

Abstract
The eye is an immunologically privileged organ whose Ags serve as targets for experimental autoimmune uveitis (EAU), a model for human uveitis. We used a hydrodynamic i.v. injection of naked DNA to express the uveitogenic retinal Ag interphotoreceptor retinoid-binding protein (IRBP) in the periphery, thus revoking its immune-privileged status. IRBP was expressed in the liver within hours of administration of as little as 10 μg of IRBP-DNA. Vaccinated mice were highly protected from EAU induced by immunization with IRBP for at least 10 wk after vaccination. Protection was partial in a reversal protocol. Mechanistic studies revealed specific hyporesponsiveness to IRBP without immune deviation, no evidence for apoptosis either by the Fas- or Bcl-2- regulated (mitochondrial) pathway and apparent lack of dependence on CD8+ cells, IL-10, or TGF-β. In contrast, depletion of CD25+ cells after vaccination and before challenge markedly abrogated protection. IRBP-specific CD4+CD25high T cells could be cultured from vaccinated mice and transferred protection to unvaccinated, EAU-challenged recipients. In vitro characterization of these cells revealed that they are Ag specific, anergic, express FoxP3, CTLA-4, and glucocorticoid-induced TNFR, and suppress by contact. Thus, expression of IRBP in the periphery by DNA vaccination results in tolerance that acts at least in part through induction of IRBP-specific, FoxP3+CD4+CD25+ regulatory T cells. DNA vaccination may offer a new approach to Ag-specific therapy of uveitis.
Experimental autoimmune uveitis (EAU),3 induced in animals by immunization with uveitogenic retinal proteins or their fragments, serves as a model for human uveitis (1). Uveitis is a potentially blinding disease in which patients frequently exhibit lymphocyte proliferative responses to retinal Ags. The eye is an immunologically privileged organ and retina-specific Ags are separated from the immune system by an efficient blood-organ barrier. Although central tolerance helps lower the threshold for autoimmunity to retinal Ags in susceptible individuals (2), peripheral tolerance to these Ags is likely to be deficient, due to their sequestered nature. This can explain the persistence in healthy animals and humans of naive retinal Ag-specific T cells that have escaped thymic deletion, and can be triggered to become uveitogenic effector T cells by chance exposure to a retinal or a cross-reactive Ag. The involvement of retinal Ags in driving human uveitis is supported by the observation that HLA class II-transgenic (Tg) mice, which present and recognize Ag on human MHC molecules, develop typical EAU pathology and respond to a uveitogenic epitope of retinal arrestin (retinal S-Ag) that is immunodominant in uveitis patients (3).
EAU induced by immunization with interphotoreceptor retinoid- binding protein (IRBP) in CFA is a Th1/Th17-driven disease in which both types of effectors are induced in parallel and both infiltrate the eyes (Ref. 4 and D. Luger, P. B. Silver, J. Tang, D. Cua, Z. Chen, Y. Iwakura, E. P. Bowman, N. M. Sgambellone, C. C. Chan, and R. R. Caspi, submitted for publication). From adoptive transfer and Ab neutralization studies, it appears that both responses are important for pathogenesis and affecting either changes the outcome of the disease (D. Luger, P. B. Silver, J. Tang, D. Cua, Z. Chen, Y. Iwakura, E. P. Bowman, N. M. Sgambellone, C. C. Chan, and R. R. Caspi, submitted for publication).
Uveitogenic T cells specific to retinal Ags are necessary and sufficient to induce EAU, making them an attractive therapeutic target. Increasingly specific therapeutic approaches to target putative uveitogenic T cells in patients are being devised, including cyclosporin A and humanized anti-IL-2R Abs (5, 6). However, therapies that target activated T cells in general may at the same time inhibit host resistance to infectious pathogens. Ag-specific therapies targeting the T cells believed to be driving the disease could provide an optimal solution. Positive prospects for Ag-specific therapy of clinical uveitis are further underscored by the results of a double-masked placebo-controlled clinical trial, where uveitis in patients who had positive responses to S-Ag and received tolerogenic oral therapy with this protein could be weaned off their conventional therapy without experiencing a relapse (7).
A significant issue in devising Ag-specific therapies for uveitis is the availability of the Ag. Native Ag is available in limiting amounts, as it must be extracted from bovine retinal tissue. Indeed, this was a significant constraint in the uveitis oral tolerance clinical trial mentioned above. No less importantly, use of components derived from the CNS of cattle is now limited by the increasing prevalence of bovine spongiform encephalopathy (“mad cow disease”) that is transmissible to humans. rAgs, which can be made in relatively large quantities in bacteria, do not have the same immunogenicity as native Ags due to posttranslational modifications that affect Ag processing and may, or may not, be efficient tolerogens (8). In addition, the cost to produce grams of protein per patient may be prohibitive.
Recently, we developed an approach using autologous B cells retrovirally transduced with an IRBP fragment to efficiently protect from, and reverse, EAU (9). This served as proof of concept that genetic therapy with Ag expressed in the periphery by appropriate tolerogenic APC can provide a feasible approach to therapy. However, viral vectors can be immunogenic and present their own hazards and the need for large numbers of autologous B cells can be limiting.
In vivo transduction with naked DNA has the potential to overcome all the limitations listed above. DNA vaccines encoding autoantigens and delivered by multiple i.m. injections, with or without a coadministered regulatory cytokine, have been used to elicit protection in models of autoimmune diabetes, encephalomyelitis, and arthritis (10 –14). In this study, we report a new approach to protective DNA vaccination using the model of EAU, in which DNA vaccination has not been explored previously. Vaccination is accomplished by hydrodynamic delivery, i.e., rapid injection of an expression plasmid encoding the protein of interest in a large volume through the tail vein, which is a highly efficient method of achieving expression of Tg proteins in vivo (15). Protective tolerance in the EAU model is induced after a single administration of as little as 10 μg of plasmid DNA, without the need for coadministration of a deviating cytokine. The data suggest that tolerance is long lived, and is mediated at least in part by regulatory T cells (T-regs).
Materials and Methods
Mice
B10.RIII mice (H-2r) were purchased from The Jackson Laboratory. Day 28-thymectomized B10.RIII mice from The Jackson Laboratory were used in some experiments. Bcl-2lck-Tg mice, which express the human Bcl-2 transgene under the lck promoter, developed by Dr. S. Korsmeyer (Howard Hughes Medical Institute, Harvard Medical School, Boston, MA) on the C3H background were bred in-house. These were mated to wild-type (WT) B10.RIII to obtain (Bcl-2lckC3H × B10.RIII)F1 hybrids. B10.RIII mice homozygous for the lpr mutation (B10.RIIIlpr/lpr) were derived by backcrossing C57BL/6lpr/lpr mice onto the B10.RIII background for 10 generations and were bred in-house. All mice were used between the ages of 10–12 wk. Treatment of animals was in compliance with institutional guidelines and all animal study protocols were approved by an institutional review board.
Antigens
Human p161–180 (sequence SGIPYIISYLHPGNTILHVD), encoding a major pathogenic epitope present in the first homologous repeat of IRBP, was synthesized on a peptide synthesizer (model 432A; Applied Biosystems) using F-moc chemistry. Peptide 161–180 encodes a dominant pathogenic IRBP epitope for the H-2r haplotype (9, 16).
IRBP-DNA construct
Human repeat 1 of IRBP (which includes the 161–180 epitope) was PCR amplified from the EcR1 plasmid (17) using proof reading Pfu enzyme (Stratagene). The primers were: forward, 5′-CACCATGGGCCCCACACACCTGTT-3′, and reverse 5′-GAATTCCTAGCGCAGAGTGAGGATGGCCAGGG-3′. During PCR, start (ATG) and stop (TAG) codons were inserted in-frame with open reading frame of human repeat 1 at 5′ and 3′, respectively. The resulting 1-kb PCR fragment was cloned into a shuttle vector (pcDNA3.1 Directional Topo Expression kit; Invitrogen Life Technologies). After confirming the orientation by restriction digest, a 1-kb HindIII/XhoI fragment was cloned into pcDNA 3.1+ vector (Invitrogen Life Technologies) that contains a CMV promoter, an SV40 splice site, a polyadenylation signal, and an ampicillin-resistance gene. A positive clone, confirmed by restriction mapping, is referred to as “IRBP DNA” and the control as “Vector DNA” (pcDNA3.1 without an insert). To confirm the expression of IRBP, COS-7 cells (American Type Culture Collection) were transfected with IRBP or vector DNA constructs with the aid of lipofectamine (Invitrogen Life Technologies). Expression of protein was confirmed by Western blotting using monoclonal anti-IRBP Ab clone H3B5, which was donated by Dr. L. Donoso (Wills Eye Hospital, Philadelphia, PA) (18). Milligram quantities of both IRBP and vector DNA constructs were prepared by the Qiagen Endotoxin Free Mega kit. Concentration of DNA was determined by OD260 nm on a UV spectrophotometer (Molecular Devices).
Vaccination
Mice were vaccinated using the hydrodynamic protocol (15). Each 22-g mouse was injected rapidly (<7 s) in the tail vein with 10–40 μg of IRBP DNA or vector DNA diluted in 2.0 ml of Ringer’s solution. In some experiments where maximal expression of IRBP was desired, mice were given 75–200 μg DNA, as specified.
Western blotting
B10.RIII mice were vaccinated with 10–40 μg of IRBP DNA or injected with vector DNA by the hydrodynamic protocol described above. Liver, spleen, kidneys, and lungs were harvested from each mouse 8, 12, 24, and 50 h later and frozen until processing. Tissue was homogenized through a 21-g needle at 4°C in sodium phosphate buffer containing protease inhibitors, incubated on ice for 30 min, and centrifuged. Supernatants were collected and quantified using the Coomassie Plus Bradford assay kit (Pierce). A total of 20 μg of protein was resolved on a 10% SDS-PAGE gel (Invitrogen Life Technologies) and transferred to a nitrocellulose membrane (Whatman) using a semidry transfer cell apparatus (Bio-Rad). The membrane was blocked for 2 h in 5% nonfat dry milk (Santa Cruz Biotechnology) prepared in 0.05% Tween 20/TBS (TTBS; Quality Biological). The membrane was probed overnight at 4°C with anti-human IRBP mAb H3B5 (18) diluted 1/2,500 in 5% milk/TTBS, washed in TTBS, and then incubated for 2 h with goat anti-mouse IgG-HRP (Southern Biotechnology Associates) diluted 1/10,000 in 5% milk/TTBS. After washing again, the membrane was developed using enhanced chemiluminescence (ECL plus) Western blotting detection system (Amersham Biosciences) following the manufacturer’s instructions. Immediately afterward, the membrane was washed, blocked again, then reprobed overnight at 4°C with actin Ab (Santa Cruz Biotechnology) diluted 1/3,000 in 5% milk/TTBS. Following washing, the membrane was incubated with goat anti-rabbit IgG (H plus L) HRP-F(ab′)2 (Zymed Laboratories) diluted 1/50,000 in 5% milk/TTBS for 2 h, washed again, and developed by the chemiluminescence detection protocol.
