that of corticobasal syndrome (68, 80-83). Cases with corticobasal syndrome often have prominent parietal atrophy and often right-greater-than-left asymmetry, whereas those with aphasia syndromes often have left-greater-than-right temporal atrophy, both on imaging and on gross pathologic examination (68, 70, 74, 76, 83, 84). Most report that motor neuron disease (MND) is absent clinically and pathologically in cases with PGRN mutations, but a few studies describe sequence variation or missense mutations of uncertain pathogenicity, but which may affect PGRN protein levels or modify the disease in ALS, causing younger age at onset, shorter survival, or both (85-87). Some cases also have pathologic AD, unusual tau pathology, or α-synuclein pathology (69, 88, 89).
A clinicopathologic correlation paper of 12 Northwestern Cognitive Neurology and Alzheimer Disease Center cases with pathologic FTLD-U or FTLD-MND analyzed for PGRN mutations was recently published (71). All had clinical cognitive impairment: 7 had clinical behavioral variant FTD, 4 had primary progressive aphasia (PPA), and 1 had “dementia.” Three also had clinical and pathologic ALS; final pathologic diagnosis in these cases was FTLD-MND. Based on pathologic and genetic results, the cases can be separated into 3 groups: cases with FTLD-U and PGRN mutations (Group 1), cases with FTLD-U but without PGRN mutations (Group 2), and cases of FTLD-MND without PGRN mutations (Group 3; Table 2). Two of the Group 1 cases had the p.Arg493X mutation that was found to be the most common PGRN mutation in a study of 3,405 neurodegenerative diseases (90). Additionally, 1 case had the p.Ser226TrpfsX28 mutation, and 1 had the p.Ala237TrpfsX4 mutation, the same as that reported for the HDDD1 family (69). Unlike the HDDD1 family, however, this p.Ala237TrpfsX4 mutation case did not have AD pathology. On the other hand, disease duration was only 8 years, and age at death was only 61 years. One case in Group 3 (FTLD-MND group) had a likely silent polymorphism, c.708C>T (p.Asn236Asn).
TABLE 2.
Clinicopathologic Correlations in 12 Patients
| Group | n | Clinical Diagnoses | Family History | Sex | Average Age at Onset, years | Average Duration, years | NIIs |
|---|---|---|---|---|---|---|---|
| 1 | 4 | 2 FTDbv | +/+ | M/M | 56.5 | 5.0 | +/+ |
| 2 PPA | +/- | M/F | +/+ | ||||
| 2 | 5 | 3 FTDbv | +/+/+ | M/M/F | 65.4 | 7.6 | +/+/- |
| 1 PPA | - | F | + | ||||
| 1 “dementia” | Unknown | M | + | ||||
| 3 | 3 | All FTD with ALS | +/-/+ | M/M/F | 62 | 3.3 | +/-/- |
Group 1, pathologic FTLD-U with PGRN mutations; Group 2, pathologic FTLD-U without PGRN mutations; Group 3, pathologic FTLD-MND without PGRN mutations.
ALS, amyotrophic lateral sclerosis; F, female; FTDbv, FTD behavioral variant; FTLD, frontotemporal lobar degeneration; FTLD-U, FTLD with ubiquitinated inclusions; M, male; MND, motor neuron disease; NIIs, neuronal intranuclear inclusions; PGRN, progranulin; PPA, primary progressive aphasia.
With regard to clinical data, this study found no difference between groups in clinical diagnoses or family history. The Group 1 cases with PGRN mutations were split evenly in clinical diagnoses: 2 presented with PPA and 2 with FTDV. With regard to neuropathologic features, there was no difference between groups in regional neuronal loss and gliosis, superficial neocortical microvacuolation, or simple presence of neuronal intranuclear inclusions (NIIs). Group 1 cases had greater caudate atrophy, and, similar to what others have found, Group 1 cases with PGRN mutations had more frontal and temporal cytoplasmic inclusions (CIs) and dystrophic neurites and higher densities of frontal and striatal NIIs (63, 68, 73, 91, 92). Group 3 cases had more dentate gyrus CIs. Ubiquitinated inclusions in all cases labeled with antibodies to TAR-DNA binding protein-43 (TDP-43).
Since the publication of this article, 11 additional Northwestern Cognitive Neurology and Alzheimer Disease Center cases have been analyzed for PGRN mutations for a current total of 23. Breakdown of the pathology in these 23 cases is as follows: 12 have FTLD-U alone, 7 have FTLD-MND, 1 has FTLD-U with MND pathology but no clinical MND, and 3 have FTLD-U with AD pathology, 2 of these sufficient for the pathologic diagnosis of AD by National Institute on Aging/Reagan criteria (93). Unlike the HDDD1 family (69), none of the cases with combined FTLD-U and AD pathology had PGRN mutations. The only additional mutations found in this group (both in FTLD-U cases) are 2 with the IVS6+2 del TGAG mutation that has not yet been proven to be pathogenic.
TDP-43 Is the Major Protein Component of Ubiquitinated Inclusions in FTLD-U
In the fall of 2006, 2 months after the first PGRN mutation articles were published, Neumann et al (94) identified TDP-43 as the major protein component of the ubiquitinated inclusions in FTLD-U; this was swiftly confirmed by Arai et al (95). Identification of this protein had previously been hampered by the relative scarcity of the inclusions and their very small size. In preliminary work, however, monoclonal antibodies were generated to the urea-soluble fraction prepared from homogenates of FTLD-U brains, and these were shown to variably label ubiquitinated cytoplasmic and intranuclear inclusions and dystrophic neurites in subsequent sections from FTLD-U brains (91). Neumann et al (94) performed 2-dimensional polyacrylamide gel electrophoresis on the urea-soluble fraction from FTLD-U brain homogenate and identified spots labeled with certain monoclonal antibodies. They identified the same spots on duplicate Coomassie blue-stained 2-dimensional polyacrylamide gel electrophoresis gels, excised the spots, analyzed them by liquid chromatographyYtandem mass spectrometry, and identified them as amino acid residues belonging to TDP-43. The monoclonal antibodies strongly labeled inclusions in FTLD-U brains, and immunoblots showed that pathologic TDP-43 in FTLD-U has signature 25-kd C-terminal breakdown or cleavage fragments, an approximately 45-kd variant, and a high molecular weight smear. Dephosphorylation of urea fractions collapsed the 45-kd fraction into a 43-kd band and separated the 2 C-terminal fragments into at least 4 TDP-43-immunolabeled bands (94). Frontotemporal lobar degeneration with ubiquitinated inclusion TDP-43 was shown to be ubiquitinated (94). Thus, in FTLD-U, TDP-43 is abnormally phosphorylated, ubiquitinated, and enzymatically hydrolyzed in a manner that produces 2 abnormal C-terminal products of 23 and 27 kd.
