

Abstract
Background
In the present work the central carbon metabolism of Dinoroseobacter shibae and Phaeobacter gallaeciensis was studied at the level of metabolic fluxes. These two strains belong to the marine Roseobacter clade, a dominant bacterial group in various marine habitats, and represent surface-associated, biofilm-forming growth (P. gallaeciensis) and symbiotic growth with eukaryotic algae (D. shibae). Based on information from recently sequenced genomes, a rich repertoire of pathways has been identified in the carbon core metabolism of these organisms, but little is known about the actual contribution of the various reactions in vivo.
Results
Using 13C labelling techniques in specifically designed experiments, it could be shown that glucose-grown cells of D. shibae catabolise the carbon source exclusively via the Entner-Doudoroff pathway, whereas alternative routes of glycolysis and the pentose phosphate pathway are obviously utilised for anabolic purposes only. Enzyme assays confirmed this flux pattern and link the lack of glycolytic flux to the absence of phosphofructokinase activity. The previously suggested formation of phosphoenolpyruvate from pyruvate during mixotrophic CO2 assimilation was found to be inactive under the conditions studied. Moreover, it could be shown that pyruvate carboxylase is involved in CO2 assimilation and that the cyclic respiratory mode of the TCA cycle is utilised. Interestingly, the use of intracellular pathways was highly similar for P. gallaeciensis.
Conclusion
The present study reveals the first insight into pathway utilisation within the Roseobacter group. Fluxes through major intracellular pathways of the central carbon metabolism, which are closely linked to the various important traits found for the Roseobacter clade, could be determined. The close similarity of fluxes between the two physiologically rather different species might provide the first indication of more general key properties among members of the Roseobacter clade which may explain their enormous success in the marine realm.
Background
The Roseobacter lineage, representing a group of Alphaproteobacteria [1], is found in various marine habitats where it is present in high abundance, comprising up to 25% of the total bacterial community [2]. Overall, the diverse metabolic properties of the Roseobacter clade and its ubiquitous occurrence in marine ecosystems suggest that members of this clade play an important role in global biogeochemical processes such as cycling of carbon or sulphur [3]. Members of the Roseobacter clade participate in DMSP demethylation [4], the oxidation of carbon monoxide [5] and degradation of aromatic compounds [6,7]. Typically, they use external organic substrates as carbon sources [8]. Of outstanding interest is the fact that they are able to generate energy from light (aerobic anoxygenic phototrophy) [9] and thus contribute significantly to phototrophic energy generation [10,11]. All these important traits are linked to the core part of central carbon metabolism involved in the breakdown of nutrients and the supply of metabolites and energy for various cellular requirements. Recent efforts in genome sequencing and annotation of Roseobacter members have provided a first insight into the repertoire of underlying metabolic reactions available (Figure 1) and have led to different suggestions for possible pathways that might be involved in important physiological functions [12]. As an example, a mixotrophic CO2 assimilation pathway has been proposed for R. denitrificans, in which CO2 is fixed either (i) via the combined action of pyruvate-orthophosphate dikinase and phosphoenolpyruvate carboxylase or (ii) via pyruvate carboxylase [13]. For glucose catabolism, up to three alternative routes are encoded in the genome: glycolysis, the pentose phosphate pathway and the Entner-Doudoroff pathway. At this point, it seems highly relevant to study the contribution of these potential pathways to the metabolism of bacteria in the Roseobacter clade to improve our understanding of their physiology. Our current knowledge of the in vivo fluxes through intracellular pathways among the Roseobacter lineage is still very limited.
Figure 1.

Metabolic network of the central carbon metabolism of Dinoroseobacter shibae [1]and Phaeobacter gallaeciensis [25]as predicted from the annotated genome sequence. G6P: glucose-6-phosphate; F6P: fructose-6-phosphate; GAP: glyceraldehyde-3-phosphate; PEP: phosphoenolpyruvate; PYR: pyruvate; AcCoA: acetyl-Coenzyme A; OGA: 2-oxoglutarate; SUC: succinate; FUM: fumarate; OAA: oxaloacetate; MAL: malate; 6PG: 6-phosphogluconate; KDGP: 2-keto-3-deoxy-6-phosphogluconate; pycA: pyruvate carboxylase; pckA: phosphoenolpyruvate carboxykinase; ppdK: pyruvate orthophosphate dikinase.
To address this issue, we applied metabolic flux analysis using 13C labelled isotopes to gain a first insight into the central catabolic pathways of Dinoroseobacter shibae DFL12 [1] and Phaeobacter gallaeciensis DSM 17395 [14]. These species represent two prominent members of the Roseobacter clade. P. gallaeciensis has received strong interest due to its ability to produce the antibiotic tropodithietic acid. D. shibae was isolated as a novel species from marine dinoflagellates and lives in a symbiotic relationship with eukaryotic algae [15]. Metabolic flux analysis using 13C labelled isotopes has proven a key technology in the unravelling of metabolic pathways and has recently been used to study different microorganisms mainly linked to biotechnological production processes [16-19]. No such study has yet been performed for members of the Roseobacter clade.
Results and Discussion
Cultivation profile
The cultivation profile of D. shibae on defined medium with glucose as the sole carbon source is displayed in Figure 2. After an initial adaptation phase, cells grew exponentially with a constant specific growth rate of 0.11 h-1. After 50 hours of cultivation the carbon source was depleted and cells entered a stationary phase. The biomass yield was 0.45 g cell dry mass per g glucose consumed, indicating efficient utilisation of the carbon source for growth. A similar growth profile was determined for P. gallaeciensis.
Figure 2.
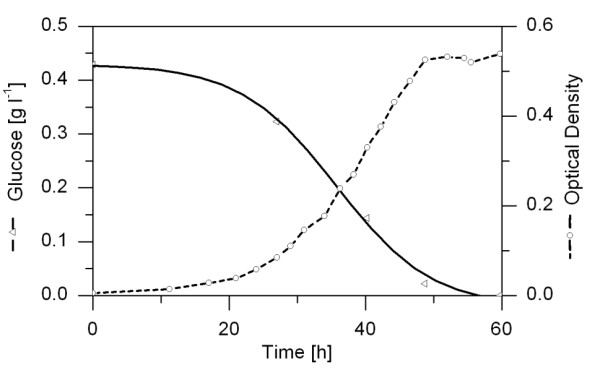
Time courses of glucose concentration and optical density during a batch cultivation of D. shibae in shake flasks under constant light.