Immunization, EAU induction, and EAU scoring
Mice were immunized subcutaneously in the thighs and base of the tail with 5–75 μg of p161–180. The Ag was emulsified in 0.2 ml in CFA supplemented to 2.5 mg/ml with Mycobacterium tuberculosis strain H37RA. In most experiments, mice were immunized 14 days after vaccination. To determine the effect on efferent-stage disease, mice were vaccinated 1 and 2 wk after immunization. For the long-term protection experiment, mice were immunized 10 wk after vaccination. Eyes were harvested for histopathology 21 days after immunization. In some experiments, EAU was induced by the adoptive transfer of a long-term CD4+ p161–180-specific Th1 cell line derived from primed IRBP-immunized B10.RIII mice. Cells were maintained essentially as described (16) by alternating cycles of stimulation with Ag on syngeneic APC and expansion in IL-2-containing medium. Freshly stimulated cells were injected i.p. into recipient mice and eyes were evaluated for disease 10 days later. Disease severity was scored in a masked fashion by one of us, who is an ophthalmic pathologist (C.-C. Chan) on a scale of 0 (no disease) to 4 (maximum disease) in half-point increments, according to a semiquantitative system described previously (19). For in vitro suppression assays, some B10.RIII mice were immunized with 50 μg of OVA in CFA for the purpose of generating OVA-primed lymph node (LN) cells.
In vivo Ab treatment
For depletion of CD8+ cells, adult-thymectomized B10.RIII mice were injected i.p. with mAb YTS169 (rat IgG2b) or SFR8-B6 mAb (specific to an epitope of HLA-Bw) as an isotype-matched control. Both Abs were purified from ascites (Harlan Bioproducts for Science). Mice received three treatments of 1.0 mg of Ab 3 days apart starting on vaccination day and were immunized 14 days after vaccination. In a pilot experiment, a fourth dose of Ab treatment resulted in lethality (possibly as a result of anaphylaxis) therefore, three Ab doses were used in subsequent experiments. Flow cytometry (see ahead) showed that treatment reduced CD8+ T cells in the spleen from 10.5 to 1.5% on average, a reduction of 86%.
To determine whether CD25+ cells were induced by vaccination, B10.RIII mice were vaccinated and were then depleted of CD25+ cells by two i.p. injections of 1.0 mg of the anti-CD25+ mAb PC61 (Harlan Bio-products for Science) given 7 and 10 days after vaccination, essentially as described by others (20). Flow cytometry (see ahead) confirmed that up to 75% of CD4+CD25+ T cells in the spleen were depleted by the treatment. Mice in these experiments were immunized 17 days after vaccination.
Anti- IL-10 (mAb JES 2A5; BioExpress) and anti-TGF-β1, -β2, -β3 (mAb 1D11, BioExpress) were administered to B10.RIII mice i.p. at 0.5 mg/mouse every other day. This regimen has been demonstrated to be an effective neutralizing dose by others for the particular Abs (21, 22). Two protocols were followed: 1) neutralization during the vaccination process in which mice began treatment one day before vaccination and were immunized 15 days later, or, 2) neutralization after the vaccination process in which mice began treatment 13 days after vaccination and were immunized one day later. A rat IgG1 (Y-13; American Type Culture Collection) served as the isotype control.
Determination of immunological responses
For Ag-specific proliferation in primary cultures, individual spleens were collected 3 wk after immunization. Triplicate 0.2-ml cultures containing 5 × 105 cells were seeded in round-bottom 96-well plates in HL-1 medium (Cambrex) supplemented with 2 mM glutamine, 100 U/ml penicillin-streptomycin (Invitrogen Life Technologies), and containing 125 μg/ml p161– 180 or 5.0 μg/ml purified protein derivative of tuberculin (PPD; Statens Serum Institute) as stimulants. To examine reversal of anergy, recombinant human IL-2 (5, 10, or 20 U; Proleukin; Midwest Medical) was added to parallel cultures at the time of initiation of the culture together with the Ag, or 24 h before adding the Ag. After 48-h stimulation with Ag, [3H]thymidine (1 μCi/well) was added for 18 h. The cultures were harvested on a PhD harvester (Cambridge Technology) and counted by liquid scintillation (PerkinElmer). The data are shown as Δ cpm (Δ cpm = mean cpm in cultures with Ag, minus the mean cpm in control cultures without Ag). Ag-specific cell frequency was assayed by ELISPOT for IL-2- and IFN- γ-producing cells in pooled LNs essentially as described (23). Spots were counted using the ImmunoSpot series 2.0 analyzer (CTL, Inc.). For cytokine analysis in supernatants, 0.2-ml cultures containing 1.0 × 106 cells were seeded in flat-bottom 96-well plates containing 125 μg/ml p161–180. Supernatants collected 48 h later were assayed for selected cytokines by Pierce multiplex SearchLight Arrays technology (Ref. 24 and www.SearchLightOnline.com). Anti-p161–180 IgG1 and IgG2a serum Ab isotypes were measured on peptide-coated ELISA plates essentially as described previously (25). The amount of each isotype was estimated from standard curves constructed by coating wells with the same goat antimouse IgG1 or goat anti-mouse IgG2a Abs and adding dilutions of Ig standards of the pertinent isotype.
Isolation and culture of CD4+ CD25high T cells
LN and spleen cells were collected from IRBP-DNA-vaccinated or nonvaccinated B10.RIII mice. CD4+ T cells were pre-enriched by negative selection using magnetic beads (CD4+ T cell isolation kit; Miltenyi Biotec). The enriched population was then stained with CD4+ PE/Cy5 (eBioscience) and CD25+ PE (clone PC61.5; eBioscience) and CD25high T cells were sorted on a FACS Aria (BD Biosciences) to 99.6%purity. Culture conditions were as described (26). Briefly, 4 × 105 T cells were seeded in each well of a 24-well plate in RPMI 1640 medium (HyClone) containing 10% FCS (HyClone), 10 ng/ml recombinant human IL-2 (PeproTech), antibiotics and other supplements as described (26). To each well was added irradiated (8000 rad) 4×106 syngeneic splenocytes and irradiated 2 × 105 bone marrow-derived dendritic cells cultured as described (27) and pulsed for 24 h with 50 μg/ml p161–180. Wells were fed with fresh medium every other day and cells were split as needed. Stimulations were performed weekly for 3 wk, with the fourth week a rest week.
In vitro characterization of T-reg function
Experiments to determine Ag specificity, suppressive activity, and contact dependency of the CD4+CD25high T cells were conducted using proliferation assays; 0.2-ml cultures were seeded in round-bottom 96-well plates containing RPMI 1640 medium supplemented as described above for CD4+CD25high T cell expansion, but with 4% fresh-frozen normal mouse serum instead of FCS. After 48 h, cultures were pulsed with [3H]thymidine for 18 h and then harvested. Data are presented as cpm.
For determination of Ag specificity of the CD4+CD25high T cells, 2.5 × 105 cultured CD4+CD25high T cells and 5 × 105 3000 rad irradiated syngeneic splenocytes as APC were cocultured with or without 125 μg/ml p161–180 as stimulant and with or without 10 ng/ml IL-2. To examine whether the CD4+CD25high T cells could suppress the proliferation of Ag-specific effector cells, 5.0 × 105 LN cells (responders), harvested 14 days after priming mice with 25 μg of p161–180 or 50 μg of OVA, were cocultured with 2.5 × 105 CD4+CD25high T cells in the presence of 125 μg/ml of p161–180 or 15 μg/ml OVA.
To investigate whether the CD4+CD25high T cells depend on cell-cell contact or a soluble factor to exert their suppressive function, 5.0 × 105 p161–180 responder cells (generated as described above) were seeded alone or together with 5.0 × 105 or 2.5 × 105 cultured CD4+CD25high T cells with or without 125 μg/ml p161–180 Ag (phase 1). After 48 h, supernatant from each culture condition was collected, centrifuged at 10,000 rpm two times and 100 μl was added to a 100-μl suspension of fresh p161–180 responder cells with peptide stimulant (phase 2) and assessed for proliferation. Additional wells from phase 1 were assayed for proliferation to ensure that the supernatant came from cultures in which suppression of responders by the CD4+CD25high T cells had occurred. In some experiments, 10–20 μg/ml anti-IL-10 mAb (mAb JES5-16E3; BD Pharmingen) and/or anti-TGF-β (clone 1D11; BioExpress), or their isotype controls; rat IgG2b and rat IgG1, respectively (BD Biosciences) were added to cocultures to determine whether these cytokines played a role in the suppressive activity. Lack of toxicity at this concentration of Ab was checked by adding the respective Abs or their isotypes to responder cells stimulated with anti- CD3/CD28 (anti-CD3-coated plates (BD Biosciences) in the presence of 2.5 μg/ml anti-CD28 (clone 37.51, purified ascites; Harlan Bioproducts)). Neutralization of IL-10 and TGF-β by the mAbs was verified by using multiplex ELISA technology (Pierce) to assay control samples containing the cytokines ± Abs.
Adoptive transfer of suppression
A total of 3 × 106 CD4+CD25high T cells, isolated by flow sorting and cultured as described above, were injected i.v. into recipients on 3 successive days. Mice were immunized 3 h after the first cell transfer. Alternatively, 5.0 × 106 cultured CD4+CD25high T cells were adoptively transferred once and animals were immunized 3 h later. Control mice did not receive cells. Eyes were evaluated by histology 21 days after immunization.
Flow cytometry
Flow cytometric analysis was performed on freshly explanted spleen cells of animals that had been treated with anti-CD25 Ab (PC61) or anti-CD8 Ab (YTS169) 1 day before uveitogenic immunization, essentially as described previously (28). Live cells were gated and 50,000 cells were analyzed using a FACSCalibur flow cytometer and CellQuest software (BD Biosciences). For determination of residual CD25+ cells, splenocytes were double-stained with anti-CD4-FITC and 7D4-PE (which binds to a different IL-2R epitope than PC61) and the percent of CD4+7D4+ cells of total CD4+ cells was calculated. For determination of residual CD8+ cells, splenocytes were double-stained with anti-CD4-FITC and anti-CD3-PE (BD Pharmingen) and the percent of CD3+CD4− cells of total CD3+ cells was calculated.