TDP-43 Function
At the time TDP-43 was identified in FTLD-U inclusions, the TDP-43 literature was quite sparse; in fact, there were only approximately a dozen articles published regarding TDP-43. Most were related to its role in human immunodeficiency virus infection and cystic fibrosis. TAR-DNA binding protein-43 is a 43-kd highly conserved and widely expressed nuclear protein encoded by the TARDBP gene on chromosome 1. Its functions are diverse and incompletely understood, but TDP-43 seems to bind to DNA, RNA, and protein. In human immunodeficiency virus infection, TDP-43 binds to the “transactive response” DNA and represses transcription in infected cells (96). In cystic fibrosis, TDP-43 is part of a complex that is involved in splicing the cystic fibrosis transmembrane conductance regulator (97). It is involved in splicing the apolipoprotein A2 gene (98). TAR-DNA binding protein-43 helps regulate expression of the mouse SP-10 gene involved in spermatogenesis (99). It also likely acts as a scaffold to link nuclear bodies (GEMS) by interacting with survival motor neuron protein (100). Mutants of human and Drosophila TDP-43 that lack the C-terminal domain are unable to affect splicing (101). Because the C-terminal fragments are aggregated in the urea-soluble fraction of human brain homogenates, it is likely that TDP-43 aggregates in FTLD-U result in loss of function, rather than a toxic gain of function. TAR-DNA binding protein-43 is likely also involved in microRNA biogenesis, apoptosis, and cell division (102), and binds to and stabilizes human low molecular weight neurofilament (hNFL) mRNA (103). Recent evidence emerged that TDP-43 resides in the dendritic-processing body of somatodendrites in the form of RNA granules colocalized with the postsynaptic protein PSD95, where it acts as a translational repressor and thus likely helps regulate neuronal plasticity (104). Restricting nuclear-cytoplasmic trafficking of TDP-43 results in accumulation of TDP-43 as insoluble aggregates (105). Finally, loss of TDP-43 results in dysmorphic nuclear shape, misregulation of the cell cycle, and apoptosis by upregulating cyclin-dependent kinase 6, resulting in increased phosphorylation of retinoblastoma protein pRB and pRb-related protein pRb2/p130 (106). There are currently approximately 60 publications regarding TDP-43 in FTLD-U and ALS, and they continue to accumulate.
TDP-43 in FTLD-U and ALS
The TDP-43 antibody labels ubiquitinated cortical, hippocampal, and striatal inclusions in FTLD-U (107-115) and lower motor neuron and striatal inclusions in ALS (107, 108, 110, 113-120); it also labels inclusions in FTLD-U white matter (121). Neurons with TDP-43-positive inclusions in either the cytoplasm or the nucleus have absence of normal nuclear TDP-43 positivity, additional information that assists in interpreting immunopositivity. Figure 6 compares ubiquitin to TDP-43 immunohistochemistry (IHC) in an FTLD-U case.
FIGURE 6.

Ubiquitin and TAR-DNA binding protein-43 (TDP-43) immunohistochemistry (IHC) in frontotemporal lobar degeneration with ubiquitinated inclusions (FTLD-U). Ubiquitin (A; ubiquitin IHC, Dako polyclonal; 60×) and TDP-43 (B; TDP-43 IHC, Proteintech) in frontal cortex of FTLD-U have similar labeling patterns; (A) Ubiquitin IHC (Dako polyclonal). (B) TDP-43 (Proteintech).
Some investigators have correlated clinical patterns with immunohistochemical TDP-43 patterns as outlined by 2 slightly different schemes (91, 92). One study showed that cases with numerous CIs (Sampathu Type 2/Mackenzie Type 3) have shorter survival, increased frequency of semantic dementia/semantic variant of PPA, and dense hippocampal inclusions; cases with numerous neurites (Sampathu Type 1/Mackenzie Type 2) have difficulty with object naming and have dense temporal and hippocampal inclusions; and cases with intranuclear inclusions (Sampathu Type 3/Mackenzie Type 1) have substantial executive deficits with dense frontal inclusions (109). PROGRANULIN mutations were found only in Sampathu Type 3/Mackenzie Type 1 cases (109), as has been reported (92, 112).
The absence of nuclear TDP-43 labeling and the presence of granular cytoplasmic TDP-43 positivity have been interpreted as being characteristic of “preinclusions” (107, 108, 112). Some interpret preinclusions to indicate that abnormal TDP-43 is prevented from relocating to the nucleus, possibly by becoming hyperphosphorylated and therefore remaining in the cytoplasm where it aggregates (107). Additionally, chromosome 9p-linked FTLD-ALS cases seem to have TDP-43-positive granular inclusions in cortical neurons that are not labeled with ubiquitin IHC, corroborating their interpretation as preinclusions that have not yet become ubiquitinated (112).
The Northwestern Cognitive Neurology and Alzheimer Disease Center participated in an international collaborative study analyzing TDP-43 immunopositivity in one of the largest group of FTLD cases published (112). TAR-DNA binding protein-43 IHC was performed using the polyclonal TDP-43 antibody (Proteintech, Chicago, IL) in 193 familial and sporadic FTLD-U and FTLD-MND cases, which included 36 with PGRN mutations, 5 with valosin-containing protein (VCP) mutations, 4 with charged multivesicular body protein 2B (CHMP2B) mutations, 7 with chromosome 9p linkage but no VCP mutation, 46 other familial, and 95 sporadic cases. These cases were compared with 49 other non-FTLD-U FTLD cases, including Pick disease, CBD, PSP, basophilic inclusion body disease, neuronal intermediate filament inclusion disease, and FTDP-17; 42 non-FTLD dementia cases, including AD, argyrophilic grain disease, tangle predominant senile dementia, Parkinson disease, DLBD, multiple systems atrophy, trinucleotide repeat disorders, and hippocampal sclerosis; and 19 normal controls. TAR-DNA binding protein-43 was positive in all the familial and sporadic FTLD-U and FTLD-MND cases except those with neuronal intermediate filament inclusion disease (112, 122-124) and those with CHMP2B mutations, as has been reported (112, 125) (see also succeeding sentences). Rare, apparently sporadic FTLD-U cases were found to be TDP-43 negative, and those were termed “atypical FTLD-U” (Fig. 7). These cases are currently being investigated for possible spontaneous CHMP2B mutations. Cases of hippocampal sclerosis were also positive with TDP-43 (Fig. 8). There were no TDP-43-positive inclusions in any of the non-FTLD-U dementias, including AD and dementia with Lewy bodies (DLB), or in the controls (112). Of particular interest to studies involving animal models of superoxide dismutase 1 (SOD1)-linked familial ALS (FALS), a large series of ALS cases was studied immunohistochemically for TDP-43. Results showed that, whereas ubiquitinated lower motor neuron inclusions in 59 sporadic ALS cases (SALS) and 11 FALS cases with SOD1 mutations excluded were labeled with TDP-43, those in 15 FALS cases with SOD1 mutations were not (Fig. 9) (119). TAR-DNA binding protein-43 negativity of lower motor neuron inclusions in 2 FALS cases with SOD1 mutations has been confirmed in another study (120).
FIGURE 7.

Typical and “atypical” frontotemporal lobar degeneration with ubiquitinated inclusions (FTLD-U). (A, B) Typical FTLD-U labeled with ubiquitin (A; 40×) and TAR-DNA binding protein-43 (TDP-43; B; 40×); both label cytoplasmic inclusions (CIs). (C, D) “Atypical” FTLD-U labeled with ubiquitin (C; 60×) and TDP-43 (D; 60×). Inclusions are ubiquitin positive but TDP-43 negative (note arrows pointing to unlabeled CIs).
FIGURE 8.

Hippocampal sclerosis, ubiquitin, and TAR-DNA binding protein-43 immunohistochemistry (IHC). (A) Ubiquitin IHC of dentate gyrus in hippocampal sclerosis shows 2 equivocal inclusions noted retrospectively (arrows; 60×). (B) TAR-DNA binding protein-43 of same case clearly shows 2 positive cytoplasmic inclusions (CIs; arrows; 40×). Note that all nuclei are labeled except those in neurons with CIs.
FIGURE 9.

Ubiquitin and TAR-DNA binding protein-43 immunohistochemistry (IHC) in familial amyotrophic lateral sclerosis (FALS) with and without superoxide dismutase 1 (SOD1) mutations. Familial amyotrophic lateral sclerosis cases without SOD1 mutation (A, B) and with SOD1 mutation (C, D). Ubiquitin (A, C) clearly labels Lewy-like bodies (A) and skein-like inclusions (C) in FALS cases with (C) and without (A) SOD1 mutations. TAR-DNA binding protein-43 (B, D) labels FALS without SOD1 mutation (B) but is negative in FALS with SOD1 mutation (D) (all magnifications: 60×).
