

Abstract
Background
In alcohol dependence, markers of inflammation are associated with increases in rapid eye movement (REM) sleep, which is thought to be a prognostic indicator of alcohol relapse. This study was undertaken to test whether blockade of biologically active tumor necrosis factor-α (TNF-α) normalizes REM sleep in alcohol-dependent adults.
Methods
In a randomized, placebo-controlled, double-blind, crossover trial, 18 abstinent alcohol-dependent male adults received a single dose of etanercept (25 mg) versus placebo in a counterbalanced order. Polysomnographic sleep was measured at baseline and for 3 nights after the acute dose of etanercept or placebo.
Results
Compared with placebo, administration of etanercept produced significant decreases in the amount and percentage of REM sleep. Decreases in REM sleep were robust and approached low levels typically found in age-comparable control subjects. Individual differences in biologically active drug as indexed by circulating levels of soluble tumor necrosis factor receptor II negatively correlated with the percentage of REM sleep.
Conclusions
Pharmacologic neutralization of TNF-α activity is associated with significant reductions in REM sleep in abstinent alcohol-dependent patients. These data suggest that circulating levels of TNF-α may have a physiologic role in the regulation of REM sleep in humans.
Keywords: Alcohol dependence, cytokine antagonism, inflammation, sleep
Sleep disturbance is one of the most prominent complaints of alcoholic patients, with more than 70% of alcohol-dependent persons reporting sleep problems that fail to resolve over the course of abstinence (1,2). Moreover, poor sleep predicts risk of relapse in recovering alcohol-dependent persons (3). It has been further demonstrated that abnormal increases in the percentage of rapid eye movement (REM) sleep are prospectively associated with risk of alcoholic relapse, which suggests that disturbances in REM sleep have prognostic implications in alcohol recovery (4,5). The neurobiological mechanisms that underlie the increases of REM sleep seen during alcohol dependence are not known, although recent evidence suggests that the production of tumor necrosis factor-α (TNF-α) and possibly other markers of inflammation may have a role. Basic observations in rodents demonstrate that TNF-α acts physiologically to regulate sleep (6,7). In alcohol dependence, it has been found that mononuclear cell production of interleukin-6 (IL-6) and TNF-α correlates with REM sleep amounts (8). Furthermore, in studies that temporally profile circulating levels of proinflammatory cytokines across the night, such increases correlate with increases in REM sleep amounts during the subsequent sleep period (9). Taken together, these studies suggest that circulating levels of TNF-α may have a negative influence on the homeostatic regulation of sleep and underlie the relative increases in REM sleep observed in alcohol dependence (10,11).
Given these associations between TNF-α and REM sleep amounts, we hypothesized that blockade of biologically active TNF-α by the antagonist etanercept (Enbrel; Immunex, Thousand Oaks, California) might normalize REM sleep in alcohol-dependent adults. Etanercept is a recombinant human protein composed of two binding domains of the p75 TNF receptor II (TNFRII) fused to the Fc portion of an immunoglobulin molecule (12). It binds specifically to TNF-α in circulation, preventing TNF binding to cell surface receptors, thereby intercepting the inflammatory cascade. Etanercept is an Food and Drug Administration-approved medication for the treatment of rheumatoid arthritis and is given in an injectable form at a subcutaneous (SC) dose of 25 mg (13). Using a randomized, double-blind, placebo-controlled, crossover design, this study tested the effects of etanercept on REM sleep amounts in alcohol dependence. Secondarily, we also explored the effects of etanercept on measures of sleep continuity and sleep architecture.
Methods and Materials
Design Overview
This randomized, placebo-controlled, double-blind, crossover trial allocated abstinent alcohol-dependent adults to receive either a single dose of etanercept (25 mg) versus placebo in a counterbalanced order. After completion of psychiatric interviews, medical evaluations, and an adaptation night in the sleep laboratory with screening for sleep apnea and nocturnal myoclonus as previously described (10,11), eligible subjects were randomized to placebo or drug as a first session. Following baseline assessment of sleep, subjects received either etanercept or placebo and underwent 3 nights of sleep evaluation. After at least 2 weeks of washout, participants underwent a second baseline sleep evaluation and were administered a second dose of either etanercept or placebo, depending on which drug they had received during the first session. Subjects then underwent by an additional 3 nights of sleep evaluation.