Pathways for glucose catabolism
The carbon core metabolism of D. shibae and P. gallaeciensis consists of three potential routes for glucose catabolism. Glucose can be alternatively catabolised via glycolysis (EMP), the pentose phosphate pathway (PPP) and the Entner-Doudoroff pathway (EDP). The use of [1-13C] glucose by each individual pathway leads to a different labelling pattern in specific fragments of alanine and serine, which can be taken as a clear differentiation of flux (Figure 3). For D. shibae the corresponding [M-57] fragment of serine did not show any enrichment of 13C but rather reflected the pattern resulting from the natural abundance of 13C only (Table 1). Any contribution of glycolysis to formation of this metabolite and its precursor 3-phosphoglycerate can therefore be excluded as this would lead to enrichment of 13C at the C3 position, yielding a higher fraction of M+1 labelled molecules of Ser. Thus glycolytic flux obviously was not present. The two remaining possibilities, the PPP and the ED pathway, can be differentiated by the labelling pattern of alanine, which represents the pyruvate pool in the cell. The high enrichment of 13C label in the [M-57] fragment of alanine indicates a large contribution of the ED pathway, since formation via the PPP would lead to non-labelled alanine. The [M-85] fragment of alanine, comprising only the carbon atoms C2 and C3 of pyruvate, was not enriched in 13C, showing that pyruvate was labelled only at its C1 position. This perfectly matches the isotope pattern expected for the ED pathway, whereas glycolytic flux would have resulted in label enrichment at the C3 position, and further confirms the flux distribution.
Figure 3.

Theoretical labelling pattern of the C3 pool (GAP and PYR) derived from 99% [1-13C] glucose depending on activities in the carbon core metabolism. The three major routes for glucose catabolism are presented: glycolysis (solid lines), the pentose phosphate pathway (dashed lines) and the Entner-Doudoroff pathway (dotted lines). White circles represent unlabelled (12C) carbon whereas black circles indicate labelled (13C) carbon. The numbers reflect the position of the carbon atom within the molecule. Ala: alanine; G6P: glucose 6-phosphate; 6PG: 6-phosphogluconate; KDPG: 2-keto-3-deoxy-6-phosphogluconate; F6P: fructose 6-phosphate; R5P: ribose 5-phosphate; GAP: glyceraldehyde 3-phosphate; PYR: pyruvate.
Table 1.
Selected TBDMSa-amino acid fragments used in the study derived from D. shibae and P. gallaeciensis grown on 99% [1-13C] glucose.
| Fragment | C-Atoms | Mass isotopomer distribution (% of total pool) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| D. shibae | P. gallaeciensis | |||||||||
| M+0 | M+1 | M+2 | M+3 | M+0 | M+1 | M+2 | M+3 | |||
| Ala | M-57 | 1-3 | 50.0 ± 0.2 | 48.2 ± 0.2 | 1.8 ± 0.0 | 0.01 ± 0.01 | 49.2 ± 0.0 | 49.3 ± 0.0 | 1.5 ± 0.0 | 0.0 ± 0.0 |
| M-85 | 2-3 | 96.8 ± 0.1 | 3.2 ± 0.1 | 0.0 ± 0.0 | 97.2 ± 0.0 | 2.8 ± 0.0 | 0.0 ± 0.0 | |||
| f302 | 1-2 | 51.2 ± 0.1 | 48.2 ± 0.1 | 0.6 ± 0.0 | 50.1 ± 0.1 | 49.3 ± 0.1 | 0.6 ± 0.0 | |||
| Asp | M-57 | 1-4 | 72.4 ± 0.7 | 23.2 ± 0.5 | 4.3 ± 0.2 | 0.12 ± 0.01 | 64.2 ± 0.2 | 29.4 ± 0.1 | 6.2 ± 0.2 | 0.13 ± 0.07 |
| M-85 | 2-4 | 83.3 ± 0.6 | 16.2 ± 0.6 | 0.4 ± 0.1 | 0.10 ± 0.03 | 80.0 ± 0.1 | 19.4 ± 0.0 | 0.6 ± 0.0 | 0.04 ± 0.02 | |
| f302 | 1-2 | 82.1 ± 0.3 | 17.6 ± 0.3 | 0.2 ± 0.0 | 76.3 ± 0.1 | 23.5 ± 0.0 | 0.3 ± 0.1 | |||
| Glu | M-57 | 1-5 | 80.7 ± 0.3 | 18.4 ± 0.4 | 0.8 ± 0.1 | 0.05 ± 0.03 | 78.1 ± 0.5 | 20.8 ± 0.3 | 0.9 ± 0.2 | 0.09 ± 0.03 |
| M-85 | 2-5 | 92.1 ± 0.2 | 7.5 ± 0.2 | 0.3 ± 0.0 | 0.06 ± 0.00 | 93.6 ± 0.1 | 6.2 ± 0.1 | 0.0 ± 0.0 | 0.09 ± 0.01 | |
| f302 | 1-2 | 83.4 ± 0.2 | 16.2 ± 0.2 | 0.3 ± 0.0 | 81.2 ± 0.3 | 18.4 ± 0.1 | 0.4 ± 0.2 | |||
| Gly | M-57 | 1-2 | 96.1 ± 0.0 | 3.8 ± 0.0 | 0.1 ± 0.0 | 97.2 ± 0.1 | 2.8 ± 0.1 | 0.03 ± 0.02 | ||
| M-85 | 2 | 98.8 ± 0.1 | 1.1 ± 0.0 | 99.0 ± 0.0 | 0.9 ± 0.0 | |||||
| Phe | M-57 | 1-9 | 85.7 ± 0.6 | 13.0 ± 0.6 | 0.6 ± 0.1 | 0.08 ± 0.03 | 86.7 ± 0.9 | 11.6 ± 0.3 | 0.5 ± 0.1 | 0.02 ± 0.01 |
| f302 | 1-2 | 95.9 ± 0.3 | 4.1 ± 0.3 | 0.0 ± 0.0 | 96.7 ± 0.2 | 3.3 ± 0.2 | 0.0 ± 0.0 | |||
| Ser | M-57 | 1-3 | 95.3 ± 0.3 | 4.6 ± 0.3 | 0.0 ± 0.0 | 0.07 ± 0.03 | 96.7 ± 0.1 | 3.3 ± 0.1 | 0.0 ± 0.0 | 0.09 ± 0.02 |