Clearly, additional clinicopathologic studies are needed to better delineate the pathologic and significance of TDP-43 in FTLD-U and ALS.
Additional FTLD-U-Associated Chromosomes
Although PGRN mutations account for many of the familial FTLD-U cases, there are rare mutations in 3 other genes associated with familial FTLD-U. Most, but not all, of these have TDP-43-positive ubiquitinated inclusions (see next two sections).
Chromosome 9 Related
1) Inclusion body myopathy with Paget disease of bone and frontotemporal dementia is due to mutations in the VCP gene (126). Twelve mutations, 11 of which are pathogenic, have been identified. The ubiquitinated inclusions in inclusion body myopathy with Paget disease of bone and frontotemporal dementia are predominantly intranuclear and are not primarily composed of VCP (127) but rather of TDP-43 (128). 2) A mutation in intraflagellar transport protein 74 on chromosome 9p has been reported in only 1 family with familial FTD-ALS (129). No pathologic description is available for this family. 3) There are other as yet undefined mutation(s) on chromosome 9p associated with familial FTD-ALS (129-131). These cases have ubiquitinated inclusions that are labeled by TDP-43 (112). 4) Lastly, there is a potential locus on chromosome 9q linked to familial FTD-ALS, although this has not been replicated (132).
Chromosome 3
A large Danish familial FTLD and FTLD-ALS pedigree linked to chromosome 3 was found to have mutations in CHMP2B (see previous sentences) (133). Ten mutations have been identified, 4 of which are pathogenic. The ubiquitinated inclusions in CHMP2B-related familial FTLD-U are TDP-43 negative (112, 125).
TDP-43 in Other Disorders
The discovery that TDP-43 is the major protein component in the ubiquitinated inclusions of FTLD-U has allowed investigation into combined pathologies of FTLD-U with other disorders. For example, in the past, because AD pathology also labels with ubiquitin, cases of AD combined with FTLD-U could only be definitively identified if the FTLD-U component had intranuclear inclusions. Because only a fraction of the AD tangle pathology sometimes labels with TDP-43, combined AD/FTLD-U pathology is now known to be quite common; in some studies, it has been shown to occur in the hippocampus and amygdala in approximately 30% of AD and combined AD-DLB cases, half of pure DLB cases, and in the dentate fascia in 70% of hippocampal sclerosis cases (134-137). Results have been conflicting, however, with some studies finding no TDP-43-positive inclusions in pure DLB (136). Likewise, some have found combined TDP-43 proteinopathy and Pick disease (95, 138) with a subset of inclusions positive for both tau and TDP-43, whereas others have not (112). On the other hand, most Guamanian ALS-Parkinson disease complex cases have TDP-43 positivity (139, 140). The significance of TDP-43 positivity in hippocampus and amygdala but not in cortex in AD and DLB is unclear. Does it play a role in the patient's cognitive impairment or is it simply a sign of generalized molecular and cellular disarray in medial temporal regions? Interestingly, in an immunohistochemical analysis of TDP-43 in 5 cases of PPA with AD pathology (which might be expected to have concomitant TDP-43-positive inclusions), none did; all had AD pathology only (141). Further studies may shed light on these issues.
Gene Expression Studies in FTLD-U
Mishra et al (142) performed gene expression micro-array analysis on homogenates from superficial frontal cortex of 10 cases of FTLD-U (n = 6) and FTLD-MND (n = 4) and 6 age-matched controls. Three of the FTLD-U cases had PGRN mutations. The FTLD-MND cases that had TDP-43-positive inclusions predominantly in the dentate gyrus and not in the frontal cortex had results similar to the controls. Frontotemporal lobar degeneration with ubiquitinated inclusion cases compared with controls had downregulated synapse-related genes. This is not surprising because normal TDP-43 colocalizes with the postsynaptic protein PSD-95 (104). There was upregulation of cytoskeletal protein-associated, mitochondrial/energy-associated, and kinase family-associated genes (142). Compared with FTLD-MND, FTLD-U also had downregulation of microtubule-/axon-associated genes, including hNFL and MAP4 (142). The downregulation of hNFL is interesting in view of the recent report that TDP-43 binds to and stabilizes hNFL mRNA (103). Several ubiquitin-/proteasome-associated genes were also downregulated (142). Subsequently, another analysis of gene expression in FTLD-U frontal cortex gray matter, hippocampus, and cerebellum showed similar downregulation of synapse-related and upregulation of cytoskeletal protein-associated genes in affected regions (143). However, this study showed dysregulation of more genes than did the study by Mishra et al and identified pathways not involved in other neurodegenerative diseases such as the cell cycle pathway and transforming growth factor β signaling (143). A gene expression analysis in ALS showed downregulation of cytoskeletal protein-related and mitochondrial-/energy-associated genes, but found similar downregulation of signaling-related genes (144). This study used slices of “fresh frozen prefrontal cortex,” and it is not clear whether it was full-thickness cortex and white matter or a focused region of cortex that was analyzed (144). One might expect results of gene expression analyses in FTLD-U and ALS to be similar because FTLD-U and ALS seem to be on a clinical and pathologic spectrum (145); additional work in this area may be enlightening.
TDP-43 Gene Analysis
The 2 major recent discoveries in the FTLD field are the PGRN mutations in non-tau FTDP-17 dementias (48, 49), which have also been called FTDU-17 in recognition of the presence of ubiquitin inclusions, and the identification of TDP-43 as the major protein component of the ubiquitinated inclusions (94). Familial FTLD-U with PGRN mutations have insoluble inclusions composed of TDP-43 rather than PGRN protein, ie the insoluble aggregates in these cases are not composed of the mutated gene protein product. This is a distinct contrast from the familial taupathies with MAPT mutations which contain insoluble tau protein deposits. Until now, mutations in the TARDBP gene have not been found in FTLD-U (146-148) and FALS (148) cases with TDP-43-labeled inclusions, which has leant credibility to those reports that TDP-43 is not truly the major protein component of the ubiquitinated inclusions (149). New reports, however, describe TARDBP mutations in FALS (150) and in FALS and SALS (151). The TARDBP mutation in the autosomal dominant FALS family is a novel missense mutation, Ala-315-Thr (c.1077 G>A), in exon 6 (150). No member of this family has yet come to autopsy, but it will be crucial to examine such a case for TDP-43-positive insoluble inclusions. In the FALS and SALS cases, 3 single base substitutions in TARDBP exon 6 were identified, all near the C-terminal protein-protein interaction region of TARDBP, resulting in substitution of valine for methionine (M337V), lysine for glutamine (Q331K), and alanine for glycine (G294A) (151). The M337V mutation segregated with disease in a large autosomal dominant FALS kindred, whereas the Q331K mutation was found in screening 200 British SALS cases (absent in 500 controls) and the G294A mutation in screening 172 Australian SALS cases (absent in 372 controls) (151). Screens of an additional 390 controls revealed no TARDBP mutations (151). Chick embryos whose spinal cords were transfected with either the M337V or the Q331K mutant TARDBP gene did not develop limb or tail buds (151). It will likely not be long until TARDBP mutations are found in familial FTLD-U.