Flow cytometric analysis was performed on cultured CD4+CD25high T cells that were freshly stimulated overnight or 7–10 days after stimulation, as specified. Intracellular Foxp3+ staining was performed using the Foxp3+ staining set (Foxp3+-allophycocyanin; eBioscience) per the manufacturer’s protocol. Abs for surface marker staining included: CCR9- FITC, CCR7-PE. CD103-PE, CD25-FITC, glucocorticoid-induced TNFR (GITR)-PE, CTLA-4-PE. Mouse IgG2a-FITC, rat IgG1-PE, rat IgG2b-PE, rat IgG2a-allophycocyanin were all obtained from eBioscience. CD69-PE, TCRβ-PE, TCRβ-FITC, CD4+-PerCP-Cy5.5, hamster Ig-PE, and rat IgG2a- PE were all obtained from BD Pharmingen.
Reproducibility, data presentation, and statistical analysis
Experiments were repeated at least twice, and usually three or more times. Results were highly reproducible. Results are represented as averages of individually assayed mice. Statistical analysis of EAU scores was done using Snedecor and Cochran z test for linear trend in proportions (non-parametric, frequency based) (29). Each mouse (average of both eyes) was treated as a statistical event. Lymphocyte proliferation, cytokine, and ELISPOT data were analyzed by independent t test. Values of p < 0.05 were considered significant.
Results
IRBP protein is present in the liver within 8 h of hydrodynamic administration of IRBP DNA
Delivery of plasmid DNA to animal tissues using the hydrodynamic protocol is a highly efficient method of achieving sustained expression of Tg proteins in vivo (15). Its success is based on a rapid infusion of a large volume of vehicle containing the DNA of interest into the tail vein, which temporarily overcomes cardiac output, “backflushing” the DNA into the internal organs. The structure of the IRBP-DNA construct used in this study is described in Materials and Methods and is depicted schematically in Fig. 1a. Note that the construct has been designed without a secretion signal. We reasoned that having immunoreactive protein secreted into the circulation for a prolonged period of time might pose a problem because of the potential for anaphylaxis (30, 31) and/or deposition of immune complexes, especially in existing disease, if there are circulating Abs.
FIGURE 1.

Immunoreactive IRBP is present in the liver 8 h after vaccination. a, Diagram of IRBP-DNA plasmid. b, B10.RIII mice were injected with 40 μg of IRBP DNA or vector DNA. The liver was collected at 8, 12, 24, and 50 h time points. Tissue extracts were resolved by SDS-PAGE and probed for IRBP expression and actin by Western blotting. The control is recombinant human IRBP repeat 1 produced in bacteria. (The slightly higher molecular mass of the bacterial product is due to the presence of seven additional amino acids at the N terminus and a six-residue His tag at the C terminus (17), which are not present in the IRBP-DNA construct). Shown is one representative experiment of two.
It is important to point out that we have not observed deleterious systemic effects in the mice after the hydrodynamic procedure. Liu et al. (32) conducted multiple biochemical assays and growth studies on mice that had undergone hydrodynamic transfection to determine whether the procedure caused long term toxic effects. Their tests showed that all the results were in the normal range.
B10.RIII mice were vaccinated with the IRBP-DNA plasmid encoding the first repeat of the human IRBP molecule using the hydrodynamic protocol. Control mice were similarly injected with the empty vector. The liver, spleen, kidneys, and lungs were harvested at various time intervals, the tissue extract of each individual organ was resolved by SDS-PAGE, and probed by Western blotting with monoclonal H3B5 Ab. Recombinant first IRBP repeat which was produced in bacteria (17) was used as the positive control. IRBP protein product was detected in the liver 8 h after vaccine delivery. Expression in the tissue gradually decreased through the 24-h time point and became undetectable at 50 h. (Fig. 1b). No IRBP band was detected in the vector-injected mice. IRBP protein expression in spleen, kidney, and lung tissues from the IRBP-DNA-vaccinated mice was below the limit of detection.
The IRBP-DNA vaccine effectively protects mice from active induction of EAU, but only partly reverses disease induced by primed T cells
B10.RIII mice were vaccinated with IRBP DNA or received vector DNA, and 2 wk later were challenged for EAU by active immunization with the immunodominant IRBP p161–180, which is contained in the transgene. EAU histology of eyes collected 21 days after immunization showed that mice vaccinated with as little as 10 μg of IRBP DNA had a greatly reduced disease incidence and severity compared with vector-injected animals (p < 3 × 10−4, Fig. 2a). Eyes of vector-injected mice showed influx of inflammatory cells, subretinal hemorrhage, and damage to the photoreceptor cells in the retina as depicted in Fig. 2c (disease score 1.5 = moderate) and Fig. 2b (disease score 3 = severe). In contrast, most of the vaccine-protected animals retained a normal retinal architecture (Fig. 2d). Doses of IRBP DNA up to 100 μg resulted in a similar level of protection (see Fig. 6b, ahead, and data not shown).
FIGURE 2.
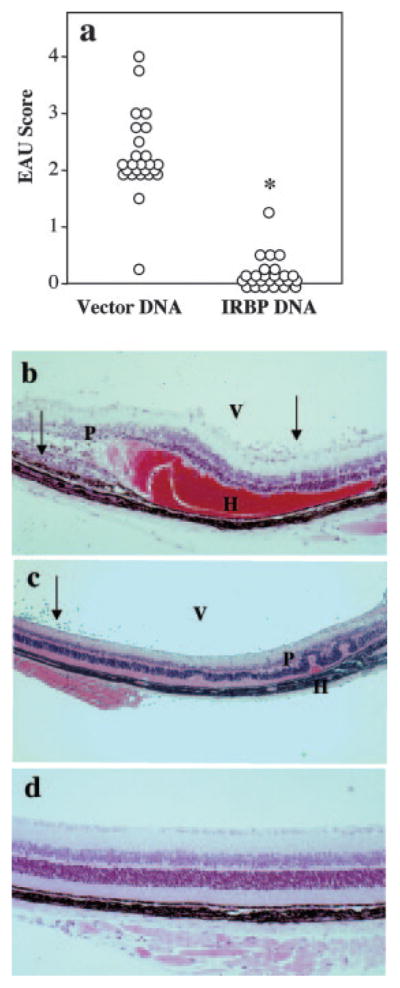
Hydrodynamic vaccination with an IRBP-DNA plasmid protects from EAU. a, EAU scores. B10.RIII mice were vaccinated with 10 μg of IRBP DNA or received 10 μg of vector DNA. Mice were challenged for EAU with 12 μg of p161–180 2 wk later and evaluated by histology after 21 days. Each circle represents the average EAU score of both eyes from one mouse (*, p < 3 × 10−4). b, Histology of eye from a mouse injected with vector DNA; EAU score of 3. Note inflammatory cells (arrows) in the vitreous (V) and retina; damage to the photoreceptor layer (P); and extensive subretinal hemorrhage (H). c, Histology of eye from a mouse injected with vector DNA; EAU score of 1–2. Note inflammatory cells (arrows) in the vitreous (V), damage to the photoreceptor layer (P), and small subretinal hemorrhage (H). d, Histology of eye from a mouse vaccinated with IRBP DNA shows normal retinal architecture, typical of a healthy eye.
We next tested whether IRBP-DNA vaccination can reverse an ongoing disease process. First, EAU was induced by the adoptive transfer of effector cells from a long-term uveitogenic Th1 cell line specific to p161–180 that (depending on the number of cells injected) can induce severe EAU in naive recipients within 4–6 days after infusion. B10.RIII mice were vaccinated with IRBP DNA or injected with vector DNA, and were infused i.p. 12 days later with freshly activated pathogenic Th1 line cells. Results showed that severity of EAU induced by the line (Fig. 3a) was reduced to about half (p < 0.04). In a second approach, EAU was induced by active immunization and the vaccine was administered 7 and 14 days later. At the 7-day time point, uveitogenic effector cells have already been primed and can transfer EAU (9). Again, a partial but significant reversal of disease was observed (p < 0.024, Fig. 3b). An additional vaccination on day 9 did not substantially improve protection (p < 0.017, data not shown). These results suggest that although the regulatory mechanisms elicited by IRBP-DNA vaccination are able to significantly inhibit an ongoing disease process, they target the priming of new effector T cells more efficiently than the functionally mature effector.
FIGURE 3.

Hydrodynamic vaccination of B10.RIII mice with IRBP DNA partly reverses an ongoing disease process. Bars shown represent EAU scores ± SE. EAU incidence (positive/total) is shown inside each bar. a, Mice were vaccinated with 100 μg of IRBP DNA or injected with vector twice 1 wk apart, then injected i.p. 12 days later with 1 × 105 p161–180-specific T cell line (*, p < 0.04). EAU was scored 10 days after cell transfer. b, Mice were immunized with 14 μg of p161–180 and vaccinated with 100 μg of IRBP DNA or injected with vector on days 7 and 14 after immunization. EAU was scored on day 21 (*, p < 0.024). Data represent scores from two independent experiments.
IRBP-DNA-vaccinated mice have reduced immunological responses, but no immune deviation
B10.RIII mice were given a hydrodynamic injection of IRBP DNA or vector DNA, and were challenged for EAU by immunization with p161–180 2 wk later. Cellular responses to p161–180 were examined 21 days after immunization in individual animals. The data showed that mice vaccinated with as little as 10 μg of IRBP DNA had significantly lower proliferative responses compared with those of animals injected with vector DNA (p < 0.001, Fig. 4a). Vaccination affected only responses to IRBP. Responses to PPD, to which the mice develop immunity because it is a component of CFA, were unaltered (Fig. 4a). The reduced proliferative responses to IRBP were not restored by adding IL-2 to the cultures, starting either at the time of, or 24 h preceding, antigenic stimulation (data not shown).
FIGURE 4.