Control subjects were not eligible for enrollment per University of California, Los Angeles (UCLA) Internal Review Board (IRB) guidelines. Hence, to determine whether changes in REM sleep following etanercept administration generated levels comparable to normal volunteers, age- and sex-comparable laboratory control subjects with no psychiatric disorder (n = 119 (14) were identified from prior studies (9–11,15–18).
Procedures
Participants who responded to the advertisement (N = 55) between October 2006 and June 2007 underwent assessment phases as previously described (9–11,15–17). Of the 55 subjects evaluated, 14 were excluded because of medical issues (e.g., positive tuberculin skin test), body mass index (BMI) > 30, or both; 5 due to comorbid psychiatric disorders (e.g., current major depressive disorder); 6 for failure to maintain abstinence for 2 weeks before testing; and 4 due to other comorbid, current substance dependence. Of the remaining 25 subjects, 4 additional subjects declined to participate. Details regarding recruitment methods, as well as inclusion and exclusion criteria are found in Supplement 1.
Twenty-one subjects were randomized and admitted to the UCLA General Clinical Research Center for sleep evaluation (Figure 1). Given the experimental nature of this study and concern about adverse side effects associated with the administration of etanercept in this population, IRB guidelines specified that the first five participants be directly allocated to receive active drug followed by placebo. However, drug allocation remained blinded for these five subjects, as well as for the staff who were charged with assessing them or scoring sleep records; only the study physician (M.R.I.), the statistician who generated the randomization schedule, and the pharmacist were aware of active drug assignment. After no adverse events were observed, the remaining 16 subjects were randomly allocated to etanercept or placebo in a counterbalanced order, taking into account the prior schedule. Before administration of etanercept or placebo, three subjects were excluded because of recent use of other substances (i.e., positive urine tests). In the interval between sessions, an additional four participants reported using alcohol and were excluded. Procedures for polysomnographic assessment (19) and assay of soluble TNFRII (sTNFRII) and soluble TNF-α are previously described (9) and also found in Supplement 1.
Figure 1.

Participant flow and distribution of subjects in study. BMI, body mass index; ETOH, ethyl alcohol.
Statistical Analysis
Given correlational evidence that proinflammatory cytokines are associated with increases in REM sleep amounts (9), the primary outcome of interest was change in amounts of REM sleep following administration of etanercept versus placebo. To determine whether REM sleep amounts before and after etanercept differed from levels found in age and sex comparable laboratory control subjects (n = 119), whose electroencephalographic sleep measures had been previously reported (9–11,15–18), planned comparisons were conducted. Secondary exploratory outcomes were changes in sleep continuity measures, along with other measures of sleep architecture. We estimated on the basis of our prior correlational data (9), as well as findings that have examined the effects of another TNF-α antagonist, infliximab, on sleep architecture (20), that the enrollment of 15 patients would provide the study with a statistical power of more than 80% (α = .05) to detect a difference in REM sleep amounts.
The general effects of drug administration were assessed using a mixed models condition (etanercept vs. placebo) × night (baseline, experimental nights 1–3) × order (etanercept, first or second session) repeated-measures analysis of variance (rANOVA) for REM sleep (amounts, percentage). Secondary covariate analyses separately examined the contribution of liver enzymes, predrug TNF-α levels, direct allocation assignment, and completer status on the effects of etanercept on REM sleep. Further exploratory analyses examined the effects of etanercept on sleep continuity outcomes (i.e., total sleep time, sleep latency, sleep efficiency), other sleep measures (i.e., Stages 1–4 sleep, REM density, REM duration), and the relationship between circulating sTNFRII levels and REM sleep. All analyses used an intention to treat approach with inclusion of subjects who had at least one session.