Links Between PGRN and TDP-43
Given that most familial FTLD-U cases with known mutations have PGRN mutations, and that the aggregated protein in these brains is composed primarily of TDP-43, it is important to understand the relationship between PGRN and TDP-43 and the roles they play in neurodegeneration. In one study, in which PGRN missense mutations were shown to result in very low PGRN protein levels, investigators asked whether reduced PGRN expression would induce accumulation or relocalization of TDP-43 fragments (52). PROGRANULIN expression was downregulated in human cell lines and in zebrafish, but neither TDP-43 relocalization nor proteolytic processing to C-terminal fragments occurred (52). Zhang et al (152) recently reported that PGRN mediates caspase-dependent cleavage of TDP-43, generating 25- and 35-kd fragments. Suppression of PGRN expression (which is similar to PGRN haploinsufficiency related to mutations) results in accumulation of TDP-43 fragments and this can be inhibited by caspase inhibitors (152). Because SLPI, binding to PGRN, inhibits elastase-mediated proteolysis of PGRN (61), the authors speculate that the mechanism involves a complex between SLPI, PGRN, caspase 3, and TDP-43; decreased PGRN and therefore decreased SLPI might free caspase 3 activity to cleave TDP-43, thereby resulting in a cascade of intracellular events that lead to FTLD-U (152). Staurosporine, a protein kinase inhibitor, also induces caspase cleavage and redistribution of TDP-43 from the nucleus to the cytoplasm, which correlates with the findings in FTLD-U and ALS (152). The results suggest a potential role for PGRN in normal TDP-43 function and a link between the two in FTLD-U disease (152).
CONCLUSIONS AND FUTURE DIRECTIONS
The pathology of frontal lobe dementia of the non-Alzheimer type, now preferably termed frontotemporal lobar degeneration, is currently divided into the same 2 major categories, tauopathies and ubiquitinopathies. There now are at least 13 different subtypes (Table 3) (153, 154). Much has been learned in 20 years, and the FTD subtype diagram has become much more complex (Fig. 10; compared with Fig. 1). There has been exciting recent progress, but much work clearly remains to elucidate the interactions between PGRN and TDP-43 and the roles they play in FTLD-U and ALS. Individuals at risk must be identified early to prevent or halt progression of the disease. Can TDP-43 be measured in cerebrospinal fluid? Can ligands similar to the Pittsburgh B compound that allow in vivo imaging of amyloid plaques be developed to image FTLD-U pathology? In view of the role of PGRN in tumorigenesis, do individuals with PGRN mutations and resulting haploinsufficiency have a decreased incidence of cancer? In view of its role in inflammation, do those with PGRN mutations or FTLD-U without mutations have an increased or decreased incidence of autoimmune or chronic inflammatory disorders? Progranulin is upregulated in activated microglial cells, but is it upregulated in neurons, and, if so, is this beneficial or deleterious to the neuron or to the CNS in general? What is the significance, if any, of TDP-43-positive inclusions in medial temporal regions in AD? Do they contribute to cognitive impairment or are they simply markers of general molecular and cellular dysfunction in these regions in AD? TARDBP mutations have now been identified in ALS—are TARDBP mutations also found in FTLD-U? The 2 discoveries of mutations in PGRN and TDP-43 protein in FTLD-U inclusions occurring only months apart offer opportunities to explore and answer these questions and learn more regarding the neurobiology of the brain in the process.
TABLE 3.
Frontotemporal Lobar Degeneration: Pathologic Subtypes, 2008
| Tauopathies | Ubiquitinopathies |
|---|---|
| Pick disease | FTLD-U/TDP-43 proteinopathy |
| Corticobasal degeneration | FTLD-MND |
| Progressive supranuclear palsy | FTDP-17 with PGRN mutations |
| FTDP-17 with MAPT mutations | FTLD-U with VCP mutations |
| Sporadic MSTD | FTLD-U with CHMP2B mutations |
| Tauopathies, unclassifiable | FTLD-MND linked to chromosome 9p |
| Atypical FTLD-U (TDP-43 negative) |
CHMP2B, charged multivesicular body protein 2B; FTDP, familial tauopathy with dementia linked to chromosome 17; FTLD, frontotemporal lobar degeneration; FTLD-U, FTLD with ubiquitinated inclusions; MND, motor neuron disease; MSTD, multiple system tauopathy with presenile dementia; PGRN, progranulin; TDP-43, TAR-DNA binding protein-43; VCP, valosin-containing protein.
FIGURE 10.

Frontotemporal lobar degeneration (FTLD) pathologic subtypes, 2008. The pathology of frontotemporal dementia (FTD) has become increasingly complex, and pathologic diagnoses now incorporate molecular information compared with Figure 1. CBD, corticobasal degeneration; CHMP2B, charged multivesicular body protein 2B; MND, motor neuron disease; MSTD, multiple system taupathy with presenile dementia; NIFID, neuronal intermediate filament inclusion disease; PGRN, progranulin; PSP, progressive supranuclear palsy; TDP-43, TAR-DNA binding protein-43; VCP, volosin-containing protein.
ACKNOWLEDGMENTS
This article summarizes the work of hundreds of dedicated researchers. Without the generosity of families of those afflicted with these devastating disorders, none of this research would be possible.
Supported in part by Grant No. AG13854 from the National Institutes of Health.
REFERENCES
- 1.Pick A. Über die Beziehungen der senilen Hirnatrophie zur Aphasie. Prager Med Wochenschr. 1892;17:165–67. [Google Scholar]
- 2.Alzheimer A. Über eigenartige Krankheitsfȧlle des späteren Alters. Z Gesamte Neurol Psychiat. 1911;4:356–85. [Google Scholar]
- 3.Gans A. Betrachtungen über art und Ausbreitung des krankhaften Prozesses in einem Fall van Pickscher Atrophie des Stirnhins. Zeitschr f d ges Neurol u Psychiatr (Berl) 1922;80:10–28. [Google Scholar]
- 4.Brun A, Gustafson L, Risberg J, et al. Frontal lobe dementia of non-Alzheimer type. Arch Gerontol Geriatr. 1987;6:193–208. doi: 10.1016/0167-4943(87)90021-5. [DOI] [PubMed] [Google Scholar]
- 5.Knopman DS, Mastri AR, Frey WH, 2nd, et al. Dementia lacking distinctive histologic features: A common non-Alzheimer degenerative dementia. Neurology. 1990;40:251–56. doi: 10.1212/wnl.40.2.251. [DOI] [PubMed] [Google Scholar]
- 6.Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–6. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 7.Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–60. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 8.Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995;269:973–77. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 9.Spillantini MG, Goedert M, Crowther RA, et al. Familial multiple system tauopathy with presenile dementia: A disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci U S A. 1997;94:4113–18. doi: 10.1073/pnas.94.8.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murrell JR, Koller D, Foroud T, et al. Familial multiple-system tauopathy with presenile dementia is localized to chromosome 17. Am J Hum Gen. 1997;61:1131–8. doi: 10.1086/301594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spillantini MG, Murrell JR, Goedert M, et al. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95:7737–41. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foster NL, Wilhelmsen K, Sima AA, et al. Frontotemporal dementia and parkinsonism linked to chromosome 17: A consensus conference. Ann Neurol. 1997;41:706–15. doi: 10.1002/ana.410410606. [DOI] [PubMed] [Google Scholar]
- 13.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 14.Poorkaj P, Bird TD, Wijsman E, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–25. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 15.Allain H, Bentue-Ferrer D, Tribut O, et al. Drug therapy of frontotemporal dementia. Hum Psychopharmacol. 2003;18:221–25. doi: 10.1002/hup.472. [DOI] [PubMed] [Google Scholar]
- 16.Alzheimer Disease & Frontotemporal Dementia Mutation Database. Available at: http://www.molgen.ua.ac.be/ADMutations. Accessed May 12, 2008.