Cellular responses to the immunodominant peptide 161–180 are decreased in IRBP-DNA-vaccinated mice. B10.RIII mice were vaccinated with 10 μg of IRBP DNA or received 10 μg of vector DNA. Mice were challenged for EAU with 12 μg of p161–180 2 wk later. Assays were conducted 21 days after challenge. a, Proliferation to IRBP (*, p < 0.001) and PPD. Data are presented as Δ cpm ± SE of triplicate cultures. Results are averaged from 15 individually assayed mice from three separate experiments. b, ELISPOT. Pooled LNs were assayed for frequency of IL-2- and IFN-γ-producing cells (*, p < 0.04). Bars represent the mean ± SE of three experiments. Data are spots per million cells. c, Cytokine secretion. Bars represent the mean level of cytokine ± SE in supernatants (after subtraction of unstimulated control) of 15 individually assayed spleens from three experiments (*, range p < 0.0004 to p < 0.027).
Spleens were assayed for production of cytokines representing both Th1 and Th2 components of the immune response. All the tested cytokines were significantly reduced in the IRBP-DNA-vaccinated mice compared with the vector-injected mice (range, p < 0.0004 to p < 0.027; Fig. 4c). It was not possible to use the ratio of IgG1 to IgG2a Ab isotypes as a criterion for deviation, because neither the vaccinated nor the control mice produced detectable Ab titers to p161–180 within this timeframe. Frequency of Ag-specific cells was determined by ELISPOT in pooled LNs (Fig. 4b). Fewer IL-2-secreting cells as well as significantly less IFN-γ-secreting cells were present in the IRBP-DNA-vaccinated mice. These results are compatible with the interpretation that IRBP-DNA vaccine elicits hyporesponsiveness to Ag, but without evidence for immune deviation or for an IL-2-reversible form of anergy.
Protection by hydrodynamic IRBP-DNA vaccination is long lived
We next wanted to determine the length of protection afforded by IRBP-DNA vaccination. B10.RIII mice were vaccinated with IRBP DNA or vector DNA, and were rested for 10 wk after vaccination. At the end of 10 wk, all mice were challenged for EAU by immunization with p161–180. Histology of eyes collected 21 days after immunization revealed that vaccinated mice were still well protected, i.e., disease incidence as well as EAU severity were substantially decreased compared with the vector-injected animals (p < 4.0 × 10−4, Fig. 5). Thus, the protective effect persists and is about as robust as shown in Fig. 2, after a much shorter interval.
FIGURE 5.
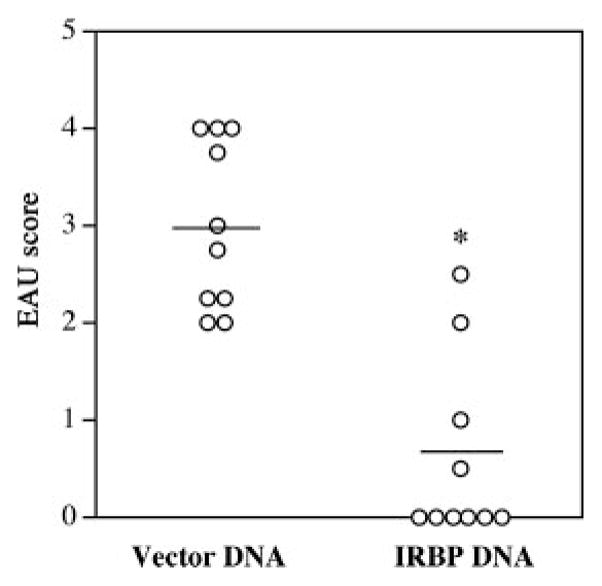
Mice vaccinated with IRBP DNA are still significantly protected from disease 10 wk after EAU challenge. B10.RIII mice were vaccinated with 10 μg of IRBP DNA or received 10 μg of vector DNA. Mice were challenged for EAU with 12 μg of p161–180 10 wk later. EAU was scored 21 days after challenge. Each dot represents the average score of both eyes from one mouse (*, p < 4.0 × 10−4). Data represents scores from two experiments. Horizontal line is the mean of the group.
Protection by IRBP-DNA vaccination does not involve IL-10, TGF-β, CD8+ cells, nor Bcl-2-dependent or Fas-regulated apoptosis
A series of experiments were conducted in an attempt to characterize the protection, that appeared to eliminate a number of mechanisms commonly associated with tolerance. Although analysis of Ag-specific responses did not reveal up-regulation of inhibitory cytokines, they could perhaps be induced in some local compartments. Because IL-10 and/or TGF-β have been associated with vaccination-induced tolerance (14, 33), we neutralized these cytokines in the mice either during, or, after the vaccination process using protocols shown by others to be effective (21, 22). Neither treatment was able to reverse the protective effects of the IRBP-DNA vaccination, failing to support a role for either of these cytokines in the mechanism of protection (data not shown).
To determine whether CD8+ cells played a role, thymectomized B10.RIII mice vaccinated with IRBP DNA or vector DNA were treated with three courses of depleting anti-CD8 Ab starting at the time of vaccination. Flow cytometry showed that CD8+ cells in the spleen were reduced by 86%. Animals were challenged for EAU by immunization with p161–180 2 wk after vaccination. Results showed that depletion of 86% of the CD8+ cell population, achieved by three 1-mg doses of anti-CD8 Ab, did not affect the ability of the vaccination to elicit efficient protection against EAU (Fig. 6a), suggesting that CD8+ cells were not necessary for the protective effect. We cannot formally exclude that a higher rate of depletion might have had some effect, however, an additional Ab dose resulted in toxicity. We next investigated whether the mechanism of protection involved apoptosis via the mitochondrial or the Fas pathways. Bcl-2lck-Tg mice (whose T cells have a defect in the mitochondrial pathway of apoptosis due to a human Bcl-2 transgene targeted to T cells by the lck promoter) were crossed to B10.RIII to obtain Tg F1 progeny. The cross was necessary for two reasons: 1) C3H mice express the rd (retinal degeneration) gene and have no photoreceptors by the time they are weaned, thereby precluding normal EAU development; 2) to develop EAU with p161– 180 we needed to retain the H-2r restriction elements. Sighted (Bcl- 2lckC3H × B10.RIII)F1 progeny were vaccinated with IRBP DNA or received vector DNA. Animals were challenged for EAU with p161– 180 2 wk after vaccination and were evaluated for disease 3 wk later. Both vector-injected Tg mice and their WT littermates developed EAU, and both were protected after IRBP-DNA vaccination, suggesting that the mitochondrial pathway does not play a major role in protection (Fig. 6b).
FIGURE 6.
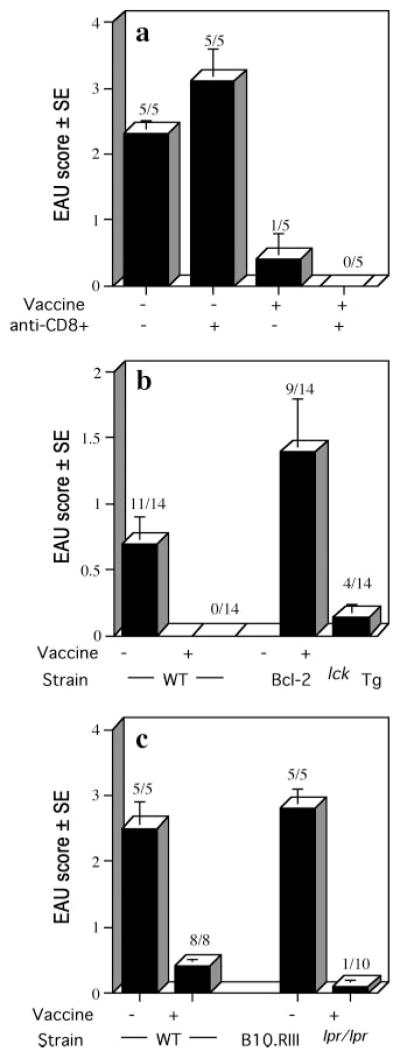
Protection by IRBP-DNA vaccination does not involve CD8+ cells nor Bcl-2- or Fas-regulated apoptosis. Bars shown represent the average EAU scores per group ± SE. Disease was scored 21 days after immunization. EAU incidence (positive/total) is shown above each bar. a, CD8+ cell depletion. Thymectomized B10.RIII mice were vaccinated with 10 μg of IRBP DNA or received 10 μg of vector DNA. Mice received three treatments of 1.0 mg of anti-CD8+ Ab or control Ig 3 days apart starting on vaccination day. Mice were challenged for EAU with 12 μg of p161–180 14 days after vaccination. b, Bcl-2lckTg (C3H × B10.RIII)F1 and WT littermates were vaccinated with 100 μg of IRBP DNA. Controls received 100 μg of vector DNA or were untreated. All mice were challenged for EAU with 70 μg of p161–180 2 wk later. Shown are average scores from two combined experiments. c, B10.RIIIlpr/lpr and WT B10.RIII mice were vaccinated with 20 or 40 μg of IRBP DNA or were untreated. Mice were challenged for EAU with 75 μg of p161–180 2 wk later. Shown are average scores from two combined experiments.
To examine the need for the Fas pathway of apoptosis, we bred the lpr gene onto the B10.RIII background. B10.RIIIllpr/lpr mutants and B10.RIII WT controls were vaccinated with IRBP DNA or received vector DNA, and were challenged for EAU with p161–180. Results showed that vector injected lpr as well as WT animals developed severe EAU, and both were protected to a similar extent by IRBP-DNA vaccination (Fig. 6c). These data suggest that Fas-dependent apoptosis does not play a major role in the mechanism of protection by the IRBP-DNA vaccine.
Depletion of CD25+ cells after IRBP-DNA vaccination partially reverses protection
To investigate a possible contribution of vaccination-induced regulatory cells to the mechanism of protection, B10.RIII mice were vaccinated with IRBP DNA or injected with vector DNA and 1 wk later were treated with PC61 Ab to deplete CD25+ cells. We reasoned that this protocol would deplete any T cells that responded to the Tg IRBP produced in the periphery as a result of IRBP-DNA vaccination. FACS analysis of representative animals showed that 75% of CD25+ T cells in the spleen were depleted by the treatment. The CD25-depleted and nondepleted animals were challenged for EAU by immunization with p161–180 10 days after depletion (= 17 days after vaccination). EAU scores of eyes collected on day 21 after immunization showed that all vector-injected mice, whether depleted or nondepleted, developed EAU. In contrast, IRBP-DNA-vaccinated mice that had been depleted of CD25+ cells showed partial, but statistically significant and reproducible, reversal of protection compared with their nondepleted control group. (p < 0.011, Fig. 7a). Individual EAU scores of animals that developed disease ranged from 1 to 4, similar to those of control (not vaccinated/nondepleted) animals. Consistent with this, examination of individual spleens for cellular responses revealed higher proliferation and significantly more IL-2-producing and IFN-γ-producing cells in CD25-depleted vs nondepleted IRBP-DNA-vaccinated mice (p < 0.03 and <0.001, respectively) (Fig. 7, b and c). In contrast, proliferation to PPD in the same mice was unaltered, demonstrating antigenic specificity of the vaccination-induced regulatory function (Fig. 7b). These results suggested that an Ag-specific, CD25+ regulatory cell population contributes to the protective effect of IRBP-DNA vaccination. The incomplete reversal of disease may reflect the fact that the PC61 treatment was unable to deplete all the CD4+CD25+ cells, or that T-reg independent pathway(s) not examined here also contribute to the mechanism of protection.