Results
Subjects fulfilled criteria for alcohol dependence in partial remission as determined by Structured Clinical Interview for DSM-IV and had a mean age of 41.4 years [SD = 9.5], education level of 13.5 years [SD = 1.8], and BMI of 25.7 [SD = 4.0]. Of the sample, 50.0% were nonwhite, and 72.2% were current smokers. All subjects were male and had liver enzymes in the laboratory normal range: aspartate aminotransferase [SGPT; mean, 28.3 U/L ± 16.8], alanine aminotransferase [SGOT; mean, 29.4 U/L ± 24.0], and bilirubin [mean, .8 μmol/L ± .4]. Pre-etanercept levels of circulating TNF-α were detectable in all subjects [mean, 1.0 pg/mL ± .4].
Amount and percentage of REM sleep showed acute decreases following administration of etanercept as compared to placebo. Among the subjects who completed at least one session, significant condition × night interactions were found for amounts and percentage of REM sleep [F(3,90.0) = 2.6, p = .05; F(3,88.4) = 2.6, p = .05; Figure 2], with decreases of REM sleep occurring during the night immediately following drug administration (p < .05; 95% confidence interval [CI] of difference vs. placebo condition: −.6 to −7.7). Furthermore, this decrease of percentage of REM sleep approached levels found in age- and sex-comparable laboratory control subjects (n = 119). At baseline, percentage of REM sleep was significantly higher in the alcohol-dependent subjects compared with laboratory controls (p < .01; 95% CI: 23.4–30.4). However, after a single dose of etanercept, percentage of REM sleep decreased in the alcohol-dependent participants and was statistically similar to levels found in control subjects (p = .08; 95% CI: 20.1–27.2). The effects of a single dose of etanercept on REM sleep were transient, and amount and percentage of REM sleep returned to levels found in the placebo condition during the second and third postinjection nights. Inclusion of liver enzyme values or predrug levels of circulating TNF-α, as covariates did not alter the effect of etanercept on REM sleep. In addition, given that the first five subjects were directly allocated to active drug, additional analyses were conducted in which the direct allocation assignment was treated as a covariate; results were similar for amounts and percentage of REM sleep [F(3,88.1) = 2.6, p = .059; F(3,88.7) = 2.6, p = .058]. Likewise, given that 14 subjects completed both sessions, additional analyses were performed in which completer status was treated as a covariate; again, results were similar for amounts and percentage of REM sleep [F(3,87.8) = 2.6, p = .06; F(3,88.4) = 2.6, p = .06]. Differences in REM sleep were similar in those who received etanercept during the first versus second session. Finally, other measures of sleep continuity, sleep architecture, and REM density and duration were not altered (all ps > .05).
Figure 2.
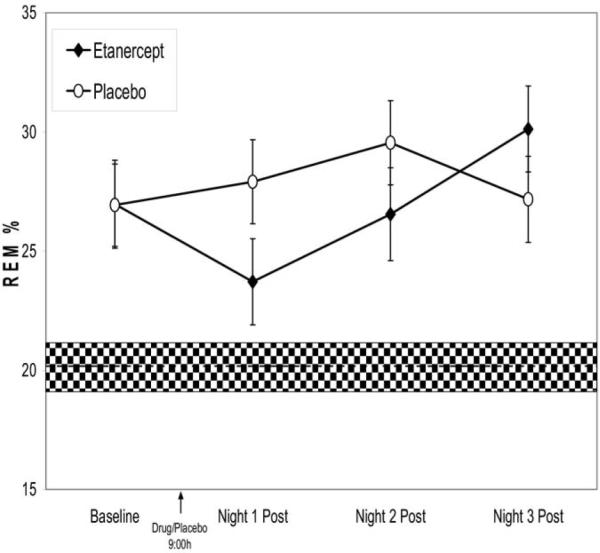
Changes in percentage of rapid eye movement (REM) sleep from baseline to three nights postdrug (etanercept ○ vs. placebo ●) in abstinent alcohol-dependent adults. The shaded area indicates the percentage of REM sleep in age comparable normal volunteers.