- 17.Clark LN, Poorkaj P, Wszolek Z, et al. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci U S A. 1998;95:13103–7. doi: 10.1073/pnas.95.22.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–17. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 19.Hasegawa M, Smith MJ, Iijima M, et al. FTDP-17 mutations N279K and S305N in tau produce increased splicing of exon 10. FEBS Lett. 1999;443:93–96. doi: 10.1016/s0014-5793(98)01696-2. [DOI] [PubMed] [Google Scholar]
- 20.D'Souza I, Poorkaj P, Hong M, et al. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci U S A. 1999;96:5598–603. doi: 10.1073/pnas.96.10.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grover A, Houlden H, Baker M, et al. 5' splice site mutations in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure that regulates alternative splicing of exon 10. J Biol Chem. 1999;274:15134–43. doi: 10.1074/jbc.274.21.15134. [DOI] [PubMed] [Google Scholar]
- 22.Yen SH, Hutton M, DeTure M, et al. Fibrillogenesis of tau: Insights from tau missense mutations in FTDP-17. Brain Pathol. 1999;9:695–705. doi: 10.1111/j.1750-3639.1999.tb00551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A. 1986;83:4044–48. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pollock NJ, Mirra SS, Binder LI, et al. Filamentous aggregates in Pick's disease, progressive supranuclear palsy, and Alzheimer's disease share antigenic determinants with microtubule-associated protein, tau. Lancet. 1986;2:211. doi: 10.1016/s0140-6736(86)92212-9. [DOI] [PubMed] [Google Scholar]
- 25.Wakabayashi K, Oyanagi K, Makifuchi T, et al. Corticobasal degeneration: Etiopathological significance of the cytoskeletal alterations. Acta Neuropathol. 1994;87:545–53. doi: 10.1007/BF00293314. [DOI] [PubMed] [Google Scholar]
- 26.Oyanagi K, Makifuchi T, Ohtoh T, et al. Amyotrophic lateral sclerosis of Guam: The nature of the neuropathological findings. Acta Neuropathol. 1994;88:405–12. doi: 10.1007/BF00389491. [DOI] [PubMed] [Google Scholar]
- 27.Tolnay M, Spillantini MG, Goedert M, et al. Argyrophilic grain disease: Widespread hyperphosphorylation of tau protein in limbic neurons. Acta Neuropathol. 1997;93:477–84. doi: 10.1007/s004010050642. [DOI] [PubMed] [Google Scholar]
- 28.Kertesz A, Kawarai T, Rogaeva E, et al. Familial frontotemporal dementia with ubiquitin-positive, tau-negative inclusions. Neurology. 2000;54:818–27. doi: 10.1212/wnl.54.4.818. [DOI] [PubMed] [Google Scholar]
- 29.Rosso SM, Kamphorst W, de Graaf B, et al. Familial frontotemporal dementia with ubiquitin-positive inclusions is linked to chromosome 17q21-22. Brain. 2001;124:1948–57. doi: 10.1093/brain/124.10.1948. [DOI] [PubMed] [Google Scholar]
- 30.Rademakers R, Cruts M, Dermaut B, et al. Tau negative frontal lobe dementia at 17q21: Significant finemapping of the candidate region to a 4.8 cM interval. Mol Psych. 2002;7:1064–74. doi: 10.1038/sj.mp.4001198. [DOI] [PubMed] [Google Scholar]
- 31.van Swieten JC, Rosso SM, van Herpen E, et al. Phenotypic variation in frontotemporal dementia and parkinsonism linked to chromosome 17. Dement Geriatr Cogn Disord. 2004;17:261–64. doi: 10.1159/000077150. [DOI] [PubMed] [Google Scholar]
- 32.Mackenzie IR, Baker M, West G, et al. A family with tau-negative frontotemporal dementia and neuronal intranuclear inclusions linked to chromosome 17. Brain. 2006;129:853–67. doi: 10.1093/brain/awh724. [DOI] [PubMed] [Google Scholar]
- 33.van der Zee J, Rademakers R, Engelborghs S, et al. A Belgian ancestral haplotype harbours a highly prevalent mutation for 17q21-linked tau-negative FTLD. Brain. 2006;129:841–52. doi: 10.1093/brain/awl029. [DOI] [PubMed] [Google Scholar]
- 34.Okamoto K, Hirai S, Yamazaki T, et al. New ubiquitin-positive intraneuronal inclusions in the extra-motor cortices in patients with amyotrophic lateral sclerosis. Neurosci Lett. 1991;129:233–36. doi: 10.1016/0304-3940(91)90469-a. [DOI] [PubMed] [Google Scholar]
- 35.Wightman G, Anderson VE, Martin J, et al. Hippocampal and neocortical ubiquitin-immunoreactive inclusions in amyotrophic lateral sclerosis with dementia. Neurosci Lett. 1992;139:269–74. doi: 10.1016/0304-3940(92)90569-s. [DOI] [PubMed] [Google Scholar]
- 36.Cooper PN, Jackson M, Lennox G, et al. Tau, ubiquitin, and alpha B-crystallin immunohistochemistry define the principal causes of degenerative frontotemporal dementia. Arch Neurol. 1995;52:1011–15. doi: 10.1001/archneur.1995.00540340103019. [DOI] [PubMed] [Google Scholar]
- 37.Jackson M, Lowe J. The new neuropathology of degenerative frontotemporal dementias. Acta Neuropathol. 1996;91:127–34. doi: 10.1007/s004010050403. [DOI] [PubMed] [Google Scholar]
- 38.Woulfe J, Kertesz A, Munoz DG. Frontotemporal dementia with ubiquitinated cytoplasmic and intranuclear inclusions. Acta Neuropathol. 2001;102:94–102. doi: 10.1007/s004010000346. [DOI] [PubMed] [Google Scholar]
- 39.Lipton AM, White CL, 3rd, Bigio EH. Frontal lobe dementia with ubiquitinated inclusions predominates in 40 cases of frontotemporal degeneration. J Neuropathol Exp Neurol. 2001;60:514. [Google Scholar]
- 40.Lipton AM, White CL, 3rd, Bigio EH. Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol. 2004;108:379–85. doi: 10.1007/s00401-004-0900-9. [DOI] [PubMed] [Google Scholar]
- 41.Josephs KA, Holton JL, Rossor MN, et al. Frontotemporal lobar degeneration and ubiquitin immunohistochemistry. Neuropathol Appl Neurobiol. 2004;30:369–73. doi: 10.1111/j.1365-2990.2003.00545.x. [DOI] [PubMed] [Google Scholar]
- 42.Josephs KA, Jones AG, Dickson DW. Hippocampal sclerosis and ubiquitin-positive inclusions in dementia lacking distinctive histopathology. Dement Geriatr Cogn Disord. 2004;17:342–45. doi: 10.1159/000077168. [DOI] [PubMed] [Google Scholar]
- 43.Shi J, Shaw CL, Du Plessis D, et al. Histopathological changes underlying frontotemporal lobar degeneration with clinicopathological correlation. Acta Neuropathol. 2005;110:501–12. doi: 10.1007/s00401-005-1079-4. [DOI] [PubMed] [Google Scholar]
- 44.Mackenzie IR, Shi J, Shaw CL, et al. Dementia lacking distinctive histology (DLDH) revisited. Acta Neuropathol. 2006;112:551–59. doi: 10.1007/s00401-006-0123-3. [DOI] [PubMed] [Google Scholar]
- 45.Kumar-Singh S, Van Broeckhoven C. Frontotemporal lobar degeneration: Current concepts in the light of recent advances. Brain Pathol. 2007;17:104–14. doi: 10.1111/j.1750-3639.2007.00055.x. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKhann GM, Albert MS, Grossman M, et al. Clinical and pathological diagnosis of frontotemporal dementia. Arch Neurol. 2001;58:1803–9. doi: 10.1001/archneur.58.11.1803. [DOI] [PubMed] [Google Scholar]
- 47.Trojanowski JQ, Dickson D. Update on the neuropathological diagnosis of frontotemporal dementias. J Neuropathol Exp Neurol. 2001;60:1123–26. doi: 10.1093/jnen/60.12.1123. [DOI] [PubMed] [Google Scholar]
- 48.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–19. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 49.Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–24. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 50.Mukherjee O, Pastor P, Cairns NJ, et al. HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol. 2006;60:314–22. doi: 10.1002/ana.20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mukherjee O, Wang J, Gitcho M, et al. Molecular characterization of novel progranulin (GRN) mutations in frontotemporal dementia. Hum Mutat. 2008;29:512–21. doi: 10.1002/humu.20681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shankaran SS, Capell A, Hruscha AT, et al. Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem. 2008;283:1744–53. doi: 10.1074/jbc.M705115200. [DOI] [PubMed] [Google Scholar]