FIGURE 7.

Depletion of CD25 T cells after IRBP-DNA vaccination partially abrogates protection and partially reverses inhibition of Ag-specific cellular responses. B10.RIII mice were vaccinated with 10 μg of IRBP DNA or received 10 μg of vector DNA. Anti-CD25+ Ab was administered 7 and 10 days later. Mice were challenged for EAU with 12 μg of p161–180 17 days after vaccination. Histopathology and cellular responses were assayed 21 days after challenge. Shown are results from two separate experiments. a, EAU scores of mice vaccinated with IRBP DNA and depleted with anti-CD25+ Ab are significantly higher than nondepleted IRBP-DNA-vaccinated mice (***, p < 0.011). (Nonvaccinated/nondepleted vs vaccine/depleted, *, p < 0.023; nonvaccinated/depleted vs vaccine/depleted, **, p < 0.005). Each bar represents the average scores ± SE. EAU incidence (positive/total) is shown above each bar. b, Proliferation. IRBP-DNA-vaccinated and depleted mice have higher responses than nondepleted IRBP-DNA-vaccinated mice (*, p < 0.052). PPD proliferation of the same mice is similar among the groups. Data are presented as Δ cpm ± SE of triplicate cultures. Each bar represents the average of the responses of seven to nine individually assayed spleens from mice representative of the group. c, ELISPOT. IRBP-DNA-vaccinated and depleted mice had significantly higher frequency of IL-2- and IFN-γ-producing cells than nondepleted IRBP-DNA-vaccinated mice (*, p < 0.03, and p < 0.001, respectively). Data are spots per million cells. Each bar represents the average of the responses of seven to nine individually assayed spleens of mice representative of the group ± SE.
CD4+CD25high T cells cultured from IRBP-DNA-vaccinated donors express FoxP3, CTLA-4, and GITR, and protect recipients from EAU
If indeed regulatory cells are functionally involved in protection, their suppressive function should be transferable. To examine this, whole splenocytes from IRBP-DNA-vaccinated donors as a source of putative T-regs were cotransferred with an equal number of splenocytes from naive donors as a source of effector cells, into sublethally irradiated recipients using the protocol described by Bynoe et al. (34). However, the recipients were not protected from a subsequent EAU challenge (data not shown). Similarly, an immunomagnetically enriched population of CD4+CD25+ cells from IRBP-DNA-vaccinated animals was unable to transfer suppression (data not shown). This could have been due to a number of reasons: 1) the donors are WT mice with a polyclonal repertoire, and the frequency of IRBP-specific T-regs is low; 2) vaccination-induced T-regs may preferentially home to tissues other than spleen; 3) under conditions of homeostatic proliferation T-regs might lose their anergic and suppressive phenotype or might require expression of IRBP in liver for continued maintenance; 4) along with T-regs, vaccination might also prime effector cells that are kept in check in their original milieu, but might be released from suppression in a new host. It was therefore necessary to isolate only those CD4+CD25+ cells that had the highest expression of CD25, as these would be more likely to be enriched for regulatory cells (CD25high) (35). Because it was not feasible to isolate CD25high cells in sufficient number and purity to perform a direct transfer into recipients, the isolated CD4+CD25high cells were first expanded in culture. A total of 1.4 × 106 CD4+CD25high T cells were isolated from pooled spleens and LN cells of 10 IRBP-DNA-vaccinated donors by flow sorting. The cells expanded rapidly in culture with IRBP p161–180 under conditions described by Suffia et al. (26) consisting of weekly stimulation with p161–180 on bone marrow-derived dendritic cells in presence of IL-2. CD4+CD25high cells from unvaccinated donors failed to expand in culture.
Phenotypic characterization of the cultured cells by flow cytometry 7 days after stimulation revealed a phenotype typically associated with T-reg cells (36 –38): 90% of the CD4+ cells expressed FoxP3. Gating on FoxP3+ cells, 100% of the population also expressed CD25 and GITR, and 98.3% expressed CTLA-4. In addition, 60% of FoxP3+ cells expressed CD69, increasing to 88% 1 day after stimulation (data not shown). There was no expression of CD103, CCR7, and CCR9, chemokine receptors which have been reported to be associated with specific T-reg-homing patterns by others (Fig. 8a) (39, 40).
FIGURE 8.

CD4+CD25high T cells from IRBP-DNA-vaccinated donors express FoxP3, CTLA-4, and GITR and protect recipients from EAU after adoptive transfer. CD4+CD25high T cells were cultured as described in Materials and Methods. a, Flow cytometry. Cells 7 days after stimulation were stained for intracellular FoxP3 expression (90.4% of total CD4+ cells) and for the following additional surface markers (shown gated on the FoxP3 population): CD25 (100%); CTLA-4 (98.3%); GITR (100%); CD103 (1.2%); CCR7 (4.4%); CCR9 (10.2%); CD69 (57.8%). Open histogram, Specific Ab; solid histogram, isotype control. Shown is a representative experiment of two. b, Adoptive transfer. Left panel, A total of 3.0 × 106 cultured CD4+CD25high T cells were adoptively transferred iv to B10.RIII recipients on 3 successive days. Mice were immunized 3 h after the first cell transfer with 5.0 μg of p161–180 (*, p < 0.002). Right panel, A total of 5.0 × 106 cultured CD4+CD25high T cells were adoptively transferred one time and immunized with 7.0 μg of p161–180 3 h later. Disease was scored 3 wk after immunization (*, p < 0.05). Control mice did not receive cells. Bars shown represent the average EAU scores ± SE per group of recipients. EAU incidence (positive/total) is shown at the end of each bar.
To test suppressor function in vivo, the cultured CD25high T cells were adoptively transferred into naive recipients who were then challenged with IRBP p161–180. Recipients were significantly protected from EAU (Fig. 8b), confirming presence of functional regulatory cells in the CD4+CD25high population derived from IRBP-DNA-vaccinated mice. Protection could be achieved not only under conditions of moderate (Fig. 8b, Expt. 1, p < 0.002) but also under conditions of severe disease (Fig. 8b, Expt. 2, p < 0.05).
The CD4+CD25high T-regs from IRBP-DNA-vaccinated mice specifically respond to p161–180, are anergic, and exhibit Ag-specific and bystander suppressive activity
As mentioned, CD4+CD25high cells isolated from unvaccinated mice failed to expand in culture with p161–180 and APC, suggesting that the CD4+CD25high cells from IRBP-DNA-vaccinated donors were elicited and expanded in an Ag-specific fashion. To directly examine their Ag specificity, we used a standard lymphocyte proliferation assay, with or without added IL-2. In the presence of IL-2, CD25high T cells proliferated vigorously to APC plus p161–180, but not to APC alone, demonstrating Ag specificity. If IL-2 was omitted from the culture, the cells failed to proliferate, demonstrating behavior consistent with anergy typically associated with T regs (Fig. 9a).
FIGURE 9.

The CD4+CD25high T cells from IRBP-DNA-vaccinated mice are Ag specific and anergic. a, The CD4+CD25high T cells respond specifically to p161–180 Ag and do not proliferate in the absence of IL-2. A total of 2.5 × 105 cultured CD4+CD25high T cells and 5 × 105 irradiated splenocytes as APC were cultured with or without 125 μg/ml p161–180 as stimulant and with or without 10 ng/ml IL-2 (*, p < 3.5 × 10−5). Data are presented as cpm ± SE. Shown is one representative experiment of two. b, The CD4+CD25high T cells significantly suppress the proliferation of p161–180 responder cells. A total of 5.0 × 105 p161–180-primed LN cells (responders) were cultured alone or cocultured with 2.5 × 105 CD4+CD25high T cells with 125 μg/ml p161–180 as stimulant (*, p < 0.005). Data are presented as cpm ± SE of triplicate cultures. Shown is a representative experiment of two. c, The CD4+CD25high T cells exhibit bystander suppression. A total of 5.0 × 105 OVA-primed LN cells (responders) were cultured alone or cocultured with 2.5 × 105 CD4+CD25high T cells with 15 μg/ml OVA as stimulant in the presence or absence of p161–180 Ag. Data are presented as cpm ± SE of triplicate cultures (*, p < 0.001). Shown is a representative experiment of two.
To determine whether the CD4+CD25high cells can suppress proliferative responses of target cells in vitro, they were cocultured with p161–180-specific lymphocytes as responders and p161–180 peptide as stimulant. Although it would have been best to use naive target cells, as DNA vaccination preferentially targets naive T cells (Figs. 2 and 3) an IRBP TCR-Tg mouse is not yet available. Therefore, we generated primed Ag-specific T cells from peptide-challenged mice to serve as the responder population and cocultured them with the T-regs. Results showed that the CD25high T cells were able to significantly reduce the proliferative capability of the p161–180 responders (Fig. 9b, p < 0.005).
We next investigated whether the CD4+CD24high cells could also suppress the proliferation of cells specific to a different Ag. For these experiments, cocultures consisted of the CD4+CD25high cells plus OVA-primed LN cells as the responder population. Results showed that with OVA alone as stimulant, the responders proliferated in the presence of the T regs. However, in the presence of their specific Ag, p161–180 peptide, the T-regs were able to significantly suppress the proliferation of OVA-specific cells to the OVA (Fig. 9c, p < 0.001). These results further support the data that the CD4+CD25high cells are p161–180 specific for their function, and are consistent with studies reporting that once triggered through the TCR, the efferent phase of suppression is not Ag specific (41, 42).
Inhibition by the CD4+CD25high T cells does not require IL-10 or TGF-β but requires cell contact
T-regs can inhibit by direct contact and/or by soluble mediators, chiefly IL-10 and TGF-β (43). We therefore examined the requirements for suppression using responder cells from p161–180- primed mice, as above. Responder cells and CD4+CD25high T cells were cocultured in the presence of p161–180 ± anti-IL-10 and/or anti-TGF-β. Results showed that neutralization of these cytokines individually (Fig. 10a) or together (data not shown) did not alter suppressive function. These results support the in vivo treatments described above, in which blockade of IL-10 or TGF-β was unable to reverse protection by the IRBP-DNA vaccine.