Responses of REM sleep to a single dose of etanercept showed individual variability, and further analyses explored whether individual differences in biological active drug as indexed by circulating levels of sTNFRII were associated with percentage of REM sleep. Circulating concentrations of sTNFRII obtained 24 hours after drug administration were negatively correlated with percentage of REM sleep (Figure 3; y = −8262.4x + 382518; R2 = .48; r = −.69, p < .01).
Figure 3.
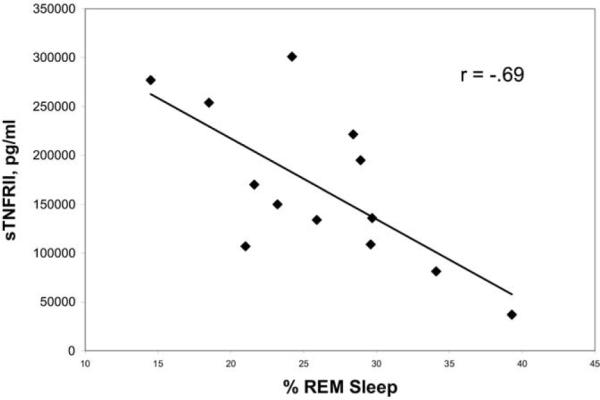
Association between biologically active etanercept as indexed by circulating levels of sTNFRII and percentage of rapid eye movement (REM) sleep during the first night after drug administration.
Discussion
Although the inflammatory response to sleep disturbance has been described (21,22), much less is known about the reciprocal action of proinflammatory cytokines on regulation of sleep in humans. This randomized, placebo-controlled study demonstrates that neutralizing TNF-α activity is associated with a significant reduction of REM sleep in abstinent alcohol-dependent patients. The reduction in percentage of REM sleep was robust; levels of REM sleep approached those typically seen in healthy volunteers without a history of alcohol dependence. Moreover, individual biological variability in the degree of TNF antagonism, as reflected by circulating levels of sTNFRII, correlated with declines in REM sleep. Taken together, these data support the hypothesis that circulating levels of TNF-α may have a physiologic role in the regulation of REM sleep amounts in humans.
These observations further suggest that inhibition of proinflammatory cytokine signaling might represent a viable strategy for targeting sleep disturbance, especially in patients with evidence of increased inflammatory activity. Antagonists of TNF-α (i.e., etanercept, infliximab) reportedly reduce depressive symptoms in the context of treating psoriasis (23). Moreover, one prior study reported that acute administration of infliximab resulted in improvements in sleep continuity and depth in rheumatoid arthritis patients, although this trial was uncontrolled and did not include a placebo condition (20).
Studies in laboratory animals demonstrate that cytokine-induced sickness behaviors, of which sleep disturbance is one component, can be reversed by administering specific cytokine antagonists (e.g., IL-1ra) or anti-inflammatory cytokines (e.g., IL-10) directly into the brain (24). Whereas in animals, proinflammatory cytokines are thought to promote increases of NREM sleep with reciprocal decreases of REM sleep (6), other data show that mice lacking the TNF 55 kD receptor that signals the effects of TNF-α have decreases of NREM as well as REM sleep (25). In six persons who were infected with human immunodeficiency virus (HIV), circulating levels of TNF-α were found to correlate with delta sleep amplitude as assessed by spectral analyses (26). However, these data contrast with other findings showing a relationship between inflammatory activity and increases of REM, but not delta, sleep in humans (8,9,27,28). In this study, the absence of an effect of TNF antagonism on delta sleep is not surprising, given our prior findings that alcohol dependence may be associated with a defect in the homeostatic regulation of delta sleep, because alcohol-dependent patients fail to show an increase in delta sleep during recovery sleep after sleep deprivation (10).