- 53.Bronner IF, Rizzu P, Seelar H, et al. Progranulin mutations in Dutch familial frontotemporal lobar degeneration. Eur J Hum Gen. 2007;15:369–74. doi: 10.1038/sj.ejhg.5201772. [DOI] [PubMed] [Google Scholar]
- 54.van der Zee J, Le Ber I, Maurer-Stroh S, et al. Mutations other than null mutations producing a pathogenic loss of progranulin in frontotemporal dementia. Hum Mutat. 2007;28:416. doi: 10.1002/humu.9484. [DOI] [PubMed] [Google Scholar]
- 55.Gijselinck I, van der Zee J, Engelborghs S, et al. Progranulin locus deletion in frontotemporal dementia. Hum Mutat. 2008;29:53–58. doi: 10.1002/humu.20651. [DOI] [PubMed] [Google Scholar]
- 56.Rademakers R, Hutton M. The genetics of frontotemporal lobar degeneration. Curr Neurol Neurosci Rep. 2007;7:434–42. doi: 10.1007/s11910-007-0067-6. [DOI] [PubMed] [Google Scholar]
- 57.He Z, Bateman A. Progranulin gene expression regulates epithelial cell growth and promotes tumor growth in vivo. Cancer Res. 1999;59:3222–29. [PubMed] [Google Scholar]
- 58.He Z, Bateman A. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med. 2003;81:600–12. doi: 10.1007/s00109-003-0474-3. [DOI] [PubMed] [Google Scholar]
- 59.Ahmed Z, Mackenzie IRA, Hutton ML, et al. Progranulin in frontotemporal lobar degeneration and neuroinflammation. J Neuro-inflammation. 2007;4:7. doi: 10.1186/1742-2094-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eriksen JL, Mackenzie IRA. Progranulin: Normal function and role in neurodegeneration. J Neurochem. 2008;104:287–97. doi: 10.1111/j.1471-4159.2007.04968.x. [DOI] [PubMed] [Google Scholar]
- 61.Zhu J, Nathan C, Jin W, et al. Conversion of proepithelin to epithelins: Roles of SLPI and elastase in host defense and wound repair. Cell. 2002;111:867–68. doi: 10.1016/s0092-8674(02)01141-8. [DOI] [PubMed] [Google Scholar]
- 62.Daniel R, He Z, Carmichael KP, et al. Cellular localization of gene expression for progranulin. J Histochem Cytochem. 2000;48:999–1009. doi: 10.1177/002215540004800713. [DOI] [PubMed] [Google Scholar]
- 63.Mackenzie IRA, Baker M, Pickering-Brown S, et al. The neuro-pathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain. 2006;129:3081–90. doi: 10.1093/brain/awl271. [DOI] [PubMed] [Google Scholar]
- 64.Gass J, Cannon A, Mackenzie IR, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15:2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- 65.Le Ber I, van der Zee J, Hannequin D, et al. Progranulin null mutations in both sporadic and familial frontotemporal dementia. Hum Mutat. 2007;28:846–55. doi: 10.1002/humu.20520. [DOI] [PubMed] [Google Scholar]
- 66.Pickering-Brown SM, Rollinson S, et al. Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: Comparison with patients with MAPT and no known mutations. Brain. 2008;131:721–31. doi: 10.1093/brain/awm331. [DOI] [PubMed] [Google Scholar]
- 67.Boeve BF, Baker M, Dickson DW, et al. Frontotemporal dementia and parkinsonism associated with the IVSI + IG→YA mutation in progranulin: A clinicopathologic study. Brain. 2006;129:3103–14. doi: 10.1093/brain/awl268. [DOI] [PubMed] [Google Scholar]
- 68.Beck J, Rohrer JD, Campbell T, et al. A distinct clinical, neuro-psychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain. 2008;131:706–20. doi: 10.1093/brain/awm320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Behrens MI, Mukherjee O, Tu P-h, et al. Neuropathologic heterogeneity in HDDD1: A familial frontotemporal lobar degeneration with ubiquitin-positive inclusions and progranulin mutation. Alzheimer Dis Assoc Disord. 2007;21:1–7. doi: 10.1097/WAD.0b013e31803083f2. [DOI] [PubMed] [Google Scholar]
- 70.Huey ED, Grafman J, Wasserman EM, et al. Characteristics of frontotemporal dementia patients with a progranulin mutation. Ann Neurol. 2006;60:374–80. doi: 10.1002/ana.20969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Davion S, Johnson N, Weintraub S, et al. Clinicopathologic correlation in PGRN mutations. Neurology. 2007;69:1113–21. doi: 10.1212/01.wnl.0000267701.58488.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mackenzie IRA. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol. 2007;114:49–54. doi: 10.1007/s00401-007-0223-8. [DOI] [PubMed] [Google Scholar]
- 73.Josephs KA, Ahmed Z, Katsuse O, et al. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol. 2007;66:142–51. doi: 10.1097/nen.0b013e31803020cf. [DOI] [PubMed] [Google Scholar]
- 74.Kelley BJ, Haidar W, Boeve BF, et al. Prominent phenotypic variability associated with mutations in progranulin. Neurobiol Aging. 2007 Oct 18; doi: 10.1016/j.neurobiolaging.2007.08.022. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Le Ber I, Camuzat A, Hannequin D, et al. Phenotype variability in progranulin mutation carriers: A clinical, neuropsychological, imaging and genetic study. Brain. 2008;131:732–46. doi: 10.1093/brain/awn012. [DOI] [PubMed] [Google Scholar]
- 76.Mesulam M, Johnson N, Krafft TA, et al. Progranulin mutations in primary progressive aphasia: The PPA1 and PPA3 families. Arch Neurol. 2007;64:43–47. doi: 10.1001/archneur.64.1.43. [DOI] [PubMed] [Google Scholar]
- 77.Snowden JS, Pickering-Brown SM, Mackenzie IR, et al. Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain. 2006;129:3091–102. doi: 10.1093/brain/awl267. [DOI] [PubMed] [Google Scholar]
- 78.Lladó A, Sánchez-Valle R, René R, et al. Late-onset frontotemporal dementia associated with a novel PGRN mutation. J Neural Transm. 2007;114:1051–54. doi: 10.1007/s00702-007-0716-6. [DOI] [PubMed] [Google Scholar]
- 79.Snowden J, Neary D, Mann D. Frontotemporal lobar degeneration: Clinical and pathological relationships. Acta Neuropathol. 2007;114:31–38. doi: 10.1007/s00401-007-0236-3. [DOI] [PubMed] [Google Scholar]
- 80.López de Munain A, Alzualde A, Gorostidi A, et al. Mutations in progranulin gene: clinical, pathological, and ribonucleic acid expression findings. Biol Psychiatry. 2007 Oct 19; doi: 10.1016/j.biopsych.2007.08.015. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 81.Benussi L, Binetti G, Sina E, et al. A novel deletion in progranulin is associated with FTDP-17 and CBS. Neurobiol Aging. 2008;29:427–35. doi: 10.1016/j.neurobiolaging.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 82.Masellis M, Momeni P, Meschino W, et al. Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain. 2006;129:3115–23. doi: 10.1093/brain/awl276. [DOI] [PubMed] [Google Scholar]
- 83.Spina S, Murrell JR, Huey ED, et al. Corticobasal syndrome associated with the A9D progranulin mutation. J Neuropathol Exp Neurol. 2007;66:892–900. doi: 10.1097/nen.0b013e3181567873. [DOI] [PubMed] [Google Scholar]
- 84.Whitwell JL, Jack CR, Jr, Baker M, et al. Voxel-based morphometry in frontotemporal lobar degeneration with ubiquitin-positive inclusions with and without progranulin mutations. Arch Neurol. 2007;64:371–76. doi: 10.1001/archneur.64.3.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Spina S, Murrell JR, Huey ED, et al. Clinicopathologic features of frontotemporal dementia with progranulin sequence variation. Neurology. 2007;68:820–27. doi: 10.1212/01.wnl.0000254460.31273.2d. [DOI] [PubMed] [Google Scholar]