| M-85 | 2-3 | 97.7 ± 0.1 | 2.3 ± 0.1 | 0.0 ± 0.0 | 98.0 ± 0.1 | 2.0 ± 0.1 | 0.0 ± 0.0 | |||
| f302 | 1-2 | 95.6 ± 0.0 | 3.9 ± 0.0 | 0.5 ± 0.0 | 96.8 ± 0.1 | 2.8 ± 0.0 | 0.4 ± 0.0 | |||
| Tyr | M-57 | 1-9 | 86.2 ± 0.7 | 12.8 ± 0.1 | 0.5 ± 0.3 | 0.06 ± 0.09 | 87.7 ± 0.2 | 11.4 ± 0.4 | 0.5 ± 0.0 | 0.08 ± 0.06 |
| f302 | 1-2 | 96.1 ± 0.2 | 3.9 ± 0.2 | 0.0 ± 0.0 | 97.3 ± 0.4 | 2.7 ± 0.4 | 0.0 ± 0.0 | |||
a tert-butyldimethylsilyl; fragmentation patterns are described elsewhere [27]
Interestingly, P. gallaeciensis showed almost identical characteristics and obviously also uses mainly the ED pathway during growth on glucose. The quantification of relative flux (Eqs. 2 and 3) revealed that the use of the ED pathway amounts to >99%, whereas glycolysis and PPP contribute only <1% (Table 2). Compared to other microorganisms such as E. coli [20], B. subtilis [21], B. megaterium [18] or C. glutamicum [22] grown on glucose, this is a rather unusual flux pattern. Most organisms use glycolysis and the pentose phosphate pathway concomitantly but at varying ratios (Table 2). Exclusive utilisation of the ED pathway, as found here, has been previously observed in selected species of Pseudomonas or Arthrobacter where this behaviour was attributed to a lack of phosphofructokinase [23,24]. Among the two microorganisms studied, D. shibae does contain a gene encoding for this enzyme, whereas P. gallaeciensis does not. For both Roseobacter species, in contrast to E. coli as positive control, phosphofructokinase activity could not be detected, clearly explaining the lack of glycolytic flux (Figure 4B). While this matches with the genomic repertoire of P. gallaeciensis, we conclude at this stage that the phosphofructokinase in D. shibae is either not expressed, might have another function or even is a non-functional protein. The flux pattern for both organisms is supported by enzymatic assays showing high in vitro activity of 6-phosphogluconate dehydratase and 2-dehydro-3-deoxyphosphogluconate aldolase, the two key enzymes in the Entner-Doudoroff pathway (Figure 4A).
Table 2.
Comparison of catabolic pathway activity and origins of metabolic intermediates in central carbon metabolism of D. shibae, P. gallaeciensis and other bacteria derived from carbon labelling experiments.
| Pathway activity/Fractional pool composition [%] | ||||||
|---|---|---|---|---|---|---|
| D. shibaea | P. gallaeciensisa | B. subtilis [21] | B. megaterium [18] | C. glutamicum [35] | E. coli [20] | |
| Glycolysis | < 1 | < 1 | 27 | 46 | 49 | 73 |
| PPP | < 1 | < 1 | 72 | 49 | 48 | 22 |
| ED pathway | > 99 | > 99 | n.a. | n.a. | n.a. | 4 |
| PEP from PYR | 0 | 0 | 0 | 0 | 0 | 0 |
| PEP from OAA | 0 | 0 | 14 | 0 | 16 | 0 |
athis study
n.a. = not available in the organism
Figure 4.

In vitro activities of key enzymes of the different catabolic pathways for D. shibae and P. gallaeciensis. PFK: 6-phosphofructokinase; EDD: 6-phosphogluconate dehydrogenase; EDA: 2-keto-3-deoxy-6-phosphogluconate aldolase.
Pathways for PEP synthesis - contribution of pyruvate-orthophosphate dikinase and phosphoenolpyruvate carboxykinase
Based on the labelling data given above, the formation of PEP from pyruvate by pyruvate-orthophosphate dikinase or via pyruvate carboxylase and phosphoenolpyruvate carboxykinase would result in the presence of PEP with13C enrichment at position C1. However, the [f302] fragments of Phe and Tyr, each corresponding to the carbon atoms C1 and C2 of PEP, do not show significant enrichment of 13C (Table 1). The same holds for the [M-57] fragment, which corresponds to the entire carbon skeleton of Phe and Tyr and thus all precursors, that is, PEP and E4P. Flux quantification using Equations 4 and 5 confirms that PEP is solely synthesised by the reactions of lower glycolysis (Table 2). This is an interesting finding with respect to the recently suggested mixotrophic CO2 assimilation pathway for some members of the Roseobacter clade, which also involves the potential contribution of pyruvate orthophosphate dikinase (PPDK) [13]. Despite the putative gene for this protein also being annotated for the species investigated here, we could clearly demonstrate that the formation of PEP from PYR is not active in vivo under the conditions studied.