The mechanisms that bridge the link between peripheral levels of TNF-α and abnormal REM sleep amounts in alcohol dependence are not fully understood. However, prior data have found that chronic alcohol consumption is associated with increases in monocyte production, as well as circulating levels, of TNF-α (9,29,30). Furthermore, acute and chronic ethanol exposure induces production of TNF-α by liver Kupffer cells, which contribute to a significant portion of systemic TNF-α (30). In turn, circulating TNF-α triggers rapid (within minutes) increase in the expression of brain TNF-α (31); TNF receptors are thought to transport circulating TNF-α across the blood-brain barrier (32,33). Whereas liver and systemic cytokine responses decline within days after ethanol exposure, brain expression of TNF-α appears to be long lasting (31). Indeed, persistent elevations of brain TNF-α could explain abnormal REM sleep amounts in abstinent alcohol-dependent persons, even though circulating levels of TNF-α are comparable to levels found previously in normal volunteers (9).
The action of acute TNF antagonism on REM sleep was found only during the first night after drug administration, with REM sleep returning to abnormally elevated levels during the second and third nights after administration of etanercept. Given that a single subcutaneous dose of etanercept has a mean half-life of over 100 hours, it is not known why the action of TNF antagonism was short in duration. Because of the persistence of high levels of long-lived TNF/etanercept complexes in the circulation, it is not possible to assess the extent of TNF antagonism by determining circulating levels of free TNF-α post-etanercept. Given the variability of concentrations of circulating sTNFRII observed among subjects post-etanercept, initial bioavailability of the drug may be important. Future studies should assess whether administration of etanercept for a longer period will be associated with a more marked and durable improvement in REM sleep and possibly measures of sleep continuity. Twice weekly administration of etanercept for 3 weeks has been found to induce marked decreases in sleepiness in sleep apnea patients, although that study did not report polysomnographic measures (34).
Limitations of this study include small sample size and sex, along with assessment of sleep solely by visually scoring approaches without additional inclusion of spectral analyses. Despite these limitations, this controlled experimental study showed that TNF-α is a mediator of REM sleep in humans, which substantially extends previous observational studies and represents an important step in ongoing investigations to determine the mechanisms that underlie disturbed sleep in patients with substance use disorders. Further interventional studies that attempt to correct these abnormalities in inflammatory signaling will promote the understanding of the pathophysiologic mechanisms that contribute to sleep disturbance in alcohol dependence and, possibly in other morbid inflammatory conditions.
Supplementary Material
Acknowledgments
This work was supported in part by Grant Nos. AA 13239 T32-MH19925, HL 079955, AG 026364, CA 10014152, CA116778, RR00827, P30-AG028748, General Clinical Research Centers Program, the University of California, Los Angles Cousins Center at the Semel Institute for Neurosciences, and the University of California, Los Angles Claude D. Pepper Older Americans Independence Center Inflammatory Biology Core. The authors thank Sam M. Eljammal, M.D., Gina Rinetti, Ph.D., and Anand Iyer for their efforts in the sleep lab, and Jamie Moran for the assay of tumor necrosis factor-α levels.
Footnotes
The authors have no financial gain related to the outcome of this research, and there are no potential conflicts of interest.
Supplementary material cited in this article is available online.