FIGURE 10.

The CD4+CD25high T cells require cell:cell contact and not IL-10 or TGF-β to exert their suppressive activity. a, Cytokine neutralization. A total of 5.0 × 105 p161–180-primed LN cells (responders) were cultured alone or cocultured with 5.0 × 105 CD4+CD25high T cells with 125 μg/ml p161–180 as stimulant in the presence or absence of 20 μg/ml anti-IL-10 or anti-TGF-β. Data are presented as cpm ± SE of triplicate cultures (*, p < 0.02; **, p < 0.001; ***, p < 0.039). Shown is a representative experiment of two. b, Cell:cell contact dependency. A total of 5 × 105 or 2.5 × 105 CD4+CD25high T cells were seeded alone or cocultured with 5 × 105 p161–180-primed LN cells (responders) with p161–180 Ag (phase 1). After 48 h, supernatant from each culture condition was added to fresh p161–180 responder cells with peptide stimulant (phase 2). Additional wells from phase 1 and all phase 2 cultures were assayed for proliferation by [3H]thymidine uptake. Data are presented as cpm ± SE of triplicate cultures. Shown are results from two combined experiments.
To examine requirement for contact, CD4+CD25high T cells were cultured alone or with p161–180-specific responders with p161–180 Ag (phase 1, Fig. 10b). After 48 h, supernatants from each culture condition were transferred to fresh responder cells under stimulation conditions (phase 2). Although the CD4+CD25high T cells effectively inhibited responder proliferation in phase 1, the supernatants from these cocultures were unable to inhibit fresh responder cells in phase 2. Control cocultures in phase 2 demonstrated that the same fresh responder cells could be inhibited by fresh CD4+CD25high T cells. These results suggest that cell-cell contact rather than a soluble factor is critical for the suppressive effects of the CD4+CD25high T cells, in line with other studies that have reported that suppressive function of T regs is cell contact dependent (44, 45).
Discussion
In the present manuscript, we show that hydrodynamic injection of IRBP DNA affords a dramatic and long-lived protection from EAU induced by immunization with a major pathogenic epitope contained in the transgene. Ag-specific protective DNA vaccination has been attempted in a number of autoimmune disease models, with diverse approaches and even more diverse results. DNA vaccination by multiple i.m. injections of a plasmid encoding the autoantigen(s), with or without a deviating cytokine or an Ig fusion partner, has been performed in models of experimental autoimmune encephalomyelitis, type 1 diabetes, and arthritis. The results varied from protection to aggravation of disease (10 –14, 33, 46– 51). In those cases where protection was the outcome, diverse and seemingly opposing mechanisms were identified by different investigators, including elicitation of a regulatory Th2 response (11, 51), elicitation of a Th1 response (12), no change in the response (13, 46, 49), requirement or lack of requirement for a deviating cytokine to elicit protection (11, 12), induction of regulatory cytokines such as TGF-β or IFN-β, or IL-10 (13, 14, 33) or effects on costimulatory molecules (48).
Our study differs fundamentally from those other reports in that we used a hydrodynamic rather than i.m. delivery of the Ag-expression plasmid. The hydrodynamic protocol is an extremely efficient in vivo transduction method (15), requiring an order of magnitude less DNA compared with i.m. vaccination and only a single injection to elicit protective tolerance. In our hands, up to 7 i.m. injections of 100 μg of IRBP DNA are needed to achieve an equivalent level of protection from EAU to that seen after a single hydrodynamic injection of 10 μg of the same plasmid (P. B. Silver, unpublished results). Furthermore, while some investigators using i.m. DNA delivery found that covaccination with a deviating cytokine was necessary to achieve protection (11, 51), this was not required in the hydrodynamic protocol. The APC that are involved in tolerogenic Ag presentation after hydrodynamic DNA vaccination may differ from those involved in i.m. vaccination, which could have direct consequences on the mechanisms that are induced. These APC have not yet been identified, and are likely to include resident cells in several internal organs. In our hands, liver was the only organ in which IRBP was detectable by Western blotting, but others using a highly sensitive luciferase system detected expression also in kidney, spleen, lung, and heart (15). Notably, although expression of IRBP dropped below detection at 50 h, protection from EAU was still present 3 mo after vaccination. Because in the luciferase system, low-grade expression persists for >6 mo (15), it is conceivable that immunologically relevant expression of IRBP may have continued long after it was no longer detectable by Western blotting.
The DNA vaccination was more effective in prevention, than in reversal of an ongoing disease process, as represented by infusion of an effector T cell line or by delayed intervention after active immunization. This suggests that protection induced by hydrodynamic DNA transduction is more effective in preventing generation of effector T cells than in inhibiting their function after they had already been generated. However, because chronic autoimmunity is dependent on continuous recruitment of new Ag-specific T cells into the effector pool (52), a therapy that targets new effector T cell generation can be of considerable value even if its efficacy against established effectors is only partial.
Ag-specific proliferation and cytokine responses in protected mice revealed hyporesponsiveness across the board, but without evidence for immune deviation or overproduction of inhibitory cytokines that might account for the protection. Mechanistic data indicated that death of the effector cells is unlikely to be a major mechanism in the protection. Depletion of CD8 cells did not affect protection, speaking against the need for elimination of effector cells by the perforin/granzyme B pathway. Experiments with Bcl- 2-Tg mice and lpr mice, demonstrating that they could be protected as efficiently as the WT, would seem to largely exclude these two other major pathways of apoptosis. Our data do not exclude that anergy might play a role in the protection; however, we were unable to restore proliferation in vitro by adding IL-2.
In contrast, depletion of CD25+ cells after vaccination markedly abrogated protection and restored associated immunological responses. Furthermore, CD4+CD25high T cells could be isolated from vaccinated mice and expanded in the presence of p161–180. These cells were able to suppress EAU upon adoptive transfer into unvaccinated recipients. CD4+CD25high cells from unvaccinated mice could not be expanded with p161–180, suggesting that this particular T-reg population is specifically induced by the vaccine. These results lead to the conclusion that vaccination-induced CD25+ T-regs are involved in protection afforded by the IRBP vaccine, although they do not exclude additional mechanisms not tested here.
Phenotypic analysis of the CD4+CD25high cells revealed a typical pattern of molecules associated with T-regs that included FoxP3, CTLA-4, and GITR as well as CD69. Expression of these molecules has been associated with natural as well as induced T-regs (36 –38). Functional analysis in vitro revealed that, as expected of T-regs, they were Ag specific and anergic (required IL-2 to proliferate to p161–180). Furthermore, while they required their specific Ag for triggering, once activated they suppressed responder T cells specific to a different Ag in a bystander fashion. Finally, they appear to inhibit by cell-to-cell rather than by soluble mediators, including IL-10 and TGF-β. These data are consistent with earlier studies on Ag requirement and mode of action of T-regs (41, 42).
The lineage of these regulatory cells remains to be determined. One possibility is that they may be induced from CD4+CD25− T cells. The generation and/or function of “induced” T-regs has in many cases been associated with IL-10 and TGF-β (43, 53) and some DNA vaccination protocols were shown to be associated with induction of these cytokines (14, 33). However, in our hands, neutralization of IL-10 and TGF-β in vivo either during what would be the inductive phase or the expression phase of suppression did not reverse vaccination-induced protection. Furthermore, in vitro, these cytokines were not needed for suppressor function. This suggests that neither IL-10 nor TGF-β are required for generation or activity of the vaccination-induced T-regs. This is reminiscent of thymic-derived “natural” T-regs and some induced T-regs whose suppressive function is contact dependent (34, 54). In an experiment not shown here, depletion of CD25+ T cells before vaccination did not prevent development of protection, which might suggest that “natural” T-regs are not involved. However, depletion of CD25+ cells by PC61 is never complete, so it is difficult to exclude the possibility that a residual population of “natural” T-regs could have been expanded by vaccination.
Several types of regulatory cells have been described in connection to eye-related immune responses. In anterior chamber-associated immune deviation, elicited by injection of Ag into the anterior chamber of the eye, two types of regulatory cells are induced in the spleen: CD8+ cells are the efferent suppressors capable of inhibiting the function of effector T cells, whereas CD4+ regulators are afferent-acting and prevent effector cell priming, though neither has been well-characterized with regard to the mechanisms they use to effect suppression (55, 56). CD4+ efferent suppressors whose generation is dependent on the eye were described in EAU-recovered animals (57). The regulatory cells induced by vaccination appear to be most closely related to the afferent CD4+ anterior chamber-associated immune deviation-induced afferent T regs. Apparent lack of dependence on CD8+ cells and the limited efficiency of vaccination to inhibit EAU induced by mature effector cells are consistent with this, and further support the view that these T-regs are mostly afferent acting, and capable of inhibiting the priming of new uveitogenic T cells more efficiently than the function of already generated effector T cells.
In another study, we reported protection from EAU by another form of Ag-specific therapy, using B cells retrovirally transduced with a construct of p161–180 fused to mouse IgG1 H chain (9). Features common between B cell therapy and DNA vaccination are elicitation of hyporesponsiveness without evidence for immune deviation (9), and lack of an apparent need for apoptosis (R. K. Agarwal, manuscript in preparation). Unlike DNA vaccination, however, induction of T regs was not seen in B cell therapy (9). Also in contrast to DNA vaccination, B cell therapy was able to very efficiently reverse EAU induced by the same uveitogenic T cell line that was used here (R. K. Agarwal, D. W. Scott, and R. R. Caspi, unpublished observation). We hypothesize that these differences may stem from the different APC involved. In B cell therapy the transduced B cells themselves appear to be the major APC, as transduced class II-deficient B cells are unable to support induction of tolerance (58). In contrast, diverse cells may be serving as APC in the hydrodynamic protocol, including tissue-resident cells as well as migrant leukocytes. Processing and presentation of the Tg IRBP product might also be affected by the fact that we did not include a secretion signal in our construct, so that most of the protein would remain intracellular. How exactly the nonsecreted IRBP is presented for induction of tolerance is beyond the scope of the current study. Nevertheless, we hypothesize that transduced cells that undergo apoptosis during normal tissue turnover would be scavenged, and their IRBP would be processed and cross-presented by the scavenging cells. This might be an integral part of the mechanism of tolerance, as cross-presentation of Ags from ingested apoptotic cells in a noninflammatory milieu has been shown to be tolerogenic (59, 60). Alternatively, APCs could be directly transfected by plasmid DNA and present an endogenously produced Ag product in a tolerogenic fashion, which, however, we consider less likely in view of the CD8 independence of the tolerance.