Pathways for oxaloacetate synthesis - contribution of CO2 assimilation and oxidative TCA cycle
Oxaloacetate as a central metabolite can be formed by two major pathways, that is, carboxylation involving pyruvate carboxylase or via pyruvate dehydrogenase and the energy-generating reactions of the TCA cycle. The following data clearly suggest that both pathways are active simultaneously in the two Roseobacters. For the experimental setup chosen and carbon transfer in the underlying metabolic reactions, the carboxylation of pyruvate is the only reaction that leads to 13C labelled oxaloacetate (Figure 5). The label can be present in carbon positions C1 or C4, whereby single- or double-labelled molecules can be formed, depending on the incorporation of 12CO2 or 13CO2. In contrast, the alternative route via the cyclic respiratory mode of the TCA cycle yields exclusively non-labelled oxaloacetate. In all possible cases the labelled carbon atoms from either pyruvate or oxaloacetate are released in the decarboxylation steps of the TCA cycle as 13CO2. Inspection of the labelling pattern of aspartate, corresponding to the oxaloacetate backbone, immediately shows that single- and double-labelled mass isotopomers are present in significant amounts for D. shibae and P. gallaeciensis, indicating in vivo activity of pyruvate carboxylase in both strains (Table 1). However, the relative fractions of these 13C enriched mass isotopomers are relatively small, excluding sole contribution of this reaction to oxaloacetate synthesis. The dominant fraction consists of non-labelled molecules, obviously derived via the oxidative TCA cycle. We thus conclude that the cyclic respiratory mode of the TCA cycle is active in vivo in both strains. For D. shibae, which possesses a photosystem for energy generation, this mode might display an important strategy to derive energy under conditions where the photosystem is not active, for example, during the night or in deeper water regions.
Figure 5.
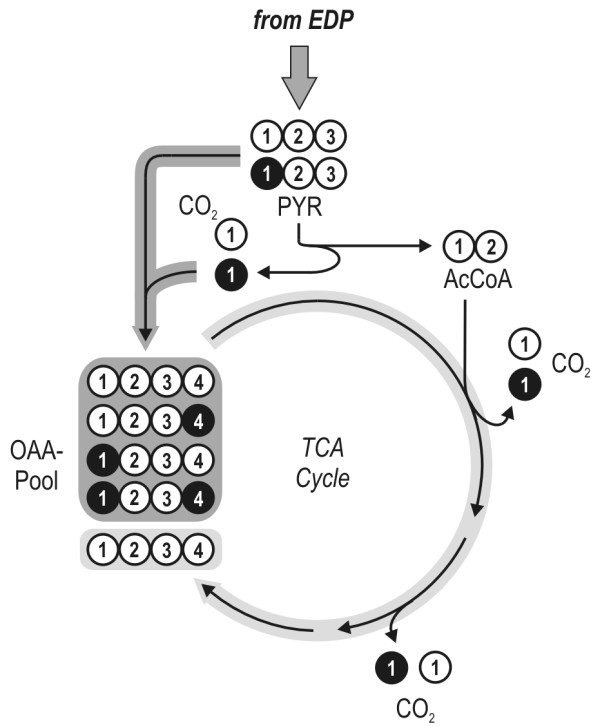
Schematic overview of the two major pathways from pyruvate towards oxaloacetate: (i) direct carboxylation via pyruvate carboxylase or (ii) pyruvate dehydrogenase and the energy forming reactions of the TCA cycle. The conserved carbon transfer of the underlying reactions yields a specific labelling pattern for oxaloacetate formed by each pathway which is presented in this figure. White circles represent 12C whereas black circles indicate labelled 13C. The numbers given reflect the position of the carbon atom within the molecule. AcCoA: acetyl-Coenzyme A; EDP: Entner-Doudoroff pathway; OAA: oxaloacetate; PYR: pyruvate; TCA: tricarboxylic acid.
Conclusion
Being one of the first metabolic studies of members of the Roseobacter clade using the 13C labelling experimental approach, a deeper insight into the activity of the important metabolic routes of D. shibae and P. gallaeciensis was achieved. Interestingly, the use of intracellular pathways is highly similar in the studied species D. shibae and P. gallaeciensis. This stands in surprising contrast to the overall differences in phenotypic behaviour exhibited by these two strains, since D. shibae is an algal-associated microorganism whereas P. gallaeciensis is free-living in marine habitats. However, this may be a first indication of more general key properties among members of the Roseobacter clade that explain their enormous success in the marine realm.
Methods
Strains, medium and growth conditions
The strains used in this study are the genome sequenced strains Dinoroseobacter shibae DFL12 [1] and Phaeobacter gallaeciensis DSM 17395 [14]. For cultivation of both strains a defined, synthetic seawater medium (minimal medium) was used [25], containing the following components per litre of medium: 4.0 g NaSO4, 0.2 g KH2PO4, 0.25 g NH4Cl, 20.0 g NaCl, 3.0 g MgCl2·6 H2O, 0.5 g KCl and 0.15 g CaCl2·2 H2O, 0.19 g NaHCO3, 1 ml trace element solution and 10 ml vitamin solution. The final glucose concentration in the medium was in the range of 0.4 to 0.9 g l-1. The trace element solution contained 2.1 g Fe(SO4)·7 H2O, 13 ml 25% (v/v) HCl, 5.2 g Na2EDTA·2 H2O, 30 mg H3BO3, 0.1 g MnCl2·4 H2O, 0.19 g CoCl2·6 H2O, 2 mg CuCl2·2 H2O, 0.144 g ZnSO4·7 H2O and 36 mg Na2MoO4·2 H2O per litre. The vitamin solution for D. shibae contained the following components per litre: 0.2 g biotin, 2.0 g nicotinic acid and 0.8 g 4-aminobenzoic acid. All solutions were sterilised separately and mixed at room temperature prior to inoculation. For carbon labelling experiments 99% [1-13C] glucose (Euriso-Top, Saint-Aubin, France) was used as substrate. The cultivations were carried out on orbital shakers at 200 rpm in 500 ml shaken flasks with a culture volume of 50 ml at 37°C (D. shibae) and 28°C (P. gallaeciensis). To ensure comparable conditions between the two microorganisms and avoid any potential influencing effects of phototrophy in D. shibae, both organisms were cultivated in the light. Under these conditions, no bacteriochlorophyll is synthesised D. shibae [1] and therefore no active photosystem is present that might affect energy metabolism and thus induce changes in the fluxes through the main metabolic pathways.