References
- 1.Brower KJ, Aldrich MS, Hall JM. Polysomnographic and subjective sleep predictors of alcoholic relapse. Alc Clin Exp Res. 1998;22:1864–1871. [PubMed] [Google Scholar]
- 2.Drummond SP, Gillin JC, Smith TL, DeModena A. The sleep of abstinent pure primary alcoholic patients: Natural course and relationship to relapse. Alcohol Clin Exp Res. 1998;22:1796–802. [PubMed] [Google Scholar]
- 3.Brower KJ. Insomnia, alcoholism and relapse. Sleep Med Rev. 2003;7:523–539. doi: 10.1016/s1087-0792(03)90005-0. [DOI] [PubMed] [Google Scholar]
- 4.Clark CP, Gillin JC, Golshan S, Demodena A, Smith TL, Danowski S, et al. Increased REM sleep density at admission predicts relapse by three months in primary alcoholics with a lifetime diagnosis of secondary depression. Biol Psychiatry. 1998;43:601–607. doi: 10.1016/s0006-3223(97)00457-5. [DOI] [PubMed] [Google Scholar]
- 5.Gillin JC, Smith TL, Irwin M, Butters N, Demodena A, Schuckit M. Increased pressure for rapid eye movement sleep at time of hospital admission predicts relapse in nondepressed patients with primary alcoholism at 3-month follow-up. Arch Gen Psychiatry. 1994;51:189–197. doi: 10.1001/archpsyc.1994.03950030025003. [DOI] [PubMed] [Google Scholar]
- 6.Opp MR, Toth LA. Neural-immune interactions in the regulation of sleep. Front Biosci. 2003;8:D768–779. doi: 10.2741/1061. [DOI] [PubMed] [Google Scholar]
- 7.Krueger JM, Toth LA. Cytokines as regulators of sleep. AnnNY Acad Sci. 1994;739:299–310. doi: 10.1111/j.1749-6632.1994.tb19832.x. [DOI] [PubMed] [Google Scholar]
- 8.Redwine L, Dang J, Hall M, Irwin M. Disordered sleep, nocturnal cytokines, and immunity in alcoholics. Psychosom Med. 2003;65:75–85. doi: 10.1097/01.psy.0000038943.33335.d2. [DOI] [PubMed] [Google Scholar]
- 9.Irwin M, Rinetti G, Redwine L, Motivala S, Dang J, Ehlers C. Nocturnal proinflammatory cytokine-associated sleep disturbances in abstinent African American alcoholics. Brain Behav Immun. 2004;18:349–360. doi: 10.1016/j.bbi.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Irwin M, Gillin JC, Dang J, Weissman J, Phillips E, Ehlers CL. Sleep deprivation as a probe of homeostatic sleep regulation in primary alcoholics. Biol Psychiatry. 2002;51:632–641. doi: 10.1016/s0006-3223(01)01304-x. [DOI] [PubMed] [Google Scholar]
- 11.Irwin M, Miller C, Gillin JC, Demodena A, Ehlers CL. Polysomno-graphic and spectral sleep EEG in primary alcoholics: An interaction between alcohol dependence and African-American ethnicity. Alc Clin Exp Res. 2000;24:1376–1384. [PubMed] [Google Scholar]
- 12.Wong M, Ziring D, Korin Y, Desai S, Kim S, Lin J, et al. TNFalpha blockade in human diseases: Mechanisms and future directions. Clin Immunol. 2008;126:121–136. doi: 10.1016/j.clim.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin J, Ziring D, Desai S, Kim S, Wong M, Korin Y, et al. TNFalpha blockade in human diseases: An overview of efficacy and safety. Clin Immunol. 2008;126:13–30. doi: 10.1016/j.clim.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.First MB, Spitzer RL, Gibbon M, Williams JB. Structured Clinical Interview for DSM-IV Axis I Disorders - Patient Edition. Version 2.0 New York State Psychiatric Institute; New York: 2006. [Google Scholar]
- 15.Irwin MR, Valladares EM, Motivala S, Thayer JF, Ehlers CL. Association between nocturnal vagal tone and sleep depth, sleep quality, and fatigue in alcohol dependence. Psychosom Med. 2006;68:159–166. doi: 10.1097/01.psy.0000195743.60952.00. [DOI] [PubMed] [Google Scholar]
- 16.Irwin MR, Ziegler M. Sleep deprivation potentiates activation of cardiovascular and catecholamine responses in abstinent alcoholics. Hypertension. 2005;45:252–257. doi: 10.1161/01.HYP.0000153517.44295.07. [DOI] [PubMed] [Google Scholar]
- 17.Irwin M, Clark C, Kennedy B, Gillin JC, Ziegler M. Nocturnal catehcolamines and immune function in insomniacs, depressed patients, and control subjects. Brain Beh Immun. 2003;17:365–372. doi: 10.1016/s0889-1591(03)00031-x. [DOI] [PubMed] [Google Scholar]