- 86.Schymick JC, Yang Y, Andersen PM, et al. Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-fronto-temporal dementia phenotypes. J Neurol Neurosurg Psych. 2007;78:754–56. doi: 10.1136/jnnp.2006.109553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sleegers K, Brouwers N, Maurer-Stroh S, et al. Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology. 2008 Jan 9; doi: 10.1212/01.wnl.0000289191.54852.75. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 88.Leverenz JB, Yu CE, Montine TJ, et al. A novel progranulin mutation associated with variable clinical presentation and tau, TDP43 and alpha-synuclein pathology. Brain. 2007;130:1360–74. doi: 10.1093/brain/awm069. [DOI] [PubMed] [Google Scholar]
- 89.Brouwers N, Nuytemans K, van der Zee J, et al. Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch Neurol. 2007;64:1436–46. doi: 10.1001/archneur.64.10.1436. [DOI] [PubMed] [Google Scholar]
- 90.Rademakers R, Baker M, Gass J, et al. Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1277C→YT (Arg493X) mutation: An international initiative. Lancet Neurol. 2007;6:857–68. doi: 10.1016/S1474-4422(07)70221-1. [DOI] [PubMed] [Google Scholar]
- 91.Sampathu DM, Neumann M, Kwong LK, et al. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169:1343–52. doi: 10.2353/ajpath.2006.060438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mackenzie IRA, Baborie A, Pickering-Brown S, et al. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: Classification and relation to clinical phenotype. Acta Neuropathol. 2006;112:539–49. doi: 10.1007/s00401-006-0138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Anonymous Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18:S1–2. Review. [PubMed] [Google Scholar]
- 94.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–33. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 95.Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–11. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 96.Ou SH, Wu F, Harrich D, et al. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69:3584–96. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Buratti E, Dork T, Zuccato E, et al. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001;20:1774–84. doi: 10.1093/emboj/20.7.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mercado PA, Ayala YM, Romano M, et al. Depletion of TDP-43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res. 2005;33:6000–10. doi: 10.1093/nar/gki897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Acharya KK, Govind CK, Shore AN, et al. cis-Requirement for the maintenance of round spermatid-specific transcription. Dev Biol. 2006;295:781–90. doi: 10.1016/j.ydbio.2006.04.443. [DOI] [PubMed] [Google Scholar]
- 100.Wang IF, Reddy NM, Shen CK. Higher order arrangement of the eukaryotic nuclear bodies. Proc Natl Acad Sci U S A. 2002;99:13583–88. doi: 10.1073/pnas.212483099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ayala YM, Pantano S, D'Ambrogio A, et al. Human, Drosophila, and C. elegans TDP43: Nucleic acid binding properties and splicing regulatory function. J Mol Biol. 2005;348:575–88. doi: 10.1016/j.jmb.2005.02.038. [DOI] [PubMed] [Google Scholar]
- 102.Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008;13:867–78. doi: 10.2741/2727. [DOI] [PubMed] [Google Scholar]
- 103.Strong MJ, Volkening K, Hammond R, et al. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci. 2007;35:320–27. doi: 10.1016/j.mcn.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 104.Wang I-F, Wu L-S, Chang H-Y, et al. TDP-43, the signature protein of FTLD-U, is a neuronal activityYresponsive factor. J Neurochem. 2008;105:797–806. doi: 10.1111/j.1471-4159.2007.05190.x. [DOI] [PubMed] [Google Scholar]
- 105.Winton MJ, Igaz LM, Wong MM, et al. Disturbance of nuclear and cytoplasmic Tar DNA binding protein (TDP-43) induces disease-like redistribution, sequestration and aggregate formation. J Biol Chem. 2008 Feb 27; doi: 10.1074/jbc.M800342200. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ayala YM, Misteli T, Baralle FE. TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc Natl Acad Sci U S A. 2008;105:3785–89. doi: 10.1073/pnas.0800546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Davidson Y, Kelley T, Mackenzie IRA, et al. Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. 2007;113:521–33. doi: 10.1007/s00401-006-0189-y. [DOI] [PubMed] [Google Scholar]
- 108.Kwong LK, Neumann M, Sampathu DM, et al. TDP-43 proteinopathy: The neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol. 2007;114:63–70. doi: 10.1007/s00401-007-0226-5. [DOI] [PubMed] [Google Scholar]
- 109.Grossman M, Wood EM, Moore P, et al. TDP-43 pathologic lesions and clinical phenotype in frontotemporal lobar degeneration with ubiquitin-positive inclusions. Arch Neurol. 2007;64:1449–54. doi: 10.1001/archneur.64.10.1449. [DOI] [PubMed] [Google Scholar]
- 110.Seelar H, Schelhaas J, Azmani A, et al. TDP-43 pathology in familial frontotemporal dementia and motor neuron disease without Progranulin mutations. Brain. 2007;130:1375–85. doi: 10.1093/brain/awm024. [DOI] [PubMed] [Google Scholar]
- 111.Higashi S, Iseki E, Yamamoto R, et al. Appearance of TDP-43 in Japanese frontotemporal lobar degeneration with ubiquitin-positive inclusions. Neurosci Lett. 2007;419:213–18. doi: 10.1016/j.neulet.2007.04.051. [DOI] [PubMed] [Google Scholar]
- 112.Cairns NJ, Neumann M, Bigio EH, et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007;171:227–40. doi: 10.2353/ajpath.2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Neumann M, Kwong LK, Sampathu DM, et al. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol. 2007;64:1388–94. doi: 10.1001/archneur.64.10.1388. [DOI] [PubMed] [Google Scholar]
- 114.Brandmeir NJ, Geser F, Kwong LK, et al. Severe subcortical TDP-43 pathology in sporadic frontotemporal lobar degeneration with motor neuron disease. Acta Neuropathol. 2008;115:123–31. doi: 10.1007/s00401-007-0315-5. [DOI] [PubMed] [Google Scholar]
- 115.Zhang H, Tan C-F, Mori F, et al. TDP-43-immunoreactive neuronal and glial inclusions in the neostriatum in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol. 2008;115:115–22. doi: 10.1007/s00401-007-0285-7. [DOI] [PubMed] [Google Scholar]
- 116.Dickson DW, Josephs KA, Amador-Ortiz C. TDP-43 in differential diagnosis of motor neuron disorders. Acta Neuropathol. 2007;114:71–79. doi: 10.1007/s00401-007-0234-5. [DOI] [PubMed] [Google Scholar]
- 117.Mackenzie IRA. The Neuropathology of FTD associated with ALS. Alzheimer Dis Assoc Disord. 2007;21:S44–49. doi: 10.1097/WAD.0b013e31815c3486. [DOI] [PubMed] [Google Scholar]
- 118.Fujita Y, Mizuno Y, Takatama M, et al. Anterior horn cells with abnormal TDP-43 immunoreactivities show fragmentation of the Golgi apparatus in ALS. J Neurol Sci. 2008 Jan 18; doi: 10.1016/j.jns.2007.12.016. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 119.Mackenzie IRA, Bigio EH, Ince PG, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–34. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 120.Tan C-F, Eguchi H, Tagawa A, et al. TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol. 2007;113:535–42. doi: 10.1007/s00401-007-0206-9. [DOI] [PubMed] [Google Scholar]