Analytics
Cell concentration was monitored by measuring the optical density (OD) at 600 nm or by gravimetry [26]. The 13C labelling pattern of the amino acids contained in the cell protein was determined as follows [27]. Cells were harvested during exponential growth phase at half-maximal optical density including a washing step in 0.9% NaCl solution, followed by lyophilisation. Subsequently, 4 mg of lyophilised cells was resuspended in 200 μl of 6 M HCl and incubated at 110°C for 24 h. The obtained hydrolysate was neutralised by addition of 6 M NaOH and cleared of insoluble matter (0.2 μm centrifugal filter device Ultrafree MC, Millipore, Bedford, MA, USA). Subsequently, 50 μl of the hydrolysate was transferred to a 2 ml sample vial, lyophilised and derivatised at 80°C for 60 min with 50 μl N, N-dimethylformamide (Carl Roth, Karslruhe, Germany) containing 0.1% (v/v) pyridine and 50 μL N-methyl-tert-butyldimethylsilyl-trifluoroacetamide (MBDSTFA, Macherey-Nagel, USA). GC/MS measurements were carried out as described earlier [27]. Subsequent MS data processing was carried out according to Fürch et al. [18] and Lee et al. [28,29].
Preparation of cell extracts for enzyme activity measurements
Cells were harvested by centrifugation at 10,000 g for 10 min, washed twice with 100 mM Tris-HCl (pH 7.0) containing 20 mM KCl, 5 mM MnSO4, 2 mM DTT and 0.1 mM EDTA, and then resuspended in the same buffer. Afterwards the cells were disrupted by sonification for 1 min using an ultrasonic disrupter (Sonifier W250 D, Branson, Danbury, USA) with an amplitude of 30%. Cell debris was removed by centrifugation. The resulting crude cell extract was immediately used to determine specific enzyme activity. All operations were carried out on ice.
Enzyme assays
Enzyme activities in crude cell extract were measured spectrophotometrically. All compounds of the reaction mixture were pipetted into a cuvette with a 1 cm light path and reactions were initiated by adding the cell extract or substrate respectively. The total protein concentration of the crude cell extract was determined using RotiQuant (Carl Roth GmbH, Karlsruhe, Germany). The overall activity of 6-phosphogluconate dehydratase (EDD) and 2-dehydro-3-deoxyphosphogluconate aldolase (EDA) was measured using a two-step reaction [30]. For this purpose 0.8 μmol 6-phosphogluconate, 1 μmol MgCl, 5 μmol Tris-HCl buffer (pH 7.65) and 100 μl of extract were incubated in a total volume of 0.5 ml for 5 min at room temperature. The reaction was stopped by dilution with 2 ml of the same buffer and then by heating in a boiling water bath for 2 min. After centrifugation, the supernatant solution was assayed for pyruvate with NADH and lactate dehydrogenase according to Peng and Shimizu [31]. The activity of 6-phosphofructosekinase (PFK) in the crude cell extract was assayed as described by Gancedo and Gancedo [32]. The reaction mix contained 50 mM imidazole HCl (pH 7.0), 0.05 mM ATP, 5 mM MgCl2, 1 mM EDTA, 0.25 mM NADH, 0.25 mM fructose 6-phosphate (F6P), 0.5 U aldolase, 0.5 U glycerolphosphate dehydrogenase and 0.5 U triosephosphate isomerase.
Metabolic flux calculations
Metabolic flux calculations were performed as described previously [18]. Briefly, metabolic flux ratio analysis was used to gain information about the flux distribution at important branch points within the network. As several alternative pathways may lead to a particular product, the fractional contribution (metabolic flux ratio) of each pathway was determined based on the molecular mass distributions of the reactants and the product according to Fischer and Sauer [33]. For the performed calculations, corrected mass spectra of selected fragments of serine, glycine, alanine, phenylalanine, tyrosine, aspartate and glutamate were used in this study (see Table 1). As the amino acids are synthesised from precursor metabolites of the central carbon metabolism with a known and well conserved carbon transition, their labelling pattern can be used to conclude the corresponding labelling pattern of their precursors [34]. To gain important information about the position of the labelling within the molecule, different fragments were considered simultaneously. In general, TBDMS-derivatised amino acids yield characteristic fragments by electron impact ionisation. The [M-57] fragment of each amino acid contains the complete carbon backbone, whereas the [M-85] fragment lacks the carbon at the C1 position that corresponds to the carbon atom of the carboxyl group of the amino acid. The third fragment considered - [f302] - always contains the C1 and C2 carbon of the corresponding amino acid.
In the case of alternative pathways yielding a specific product, the fractional contribution of each pathway can be determined concerning the mass distributions of the reactants and the product according to Eq. (1) [33].
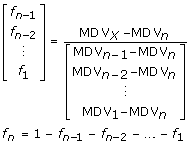 |
(1) |
In Eq. (1) index X indicates the product molecule whereas the consecutive numbers 1 through n represent reactant molecules of alternative pathways contributing to the mass distribution of the product pool. The corresponding fractional amount of each pathway f can then be calculated by considering two additional constraints: (i) all fractions must have a positive value and (ii) their sum has to equal 1. A more detailed description will be given in the following respective sections.
Theoretical framework for flux estimation
To carry out metabolic flux calculations for D. shibae and P. gallaeciensis, a metabolic network was constructed based on genome data (GenBank accession numbers NC_009952 [D. shibae] and NZ_ABIF00000000 [P. gallaeciensis]). As we focused on the central carbon metabolism, the major catabolic routes for glucose as well as the reactions linking the C3 and C4 pools were considered. In terms of glucose catabolism, the annotated genome revealed the presence of the genes encoding for glycolytic enzymes, enzymes of reactions in both the PPP and the ED pathway and TCA cycle. For D. shibae, pyruvate carboxylase (PYRCx) and phosphoenolpyruvate carboxykinase (PEPCk) were found to be the interconnecting reactions between the C3 and the C4 pool. Furthermore, a gene encoding for pyruvate orthophosphate dikinase (PPDK) is annotated, indicating a potential exchange flux between the PYR and PEP pool. A summary of all reactions considered is presented in Figure 1. To resolve the metabolic fluxes through catabolic pathways and around important branch points within the metabolic network, appropriate approaches involving the mass patterns of different amino acid fragments were developed.