In terms of clinical applicability, hydrodynamic injection can be considered as analogous to portal vein infusion. Although clearly not a trivial procedure, in severe disease, this approach is being used to deliver various therapeutic agents into liver tissue, including chemotherapeutic substances, gene constructs, and transplanted islets of Langerhans. Conventional i.v. administration of naked DNA is not practical, because most of the of DNA would be degraded in the general circulation before it reaches the tissues where it is to be expressed. However, encapsulation and stabilization technologies may well provide a solution to this in the not too distant future. It is tempting to speculate that, once the APC that are important for tolerogenesis in this approach are identified, it will be possible to direct delivery of DNA to these cells using specific Ab or other receptor-targeting technologies, thus maximizing transduction efficiency.
CD4+CD25+ T-regs are dependent on the presence of IL-2 in vivo for their maintenance and function, and a proven way to eliminate them experimentally is treatment with anti-IL-2R Abs (36, 43, 54). Paradoxically, a promising therapy, for some types of autoimmune uveitis, that is currently in clinical trials employs treatment with daclizumab, a humanized anti-IL-2R Ab (6, 61). The mechanism was hypothesized to be direct inhibition of CD25+ effector T cells, but appears to also involve induction of CD56bright regulatory NK cells (62). In view of the role of CD4+CD25+ T-regs in protective tolerance induced by DNA vaccination, these two treatment paradigms would seem to be incompatible.
In summary, we show that expression of a privileged retinal Ag in the periphery through hydrodynamic DNA vaccination affords an efficient and long-lasting protection from EAU. The protection appears to target the induction more efficiently than the expression phase of effector cell function. Development of protection does not require apoptosis and appears to act, at least in part, through CD4+CD25+ postvaccination T-regs which are Ag specific and do not require IL-10 or TGF-β. Ongoing studies are aimed at characterizing the APC and the T-regs involved in the protective effect of DNA vaccination in this model.
Footnotes
Abbreviations used in this paper: EAU, experimental autoimmune uveitis; IRBP, interphotoreceptor retinoid-binding protein; Tg, transgenic; T-reg, T regulatory cell; WT, wild type; LN, lymph node; PPD, purified protein derivative; GITR, glucocorticoid- induced TNFR.
Disclosures
The authors have no financial conflict of interest.
This work was supported by National Institutes of Health intramural funding.
References
- 1.Caspi RR. Immune mechanisms in uveitis. Springer Semin Immunopathol. 1999;21:113–124. doi: 10.1007/BF00810244. [DOI] [PubMed] [Google Scholar]
- 2.Avichezer D, Grajewski RS, Chan CC, Mattapallil MJ, Silver PB, Raber JA, Liou GI, Wiggert B, Lewis GM, Donoso LA, Caspi RR. An immunologically privileged retinal antigen elicits tolerance: major role for central selection mechanisms. J Exp Med. 2003;198:1665–1676. doi: 10.1084/jem.20030413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pennesi G, Mattapallil MJ, Sun SH, Avichezer D, Silver PB, Karabekian Z, David CS, Hargrave PA, McDowell JH, Smith WC, et al. A humanized model of experimental autoimmune uveitis in HLA class II transgenic mice. J Clin Invest. 2003;111:1171–1180. doi: 10.1172/JCI15155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang J, Zhu W, Silver PB, Su SB, Chan CC, Caspi RR. Autoimmune uveitis elicited with antigen-pulsed dendritic cells has a distinct clinical signature and is driven by unique effector mechanisms: initial encounter with autoantigen defines disease phenotype. J Immunol. 2007;178:5578–5587. doi: 10.4049/jimmunol.178.9.5578. [DOI] [PubMed] [Google Scholar]
- 5.Nussenblatt RB, Palestine AG, Chan CC. Cyclosporine therapy for uveitis: long-term followup. J Ocul Pharmacol. 1985;1:369–382. doi: 10.1089/jop.1985.1.369. [DOI] [PubMed] [Google Scholar]
- 6.Nussenblatt RB, Fortin E, Schiffman R, Rizzo L, Smith J, Van Veldhuisen P, Sran P, Yaffe A, Goldman CK, Waldmann TA, Whitcup SM. Treatment of noninfectious intermediate and posterior uveitis with the humanized anti-Tac mAb: a phase I/II clinical trial. Proc Natl Acad Sci USA. 1999;96:7462–7466. doi: 10.1073/pnas.96.13.7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nussenblatt RB, Gery I, Weiner HL, Ferris FL, Shiloach J, Remaley N, Perry C, Caspi RR, Hafler DA, Foster CS, Whitcup SM. Treatment of uveitis by oral administration of retinal antigens: results of a phase I/II randomized masked trial. Am J Ophthalmol. 1997;123:583–592. doi: 10.1016/s0002-9394(14)71070-0. [DOI] [PubMed] [Google Scholar]
- 8.Corthay A, Backlund J, Holmdahl R. Role of glycopeptide-specific T cells in collagen-induced arthritis: an example how post-translational modification of proteins may be involved in autoimmune disease. Ann Med. 2001;33:456–465. doi: 10.3109/07853890109002094. [DOI] [PubMed] [Google Scholar]
- 9.Agarwal RK, Kang Y, Zambidis E, Scott DW, Chan CC, Caspi RR. Retroviral gene therapy with an immunoglobulin-antigen fusion construct protects from experimental autoimmune uveitis. J Clin Invest. 2000;106:245–252. doi: 10.1172/JCI9168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coon B, An LL, Whitton JL, von Herrath MG. DNA immunization to prevent autoimmune diabetes. J Clin Invest. 1999;104:189–194. doi: 10.1172/JCI7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tisch R, Wang B, Weaver DJ, Liu B, Bui T, Arthos J, Serreze DV. Antigen-specific mediated suppression of β cell autoimmunity by plasmid DNA vaccination. J Immunol. 2001;166:2122–2132. doi: 10.4049/jimmunol.166.3.2122. [DOI] [PubMed] [Google Scholar]
- 12.Lobell A, Weissert R, Eltayeb S, de Graaf KL, Wefer J, Storch MK, Lassmann H, Wigzell H, Olsson T. Suppressive DNA vaccination in myelin oligodendrocyte glycoprotein peptide-induced experimental autoimmune encephalomyelitis involves a T1-biased immune response. J Immunol. 2003;170:1806–1813. doi: 10.4049/jimmunol.170.4.1806. [DOI] [PubMed] [Google Scholar]
- 13.Wefer J, Harris RA, Lobell A. Protective DNA vaccination against experimental autoimmune encephalomyelitis is associated with induction of IFNβ. J Neuroimmunol. 2004;149:66–76. doi: 10.1016/j.jneuroim.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 14.Quintana FJ, Carmi P, Mor F, Cohen IR. Inhibition of adjuvant arthritis by a DNA vaccine encoding human heat shock protein 60. J Immunol. 2002;169:3422–3428. doi: 10.4049/jimmunol.169.6.3422. [DOI] [PubMed] [Google Scholar]
- 15.Song YK, Liu F, Zhang G, Liu D. Hydrodynamics-based transfection: simple and efficient method for introducing and expressing transgenes in animals by intravenous injection of DNA. Methods Enzymol. 2002;346:92–105. doi: 10.1016/s0076-6879(02)46050-8. [DOI] [PubMed] [Google Scholar]
- 16.Silver PB, Rizzo LV, Chan CC, Donoso LA, Wiggert B, Caspi RR. Identification of a major pathogenic epitope in the human IRBP molecule recognized by mice of the H-2r haplotype. Invest Ophthalmol Vis Sci. 1995;36:946–954. [PubMed] [Google Scholar]
- 17.Lin ZY, Li GR, Takizawa N, Si JS, Gross EA, Richardson K, Nickerson JM. Structure-function relationships in interphotoreceptor retinoid- binding protein (IRBP) Mol Vis. 1997;3:17. [PubMed] [Google Scholar]
- 18.Donoso LA, Rodrigues M, Vrabec TR, Sery TW, Fong SL. IRBP: preparation and characterization of site-specific monoclonal antibodies. Curr Eye Res. 1990;9:357–362. doi: 10.3109/02713689008999623. [DOI] [PubMed] [Google Scholar]
- 19.Caspi RR. Experimental autoimmune uveoretinitis in the rat and mouse. In: Coligan JE, Kruisbeek AM, Margulies DH, Shevech EM, Strober W, editors. Current Protocols in Immunology. Unit 156. John Wiley and Sons; New York: 2003. [DOI] [PubMed] [Google Scholar]
- 20.McHugh RS, Shevach EM. Cutting edge: depletion of CD4+CD25+ regulatory T cells is necessary, but not sufficient, for induction of organ-specific autoimmune disease. J Immunol. 2002;168:5979–5983. doi: 10.4049/jimmunol.168.12.5979. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Reddy J, Ochi H, Frenkel D, Kuchroo VK, Weiner HL. Recovery from experimental allergic encephalomyelitis is TGF-β dependent and associated with increases in CD4+LAP+ and CD4+CD25+ T cells. Int Immunol. 2006;18:495–503. doi: 10.1093/intimm/dxh390. [DOI] [PubMed] [Google Scholar]
- 22.Kaya Z, Dohmen KM, Wang Y, Schlichting J, Afanasyeva M, Leuschner F, Rose NR. Cutting edge: a critical role for IL-10 in induction of nasal tolerance in experimental autoimmune myocarditis. J Immunol. 2002;168:1552–1556. doi: 10.4049/jimmunol.168.4.1552. [DOI] [PubMed] [Google Scholar]
- 23.Targoni OS, Baus J, Hofstetter HH, Hesse MD, Karulin AY, Boehm BO, Forsthuber TG, Lehmann PV. Frequencies of neuroantigen- specific T cells in the central nervous system versus the immune periphery during the course of experimental allergic encephalomyelitis. J Immunol. 2001;166:4757–4764. doi: 10.4049/jimmunol.166.7.4757. [DOI] [PubMed] [Google Scholar]