- 18.Valladares E, Motivala SJ, Ehlers C, Irwin MR. Sex differences in cardiac sympathovagal balance and vagal tone during nocturnal sleep. Sleep Med. 2008;9:310–316. doi: 10.1016/j.sleep.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 19.Rechtschaffen A, Kales A. A manual of standardized terminology, techniques and scoring system for sleep stages of human subjects. National Institute of Neurologic Diseases and Blindness; Bethesda, MD: 1968. [Google Scholar]
- 20.Zamarron F, Maceiras F, Mera A, Gomez-Reino JJ. Effects of the first infliximab infusion on sleep and alertness in patients with active rheumatoid arthritis. Ann Rheum Dis. 2004;63:88–90. doi: 10.1136/ard.2003.007831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Irwin M, Wang M, Ribiero D, Cho H, Breen E, Martinez-Maza O, Cole S. Sleep loss activates cellular inflammatory signaling. Biol Psychiatry. 2008;64:538–540. doi: 10.1016/j.biopsych.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Irwin MR, Wang M, Campomayor CO, Collado-Hidalgo A, Cole S. Sleep deprivation and activation of morning levels of cellular and genomic markers of inflammation. Arch Intern Med. 2006;166:1756–1762. doi: 10.1001/archinte.166.16.1756. [DOI] [PubMed] [Google Scholar]
- 23.Tyring S, Gottlieb A, Papp K, Gordon K, Leonardi C, Wang A, et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet. 2006;367:29–35. doi: 10.1016/S0140-6736(05)67763-X. [DOI] [PubMed] [Google Scholar]
- 24.Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang J, Wang Y, Krueger JM. Mice lacking the TNF 55 kDa receptor fail to sleep more after TNFalpha treatment. J Neurosci. 1997;17:5949–5955. doi: 10.1523/JNEUROSCI.17-15-05949.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Darko DF, Miller JC, Gallen C, White J, Koziol J, Brown SJ, et al. Sleep electroencephalogram delta-frequency amplitude, night plasma levels of tumor necrosis factor, and human immunodeficiency virus infection. Proc Natl Acad Sci USA. 1995;92:12080–12084. doi: 10.1073/pnas.92.26.12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Motivala SJ, Sarfatti A, Olmos L, Irwin MR. Inflammatory markers and sleep disturbance in major depression. Psychosom Med. 2005;67:187–194. doi: 10.1097/01.psy.0000149259.72488.09. [DOI] [PubMed] [Google Scholar]
- 28.Raison CL, Capuron L, Miller AH. Cytokinessingtheblues:Inflammation and the pathogenesis of depression. Trends Immunol. 2006;27:24–31. doi: 10.1016/j.it.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McClain CJ, Cohen DA. Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology. 1989;9:349–351. doi: 10.1002/hep.1840090302. [DOI] [PubMed] [Google Scholar]
- 30.McClain CJ, Song Z, Barve SS, Hill DB, Deaciuc I. Recent advances in alcoholic liver disease. IV. Dysregulated cytokine metabolism in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2004;287:G497–502. doi: 10.1152/ajpgi.00171.2004. [DOI] [PubMed] [Google Scholar]
- 31.Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflammation. 2008;5:10. doi: 10.1186/1742-2094-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quan N, Banks WA. Brain-immune communication pathways. Brain Behav Immun. 2007;21:727–735. doi: 10.1016/j.bbi.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 33.Pan W, Kastin AJ. TNF alpha transport across the blood-brain barrier is abolished in receptor knockout mice. Exp Neurol. 2002;174:193–200. doi: 10.1006/exnr.2002.7871. [DOI] [PubMed] [Google Scholar]
- 34.Vgontzas AN, Zoumakis E, Lin HM, Bixler EO, Trakada G, Chrousos GP. Marked decrease in sleepiness in patients with sleep apnea by etanercept, a tumor necrosis factor-alpha antagonist. J Clin Endocrinol Metab. 2004;89:4409–4413. doi: 10.1210/jc.2003-031929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.