Strategy for the estimation of glucose catabolic fluxes
In Figure 3 the theoretical labelling patterns of the C3 pool depending on the activity of the glycolysis, PPP and ED pathways are presented. It can be taken from the illustration that the combined analysis of two fragments derived from PYR (Ala [M-57] and Ala [M-85]) enables the contributions of each pathway to be resolved. The scheme for the estimation of the major catabolic pathways is shown in Figure 6. A comparison of the theoretical mass distribution pattern of the Ala [M-57] fragment derived from the activity of each pathway and the experimental data allows differentiation between the activity of the PPP and the combined flux through EMP and EDP (Eq. 2). The latter cannot be further subdivided as the resulting mass patterns for Ala [M-57] are similar for both pathways. The Ala [M-85] fragment therefore provides additional information for complete resolution of the three catabolic pathways. Its theoretical mass distribution compared to the experimental data yields the activity of the EMP pathway and the combined flux through EDP and PPP (Eq. 3).
| (2) |
Figure 6.

Strategy to estimate relative flux through major catabolic pathways. To completely resolve the contribution of each route, theoretical mass distributions of the [M-57] and [M-85] fragments of alanine were compared to the experimental data. In this schematic illustration, white circles represent unlabelled (12C) carbon whereas black circles indicate labelled (13C) carbon. The numbers given reflect the position of the carbon atom within the molecule. EDP: Entner-Doudoroff pathway; EMP: Embden-Meyerhof-Parnas pathway; PPP: pentose phosphate pathway.
Strategy for estimating fluxes around the PEP pool
The metabolic reaction network around the PEP node is presented in Figure 7. It contains all reactions for which the corresponding genes have been annotated in the KEGG database. The pathways through lower glycolysis and the reactions catalysed by phosphoenolpyruvate carboxykinase (PEPCk) and pyruvate orthophosphate dikinase (PPDK) yielding PEP from either OAA or PYR are considered. Fluxes into the PEP pool were resolved using the mass distribution patterns of the [f302] fragments (carbon atoms at position C1 and C2) of the amino acids directly connected to the PEP pool according to Equations 4 and 5.
| (4) |
Figure 7.
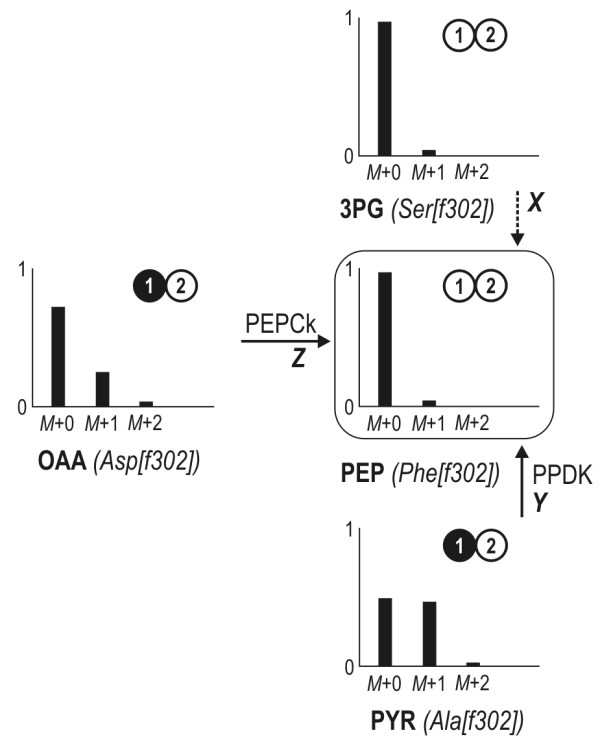
Estimation of fluxes into the PEP pool. The fractional contribution of each alternative pathway (X, Y, Z) directly follows from the mass pattern of the [f302] fragments of the precursors 3-phosphoglycerate (serine), oxaloacetate (aspartate), pyruvate (alanine) and the product PEP (phenylalanine). The corresponding isotopomer of each molecule is illustrated next to the experimental data (mass spectra). White circles represent 12C whereas black circles indicate labelled 13C. The numbers given reflect the position of the carbon atom within the molecule. PEPCk: phosphoenolpyruvate carboxykinase; PPDK: pyruvate-orthophosphate dikinase.
Abbreviations
[1-13C] glucose: Glucose labelled at C1-position; 3PG: 3-phosphoglycerate; 6PG: 6-phosphogluconate; AcCoA: Acetyl-Coenzyme A; Ala: Alanine; CLE: labelling experiment; EDA: 2-keto-3-deoxy-6-phosphogluconate aldolase; EDD: 6-phosphogluconate dehydrogenase; EDP: Entner-Doudoroff pathway; EMP: Embden-Meyerhof-Parnas; F6P: Fructose-6-phosphate; FUM: Fumarate; G6P: Glucose 6-phosphate; GAP: Glyceraldehyde 3-phosphate; GC/MS: Gas chromatography/mass spectrometry; KDPG: 2-keto-3-deoxy-6-phosphogluconate; MAL: Malate; MDV: Mass distribution vector; OAA: Oxaloacetate; OD: Optical density; OGA: 2-oxoglutarate; PEP: Phosphoenolpyruvate; PEPCk, pckA: Phosphoenolpyruvate carboxykinase; PFK: 6-phosphofructokinase; Phe: Phenylalanine; PPP: Pentose phosphate pathway; PPDK: ppdK Pyruvate orthophosphate dikinase; PYR: Pyruvate; PYRCx, pycA: Pyruvate carboxylase; R5P: Ribose 5-phosphate; Ser: Serine; SUC: Succinate; TCA: Tricarboxylic acid; Tyr: Tyrosine
Authors' contributions
TF carried out the labelling analytics and data processing, performed the flux calculations and drafted the manuscript together with CW. MP performed the cultivation experiments for D. shibae. HZ performed the cultivation experiments for P. gallaeciensis. JT assisted in method set-up for cultivation and analytics. IWD helped to draft the manuscript. RR helped to draft the manuscript. CW conceived, designed and coordinated the study and drafted the manuscript together with TF. All authors read and approved the final manuscript.