- 24.Moody MD, Van Arsdell SW, Murphy KP, Orencole SF, Burns C. Array-based ELISAs for high-throughput analysis of human cytokines. BioTechniques. 2001;31:186–190. 192–194. doi: 10.2144/01311dd03. [DOI] [PubMed] [Google Scholar]
- 25.Rizzo LV, DeKruyff RH, Umetsu DT, Caspi RR. Regulation of the interaction between Th1 and Th2 cell clones to provide help for antibody production in vivo. Eur J Immunol. 1995;25:708. doi: 10.1002/eji.1830250312. [DOI] [PubMed] [Google Scholar]
- 26.Suffia IJ, Reckling SK, Piccirillo CA, Goldszmid RS, Belkaid Y. Infected site-restricted Foxp3+ natural regulatory T cells are specific for microbial antigens. J Exp Med. 2006;203:777–788. doi: 10.1084/jem.20052056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 28.Wang P, Sun SH, Silver PB, Chan CC, Agarwal RK, Wiggert B, Kohn LD, Jamieson GA, Jr, Caspi RR. Methimazole protects from experimental autoimmune uveitis (EAU) by inhibiting antigen presenting cell function and reducing antigen priming. J Leukocyte Biol. 2003;73:57–64. doi: 10.1189/jlb.0102047. [DOI] [PubMed] [Google Scholar]
- 29.Snedecor GW, Cochran WG. Statistical Methods. 6. Iowa State University Press; Ames: 1967. [Google Scholar]
- 30.Pedotti R, Mitchell D, Wedemeyer J, Karpuj M, Chabas D, Hattab EM, Tsai M, Galli SJ, Steinman L. An unexpected version of horror autotoxicus: anaphylactic shock to a self-peptide. Nat Immunol. 2001;2:216–222. doi: 10.1038/85266. [DOI] [PubMed] [Google Scholar]
- 31.Pedotti R, Sanna M, Tsai M, DeVoss J, Steinman L, McDevitt H, Galli SJ. Severe anaphylactic reactions to glutamic acid decarboxylase (GAD) self peptides in NOD mice that spontaneously develop autoimmune type 1 diabetes mellitus. BMC Immunol. 2003;4:2. doi: 10.1186/1471-2172-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–1266. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 33.Glinka Y, De Pooter R, Croze F, Prud’homme GJ. Regulatory cytokine production stimulated by DNA vaccination against an altered form of glutamic acid decarboxylase 65 in nonobese diabetic mice. J Mol Med. 2003;81:175–184. doi: 10.1007/s00109-002-0412-9. [DOI] [PubMed] [Google Scholar]
- 34.Bynoe MS, Evans JT, Viret C, Janeway CA., Jr Epicutaneous immunization with autoantigenic peptides induces T suppressor cells that prevent experimental allergic encephalomyelitis. Immunity. 2003;19:317–328. doi: 10.1016/s1074-7613(03)00239-5. [DOI] [PubMed] [Google Scholar]
- 35.Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25+ regulatory cells from human peripheral blood express very high levels of CD25 ex vivo. Novartis Found Symp. 2003;252:67–88. doi: 10.1002/0470871628.ch6. discussion 88–91, 106–114. [DOI] [PubMed] [Google Scholar]
- 36.Ziegler SF. FOXP3: of mice and men. Annu Rev Immunol. 2006;24:209–226. doi: 10.1146/annurev.immunol.24.021605.090547. [DOI] [PubMed] [Google Scholar]
- 37.Zwar TD, I, van Driel R, Gleeson PA. Guarding the immune system: suppression of autoimmunity by CD4+CD25+ immunoregulatory T cells. Immunol Cell Biol. 2006;84:487–501. doi: 10.1111/j.1440-1711.2006.01471.x. [DOI] [PubMed] [Google Scholar]
- 38.McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, Byrne MC. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–323. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 39.Nagatani K, Sagawa K, Komagata Y, Yamamoto K. Peyer’s patch dendritic cells capturing oral antigen interact with antigen-specific T cells and induce gut-homing CD4+CD25+ regulatory T cells in Peyer’s patches. Ann NY Acad Sci. 2004;1029:366–370. doi: 10.1196/annals.1309.020. [DOI] [PubMed] [Google Scholar]
- 40.Huehn J, Hamann A. Homing to suppress: address codes for Treg migration. Trends Immunol. 2005;26:632–636. doi: 10.1016/j.it.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 41.Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000;164:183–190. doi: 10.4049/jimmunol.164.1.183. [DOI] [PubMed] [Google Scholar]
- 42.Li J, Bracht M, Shang X, Radewonuk J, Emmell E, Griswold DE, Li L. Ex vivo activated OVA specific and non-specific CD4+CD25+ regulatory T cells exhibit comparable suppression to OVA mediated T cell responses. Cell Immunol. 2006;241:75–84. doi: 10.1016/j.cellimm.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 43.Bacchetta R, Gregori S, Roncarolo MG. CD4+ regulatory T cells: mechanisms of induction and effector function. Autoimmun Rev. 2005;4:491–496. doi: 10.1016/j.autrev.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 44.Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takahashi T, Kuniyasu Y, Toda M, Sakaguchi N, Itoh M, Iwata M, Shimizu J, Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998;10:1969–1980. doi: 10.1093/intimm/10.12.1969. [DOI] [PubMed] [Google Scholar]
- 46.Lobell A, Weissert R, Storch MK, Svanholm C, de Graaf KL, Lassmann H, Andersson R, Olsson T, Wigzell H. Vaccination with DNA encoding an immunodominant myelin basic protein peptide targeted to Fc of immunoglobulin G suppresses experimental autoimmune encephalomyelitis. J Exp Med. 1998;187:1543–1548. doi: 10.1084/jem.187.9.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsunoda I, Kuang LQ, Tolley ND, Whitton JL, Fujinami RS. Enhancement of experimental allergic encephalomyelitis (EAE) by DNA immunization with myelin proteolipid protein (PLP) plasmid DNA. J Neuropathol Exp Neurol. 1998;57:758–767. doi: 10.1097/00005072-199808000-00005. [DOI] [PubMed] [Google Scholar]
- 48.Ruiz PJ, Garren H, Ruiz IU, Hirschberg DL, Nguyen LV, Karpuj MV, Cooper MT, Mitchell DJ, Fathman CG, Steinman L. Suppressive immunization with DNA encoding a self-peptide prevents autoimmune disease: modulation of T cell costimulation. J Immunol. 1999;162:3336–3341. [PubMed] [Google Scholar]
- 49.Selmaj K, Kowal C, Walczak A, Nowicka J, Raine CS. Naked DNA vaccination differentially modulates autoimmune responses in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2000;111:34–44. doi: 10.1016/s0165-5728(00)00329-5. [DOI] [PubMed] [Google Scholar]
- 50.Weaver DJ, Jr, Liu B, Tisch R. Plasmid DNAs encoding insulin and glutamic acid decarboxylase 65 have distinct effects on the progression of autoimmune diabetes in nonobese diabetic mice. J Immunol. 2001;167:586–592. doi: 10.4049/jimmunol.167.1.586. [DOI] [PubMed] [Google Scholar]
- 51.Garren H, Ruiz PJ, Watkins TA, Fontoura P, Nguyen LT, Estline ER, Hirschberg DL, Steinman L. Combination of gene delivery and DNA vaccination to protect from and reverse Th1 autoimmune disease via deviation to the Th2 pathway. Immunity. 2001;15:15–22. doi: 10.1016/s1074-7613(01)00171-6. [DOI] [PubMed] [Google Scholar]
- 52.Miller SD, Eagar TN. Functional role of epitope spreading in the chronic pathogenesis of autoimmune and virus-induced demyelinating diseases. Adv Exp Med Biol. 2001;490:99–107. doi: 10.1007/978-1-4615-1243-1_10. [DOI] [PubMed] [Google Scholar]
- 53.Levings MK, Bacchetta R, Schulz U, Roncarolo MG. The role of IL-10 and TGF-β in the differentiation and effector function of T regulatory cells. Int Arch Allergy Immunol. 2002;129:263–276. doi: 10.1159/000067596. [DOI] [PubMed] [Google Scholar]
- 54.Shevach EM, McHugh RS, Piccirillo CA, Thornton AM. Control of T-cell activation by CD4+ CD25+ suppressor T cells. Immunol Rev. 2001;182:58–67. doi: 10.1034/j.1600-065x.2001.1820104.x. [DOI] [PubMed] [Google Scholar]
- 55.Stein-Streilein J, Streilein JW. Anterior chamber associated immune deviation (ACAID): regulation, biological relevance, and implications for therapy. Int Rev Immunol. 2002;21:123–152. doi: 10.1080/08830180212066. [DOI] [PubMed] [Google Scholar]
- 56.Stein-Streilein J, Taylor AW. An eye’s view of T regulatory cells. J Leukocyte Biol. 2006;81:593–598. doi: 10.1189/jlb.0606383. [DOI] [PubMed] [Google Scholar]
- 57.Kitaichi N, Namba K, Taylor AW. Inducible immune regulation following autoimmune disease in the immune-privileged eye. J Leukocyte Biol. 2005;77:496–502. doi: 10.1189/jlb.0204114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.El-Amine M, Melo M, Kang Y, Nguyen H, Qian J, Scott DW. Mechanisms of tolerance induction by a gene-transferred peptide-IgG fusion protein expressed in B lineage cells. J Immunol. 2000;165:5631–5636. doi: 10.4049/jimmunol.165.10.5631. [DOI] [PubMed] [Google Scholar]
- 59.Pittoni V, Valesini G. The clearance of apoptotic cells: implications for autoimmunity. Autoimmun Rev. 2002;1:154–161. doi: 10.1016/s1568-9972(02)00032-0. [DOI] [PubMed] [Google Scholar]
- 60.Ferguson TA, Stuart PM, Herndon JM, Griffith TS. Apoptosis, tolerance, and regulatory T cells– old wine, new wineskins. Immunol Rev. 2003;193:111–123. doi: 10.1034/j.1600-065x.2003.00042.x. [DOI] [PubMed] [Google Scholar]
- 61.Nussenblatt RB, Peterson JS, Foster CS, Rao NA, See RF, Letko E, Buggage RR. Initial evaluation of subcutaneous daclizumab treatments for noninfectious uveitis: a multicenter noncomparative interventional case series. Ophthalmology. 2005;112:764–770. doi: 10.1016/j.ophtha.2004.12.034. [DOI] [PubMed] [Google Scholar]
- 62.Li Z, Lim WK, Mahesh SP, Liu B, Nussenblatt RB. Cutting edge: in vivo blockade of human IL-2 receptor induces expansion of CD56bright regulatory NK cells in patients with active uveitis. J Immunol. 2005;174:5187–5191. doi: 10.4049/jimmunol.174.9.5187. [DOI] [PubMed] [Google Scholar]