- 121.Neumann M, Kwong LK, Truax AC, et al. TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol. 2007;66:177–83. doi: 10.1097/01.jnen.0000248554.45456.58. [DOI] [PubMed] [Google Scholar]
- 122.Bigio EH, Lipton AM, White CL, III, et al. Frontotemporal and motor neurone degeneration with neurofilament inclusion bodies: Additional evidence for overlap between FTD and ALS. Neuropathol Appl Neurobiol. 2003;29:239–53. doi: 10.1046/j.1365-2990.2003.00466.x. [DOI] [PubMed] [Google Scholar]
- 123.Cairns NJ, Perry RH, Jaros E, et al. Patients with a novel neurorfilamentopathy: Dementia with neurofilament inclusions. Neurosci Lett. 2003;341:177–80. doi: 10.1016/s0304-3940(03)00100-9. [DOI] [PubMed] [Google Scholar]
- 124.Josephs KA, Holton JL, Rossor MN, et al. Neurofilament inclusion body disease: A new proteinopathy? Brain. 2003;126:L2291–303. doi: 10.1093/brain/awg231. [DOI] [PubMed] [Google Scholar]
- 125.Holm IE, Englund E, Mackenzie IRA, et al. A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3. J Neuropathol Exp Neurol. 2007;66:884–91. doi: 10.1097/nen.0b013e3181567f02. [DOI] [PubMed] [Google Scholar]
- 126.Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–81. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- 127.Forman MS, Mackenzie IR, Cairns NJ, et al. Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol. 2006;65:571–81. doi: 10.1097/00005072-200606000-00005. [DOI] [PubMed] [Google Scholar]
- 128.Neumann M, Mackenzie IR, Cairns NJ, et al. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP mutations. J Neuropathol Exp Neurol. 2007;66:152–57. doi: 10.1097/nen.0b013e31803020b9. [DOI] [PubMed] [Google Scholar]
- 129.Momeni P, Schymick J, Jain S, et al. Analysis of IFT74 as a candidate gene for chromosome 9p-linked ALS-FTD. BMC Neurol. 2006;6:44. doi: 10.1186/1471-2377-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vance C, Al-Chalabi A, Ruddy D, et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain. 2006;129:868–76. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- 131.Morita M, Al-Chalabi A, Andersen PM, et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006;66:839–44. doi: 10.1212/01.wnl.0000200048.53766.b4. [DOI] [PubMed] [Google Scholar]
- 132.Hosler BA, Siddique T, Sapp PC, et al. Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21-q22. JAMA. 2000;284:1664–69. doi: 10.1001/jama.284.13.1664. [DOI] [PubMed] [Google Scholar]
- 133.Skibinski G, Parkinson NJ, Brown JM, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37:806–8. doi: 10.1038/ng1609. [DOI] [PubMed] [Google Scholar]
- 134.Amador-Ortiz C, Lin W-L, Ahmed Z, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61:435–45. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Higashi S, Iseki E, Yamamoto R, et al. Concurrence of TDP-43, tau and a-synuclein pathology in brains of Alzheimer's disease and dementia with Lewy bodies. Brain Res. 2007;1184:284–94. doi: 10.1016/j.brainres.2007.09.048. [DOI] [PubMed] [Google Scholar]
- 136.Nakashima-Yasuda H, Uryu K, Robinson J, et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007;114:221–29. doi: 10.1007/s00401-007-0261-2. [DOI] [PubMed] [Google Scholar]
- 137.Probst A, Taylor KI, Tolnay M. Hippocampal sclerosis dementia: A reappraisal. Acta Neuropathol. 2007;114:335–45. doi: 10.1007/s00401-007-0262-1. [DOI] [PubMed] [Google Scholar]
- 138.Freeman SH, Spires-Jones T, Hyman BT, et al. TAR-DNA binding protein 43 in Pick disease. J Neuropathol Exp Neurol. 2008;67:62–67. doi: 10.1097/nen.0b013e3181609361. [DOI] [PubMed] [Google Scholar]
- 139.Hasegawa M, Arai T, Akiyama H, et al. TDP-43 is deposited in the Guam parkinsonism-dementia complex brains. Brain. 2007;130:1386–94. doi: 10.1093/brain/awm065. [DOI] [PubMed] [Google Scholar]
- 140.Geser F, Winton MJ, Kwong LK, et al. Pathological TDP-43 in parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol. 2008;115:133–45. doi: 10.1007/s00401-007-0257-y. [DOI] [PubMed] [Google Scholar]
- 141.Josephs KA, Whitwell JL, Duffy JR, et al. Progressive aphasia secondary to Alzheimer disease vs FTLD pathology. Neurology. 2008;70:25–34. doi: 10.1212/01.wnl.0000287073.12737.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mishra M, Paunesku T, Woloschak GE, et al. Gene expression analysis of frontotemporal lobar degeneration of the motor neuron disease type with ubiquitinated inclusions. Acta Neuropathol. 2007;114:81–94. doi: 10.1007/s00401-007-0240-7. [DOI] [PubMed] [Google Scholar]
- 143.Chen-Plotkin AS, Geser F, Plotkin JB, et al. Variations in the progranulin gene affect global gene expression in frontotemporal lobar degeneration. Hum Mol Genet. 2008 Jan 25; doi: 10.1093/hmg/ddn023. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lederer CW, Torrisi A, Pantelidou M, et al. Pathways and genes differentially expressed in the motor cortex of patients with sporadic amyotrophic lateral sclerosis. BMC Genomics. 2007;8:26. doi: 10.1186/1471-2164-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mackenzie IR, Feldman H. Ubiquitin immunohistochemistry suggests classic motor neuron disease, motor neuron disease with dementia, and frontotemporal dementia of the motor neuron disease type represent a clinicopathologic spectrum. J Neuropathol Exp Neurol. 2005;64:730–39. doi: 10.1097/01.jnen.0000174335.27708.0a. [DOI] [PubMed] [Google Scholar]
- 146.Rollinson S, Snowden JS, Neary D, et al. TDP-43 gene analysis in frontotemporal lobar degeneration. Neurosci Lett. 2007;419:1–4. doi: 10.1016/j.neulet.2007.03.044. [DOI] [PubMed] [Google Scholar]
- 147.Schumacher A, Friedrich P, Diehl-Schmid J, et al. No association of TDP-43 with sporadic frontotemporal dementia. Neurobiol Aging. 2007 Jul 3; doi: 10.1016/j.neurobiolaging.2007.05.022. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 148.Gijselinck I, Sleegers K, Engelborghs S, et al. Neuronal inclusion protein TDP-43 has no primary genetic role in FTD and ALS. Neurobiol Aging. 2007 Dec 7; doi: 10.1016/j.neurobiolaging.2007.11.002. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 149.Sanelli T, Xiao S, Horne P, et al. Evidence that TDP-43 is not the major ubiquitinated target within the pathological inclusions of amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2007;66:1147–53. doi: 10.1097/nen.0b013e31815c5edd. [DOI] [PubMed] [Google Scholar]
- 150.Gitcho MA, Baloh RH, Chakraverty S, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008 Feb 20; doi: 10.1002/ana.21344. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–72. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhang Y-J, Xu Y-F, Dickey CA, Buratti E, et al. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci. 2007;27:10530–4. doi: 10.1523/JNEUROSCI.3421-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Shaw LM, Korecka M, Clark CM, et al. Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nat Rev Drug Discov. 2007;6:295–303. doi: 10.1038/nrd2176. [DOI] [PubMed] [Google Scholar]
- 154.Cairns NJ, Bigio EH, Mackenzie IRA, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: Consensus of the consortium for frontotemporal lobar degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]