Contributor Information
Tobias Fürch, Email: t.fuerch@tu-bs.de.
Matthias Preusse, Email: m.preusse@tu-bs.de.
Jürgen Tomasch, Email: jtomasch@gmx.de.
Hajo Zech, Email: hajo.zech@icbm.de.
Irene Wagner-Döbler, Email: Irene.Wagner-Doebler@helmholtz-hzi.de.
Ralf Rabus, Email: rrabus@mpi-bremen.de.
Christoph Wittmann, Email: c.wittmann@tu-bs.de.
Acknowledgements
JT and IWD gratefully acknowledge the support of the Volkswagen Foundation under the grant VW-Vorab (ZN 2182, "Comparative functional genome analysis of representative members of the Roseobacter Clade"). We are grateful to Renate Gahl-Janssen (Oldenburg) for technical assistance. HZ and RR acknowledge support from of the Volkswagen Foundation under the grant VW-Vorab (ZN2235, "Comparative functional genome analysis of representative members of the Roseobacter clade") and the Marine Microbiology Initiative of the Moore Foundation (USA).
References
- Biebl H, Allgaier M, Tindall BJ, Koblizek M, Lunsdorf H, Pukall R, Wagner-Döbler I. Dinoroseobacter shibae gen. nov., sp. nov., a new aerobic phototrophic bacterium isolated from dinoflagellates. Int J Syst Evol Microbiol. 2005;55(Pt 3):1089–1096. doi: 10.1099/ijs.0.63511-0. [DOI] [PubMed] [Google Scholar]
- Buchan A, Gonzalez JM, Moran MA. Overview of the marine roseobacter lineage. Appl Environ Microbiol. 2005;71(10):5665–5677. doi: 10.1128/AEM.71.10.5665-5677.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard EC, Henriksen JR, Buchan A, Reisch CR, Burgmann H, Welsh R, Ye W, Gonzalez JM, Mace K, Joye SB. Bacterial taxa that limit sulfur flux from the ocean. Science. 2006;314(5799):649–652. doi: 10.1126/science.1130657. [DOI] [PubMed] [Google Scholar]
- Kiene RP, Linn LJ, Gonzalez J, Moran MA, Bruton JA. Dimethylsulfoniopropionate and methanethiol are important precursors of methionine and protein-sulfur in marine bacterioplankton. Appl Environ Microbiol. 1999;65(10):4549–4558. doi: 10.1128/aem.65.10.4549-4558.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King GM. Molecular and culture-based analyses of aerobic carbon monoxide oxidizer diversity. Appl Environ Microbiol. 2003;69(12):7257–7265. doi: 10.1128/AEM.69.12.7257-7265.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan A, Collier LS, Neidle EL, Moran MA. Key aromatic-ring-cleaving enzyme, protocatechuate 3,4-dioxygenase, in the ecologically important marine Roseobacter lineage. Appl Environ Microbiol. 2000;66(11):4662–4672. doi: 10.1128/AEM.66.11.4662-4672.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan A, Neidle EL, Moran MA. Diversity of the ring-cleaving dioxygenase gene pcaH in a salt marsh bacterial community. Appl Environ Microbiol. 2001;67(12):5801–5809. doi: 10.1128/AEM.67.12.5801-5809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurkov VV, Beatty JT. Aerobic anoxygenic phototrophic bacteria. Microbiol Mol Biol Rev. 1998;62(3):695–724. doi: 10.1128/mmbr.62.3.695-724.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béjà O, Suzuki MT, Heidelberg JF, Nelson WC, Preston CM, Hamada T, Eisen JA, Fraser CM, DeLong EF. Unsuspected diversity among marine aerobic anoxygenic phototrophs. Nature. 2002;415(6872):630–633. doi: 10.1038/415630a. [DOI] [PubMed] [Google Scholar]
- Kolber ZS, Plumley FG, Lang AS, Beatty JT, Blankenship RE, VanDover CL, Vetriani C, Koblizek M, Rathgeber C, Falkowski PG. Contribution of aerobic photoheterotrophic bacteria to the carbon cycle in the ocean. Science. 2001;292(5526):2492–2495. doi: 10.1126/science.1059707. [DOI] [PubMed] [Google Scholar]
- Kolber ZS, Van Dover CL, Niederman RA, Falkowski PG. Bacterial photosynthesis in surface waters of the open ocean. Nature. 2000;407(6801):177–179. doi: 10.1038/35025044. [DOI] [PubMed] [Google Scholar]
- Wagner-Döbler I, Ballhausen B, Berger M, Brinkhoff T, Buchholz I, Bunk B, Cypionka H, Daniel R, Drepper T, Gerdts G. The complete genome sequence of the algal symbiont Dinoroseobacter shibae: a hitchhiker's guide to life in the sea. Isme J. 2009. in press . [DOI] [PubMed]
- Swingley WD, Sadekar S, Mastrian SD, Matthies HJ, Hao J, Ramos H, Acharya CR, Conrad AL, Taylor HL, Dejesa LC. The complete genome sequence of Roseobacter denitrificans reveals a mixotrophic rather than photosynthetic metabolism. J Bacteriol. 2007;189(3):683–690. doi: 10.1128/JB.01390-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens T, Heidorn T, Pukall R, Simon M, Tindall BJ, Brinkhoff T. Reclassification of Roseobacter gallaeciensis Ruiz-Ponte et al. 1998 as Phaeobacter gallaeciensis gen. nov., comb. nov., description of Phaeobacter inhibens sp. nov., reclassification of Ruegeria algicola (Lafay et al. 1995) Uchino et al. 1999 as Marinovum algicola gen. nov., comb. nov., and emended descriptions of the genera Roseobacter, Ruegeria and Leisingera. Int J Syst Evol Microbiol. 2006;56(Pt 6):1293–1304. doi: 10.1099/ijs.0.63724-0. [DOI] [PubMed] [Google Scholar]
- Alavi MR. Predator/prey interaction between Pfiesteria piscicida and Rhodomonas mediated by a marine alpha proteobacterium. Microb Ecol. 2004;47(1):48–58. doi: 10.1007/s00248-003-1018-7. [DOI] [PubMed] [Google Scholar]
- Christensen B, Nielsen J. Metabolic network analysis of Penicillium chrysogenum using (13)C-labeled glucose. Biotechnol Bioeng. 2000;68(6):652–659. doi: 10.1002/(SICI)1097-0290(20000620)68:6<652::AID-BIT8>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Dauner M, Bailey JE, Sauer U. Metabolic flux analysis with a comprehensive isotopomer model in Bacillus subtilis. Biotechnol Bioeng. 2001;76(2):144–156. doi: 10.1002/bit.1154. [DOI] [PubMed] [Google Scholar]
- Fürch T, Hollmann R, Wittmann C, Wang W, Deckwer WD. Comparative study on central metabolic fluxes of Bacillus megaterium strains in continuous culture using 13C labelled substrates. Bioprocess Biosyst Eng. 2007;30(1):47–59. doi: 10.1007/s00449-006-0095-7. [DOI] [PubMed] [Google Scholar]
- Wittmann C, Hans M, van Winden WA, Ras C, Heijnen JJ. Dynamics of intracellular metabolites of glycolysis and TCA cycle during cell-cycle-related oscillation in Saccharomyces cerevisiae. Biotechnol Bioeng. 2005;89(7):839–847. doi: 10.1002/bit.20408. [DOI] [PubMed] [Google Scholar]
- Fischer E, Zamboni N, Sauer U. High-throughput metabolic flux analysis based on gas chromatography-mass spectrometry derived 13C constraints. Anal Biochem. 2004;325(2):308–316. doi: 10.1016/j.ab.2003.10.036. [DOI] [PubMed] [Google Scholar]
- Sauer U, Hatzimanikatis V, Bailey JE, Hochuli M, Szyperski T, Wüthrich K. Metabolic fluxes in riboflavin-producing Bacillus subtilis. Nat Biotechnol. 1997;15(5):448–452. doi: 10.1038/nbt0597-448. [DOI] [PubMed] [Google Scholar]
- Wendisch VF, de Graaf AA, Sahm H, Eikmanns BJ. Quantitative determination of metabolic fluxes during coutilization of two carbon sources: comparative analyses with Corynebacterium glutamicum during growth on acetate and/or glucose. J Bacteriol. 2000;182(11):3088–3096. doi: 10.1128/JB.182.11.3088-3096.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessie TG, Phibbs PV Jr. Alternative pathways of carbohydrate utilization in pseudomonads. Annu Rev Microbiol. 1984;38:359–388. doi: 10.1146/annurev.mi.38.100184.002043. [DOI] [PubMed] [Google Scholar]
- Lynn AR, Sokatch JR. Incorporation of isotope from specifically labeled glucose into alginates of Pseudomonas aeruginosa and Azotobacter vinelandii. J Bacteriol. 1984;158(3):1161–1162. doi: 10.1128/jb.158.3.1161-1162.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zech H, Thole S, Schreiber K, Kalhöfer D, Voget S, Schomburg D, Rabus R. Growth phase-dependent global protein and metabolite profiles of Phaeobacter gallaeciensis strain DSM 1 a member of the marine Roseobacter clade. Proteomics. 7395;9:3677–3697. doi: 10.1002/pmic.200900120. [DOI] [PubMed] [Google Scholar]
- Kiefer P, Heinzle E, Zelder O, Wittmann C. Comparative metabolic flux analysis of lysine-producing Corynebacterium glutamicum cultured on glucose or fructose. Appl Environ Microbiol. 2004;70(1):229–239. doi: 10.1128/AEM.70.1.229-239.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann C, Hans M, Heinzle E. In vivo analysis of intracellular amino acid labelings by GC/MS. Anal Biochem. 2002;307(2):379–382. doi: 10.1016/S0003-2697(02)00030-1. [DOI] [PubMed] [Google Scholar]
- Guo ZK, Lee WN, Katz J, Bergner AE. Quantitation of positional isomers of deuterium-labeled glucose by gas chromatography/mass spectrometry. Anal Biochem. 1992;204(2):273–282. doi: 10.1016/0003-2697(92)90238-3. [DOI] [PubMed] [Google Scholar]
- Lee WN, Byerley LO, Bergner EA, Edmond J. Mass isotopomer analysis: theoretical and practical considerations. Biol Mass Spectrom. 1991;20(8):451–458. doi: 10.1002/bms.1200200804. [DOI] [PubMed] [Google Scholar]
- Gardner PR, Fridovich I. Superoxide sensitivity of the Escherichia coli 6-phosphogluconate dehydratase. J Biol Chem. 1991;266(3):1478–1483. [PubMed] [Google Scholar]
- Peng L, Shimizu K. Global metabolic regulation analysis for Escherichia coli K12 based on protein expression by 2-dimensional electrophoresis and enzyme activity measurement. Appl Microbiol Biotechnol. 2003;61(2):163–178. doi: 10.1007/s00253-002-1202-6. [DOI] [PubMed] [Google Scholar]
- Gancedo JM, Gancedo C. Fructose-1,6-diphosphatase, phosphofructokinase and glucose-6-phosphate dehydrogenase from fermenting and non fermenting yeasts. Arch Mikrobiol. 1971;76(2):132–138. doi: 10.1007/BF00411787. [DOI] [PubMed] [Google Scholar]
- Fischer E, Sauer U. Metabolic flux profiling of Escherichia coli mutants in central carbon metabolism using GC-MS. Eur J Biochem. 2003;270(5):880–891. doi: 10.1046/j.1432-1033.2003.03448.x. [DOI] [PubMed] [Google Scholar]
- Szyperski T. Biosynthetically directed fractional 13C-labeling of proteinogenic amino acids. An efficient analytical tool to investigate intermediary metabolism. Eur J Biochem. 1995;232(2):433–448. doi: 10.1111/j.1432-1033.1995.tb20829.x. [DOI] [PubMed] [Google Scholar]
- Becker J, Klopprogge C, Wittmann C. Metabolic responses to pyruvate kinase deletion in lysine producing Corynebacterium glutamicum. Microb Cell Fact. 2008;7:8. doi: 10.1186/1475-2859-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]