

Abstract

Efficient [2+2] heterodimerizations of dissimilar acyclic enones can be accomplished upon visible light irradiation in the presence of a ruthenium(II) photocatalyst. Similar cycloadditions under standard UV photolysis conditions are inefficient and unselective. Nevertheless, a diverse range of unsymmetrical tri- and tetrasubstituted cyclobutane structures can be produced in good yields and excellent diastereoselectivities using this new method. The reaction is promoted by any visible light source, and efficient, gram-scale cycloadditions can be conducted upon irradiating with ambient sunlight.
The first example of a photoinitiated [2+2] enone cycloaddition was reported by Ciamician in 1908 and involved the intramolecular cyclobutanation of carvone upon prolonged exposure to intense sunlight.1 In the century since this initial discovery, [2+2] enone cycloaddition reactions promoted by UV irradiation have become recognized as an efficient method for the construction of cyclobutane-containing structures.2 Nevertheless, the synthetic utility of this reaction has been largely limited to cycloadditions of cyclic enones. Photoexcited acyclic enones undergo rapid relaxation to the ground state by cis-trans isomerization,3 an energy-wasting process that dramatically diminishes the efficiency of productive cycloaddition. Thus, efficient intermolecular photochemical [2+2] cycloadditions of acyclic enones are a long-standing unsolved problem in synthetic chemistry.
Recently, our laboratory has become interested in designing new reactions initiated by visible light photoexcitation of organometallic complexes.4 We have reported that Ru(bipy)32+ serves as an excellent photocatalyst for intramolecular anion radical-mediated [2+2] enone cycloadditions,5 which were first described by Krische and Bauld.6 In accord with this precedent, we found that only aryl enones could be used to initiate the cycloaddition, presumably due to the greater ease of generating the requisite radical anion intermediate, but that the reacting partner could be any suitable Michael acceptor (Scheme 1). In contrast to the Krische and Bauld precedent, however, efficient intermolecular dimerizations of enones could be performed under the photocatalytic conditions we had developed. We wondered, therefore, if crossed intermolecular [2+2] heterodimerizations would be possible using two dissimilar enone substrates.
Scheme 1.
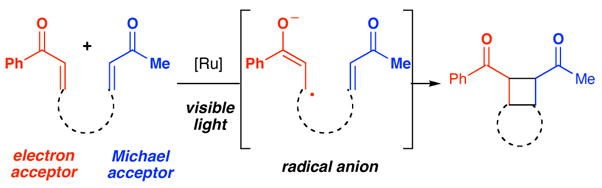
We first set out to probe the feasibility of the intermolecular heterocoupling process by studying the reaction shown in eq 1. The design of this experiment was based upon two main considerations. First, given the requirement that a reducible aryl enone serve as an electrophore in this process, we selected enone 1 to serve as the precursor to the key radical anion intermediate. Second, we recognized that the reaction partner must be a more reactive Michael acceptor than 1 in order to minimize the undesired homodimerization, but be less susceptible to one-electron reduction by the photocatalyst. Ultimately, we selected methyl vinyl ketone 2, which we expected should be less easily reduced than 1, but, due to the lack of a β-substituent, should serve as a better electrophilic reaction partner for the radical anion intermediate. In the event, we observed highly chemoselective formation of the crossed [2+2] cycloadduct 3 in 84% isolated yield (eq 1). Only trace amounts of products arising from the homocoupling of 1 and none of the product from dimerization of 2 could be observed upon NMR analysis of the reaction mixture. In addition, the cycloaddition proceeded with excellent diastereoselectivity (>10:1 dr).7
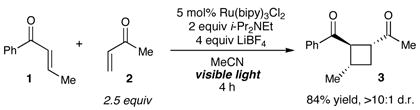 |
(1) |
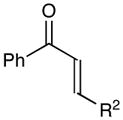
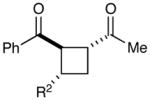
Next, we examined the scope of the aryl enone in the crossed cycloaddition, using 2 as the Michael acceptor (Table 1). Both electron-poor and electron-rich substrates are amenable to cycloaddition (entries 1–3), as are heteroaryl enones (entry 4). On the other hand, aliphatic enones are completely unreactive (entry 5), which is consistent with our design plan and our understanding of the mechanism of this process. Variation of the β-substituent is also possible; however, the reaction is sensitive to steric bulk. While reactions of enones bearing primary aliphatic substituents proceed smoothly (entry 6), secondary substituents at this position retard the reaction rate (entry 7), and reactions of enones bearing tertiary alkyl substituents fail to proceed at synthetically useful rates (entry 8). On the other hand, heteroatom-bearing substituents are easily tolerated (entry 9).
Table 1.
Effect of aryl enone structure in photocatalytic crossed cycloadditions with methyl vinyl ketone 2a
| entry | enone | cycloadduct | yieldb | d.r.b |
|---|---|---|---|---|
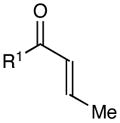 |
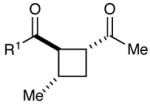 |
|||
| 1 | R1 = Ph | 84% | >10:1 | |
| 2 | R1 = 4-ClPh | 82% | >10:1 | |
| 3 | R1 = 4-MeOPh | 53% | >10:1 | |
| 4 | R1 = 2-furyl | 74% | >10:1 | |
| 5 | R1 = Et | 0% | -- | |
 |
 |
|||
| 6 | R2 = Et | 70% | 6:1 | |
| 7c | R2 = i-Pr | 64% | >10:1 | |
| 8d | R2 = t-Bu | 8% | >10:1 | |
| 9 | R2 = CH2OBn | 61% | >10:1 | |
Unless otherwise noted, reactions conducted using 2.5 equiv 2 and 5 mol% Ru(bipy)3Cl2 in the presence of LiBF4 and i-Pr2NEt in MeCN. Irradiation time using a 23 W compact fluorescent bulb at 30 cm was 4 h.
Isolated yields and diastereomer ratios are the averaged results of two reproducible experiments.
12 h irradiation.
24 h irradiation.
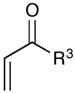
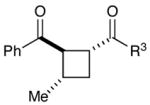
Experiments exploring the generality of the crossed reaction with respect to the Michael acceptor are summarized in Table 2. A variety of aliphatic enones are good reaction partners for the crossed [2+2] cycloaddition (e.g., entries 1–2). Acrylate esters also participate, but given their reduced electrophilicity, several additional equivalents are required to out-compete homodimerization of 1 (entry 3). Thioesters, on the other hand, are excellent acceptor enones and provide high yields of the heterocoupling product (entry 4). Substrates bearing alkyl substituents at the α-position also participate and provide access to cyclobutane structures bearing all-carbon quaternary stereocenters with high selectivity (entry 5). On the other hand, as expected, β-substituents that sterically deactivate the Michael acceptor ability of the enone cause homodimerization of 1 to predominate. Nevertheless, the heterocoupling product can be isolated when a larger excess of the acceptor enone is used (entries 6 and 7).
Table 2.
Effect of Michael acceptor structure in photocatalytic crossed cycloadditions with phenyl enone 1a
| entry | enone | cycloadduct | yieldb | d.r.b |
|---|---|---|---|---|
 |
 |
|||
| 1 | R3 = Me | 84% | >10:1 | |
| 2 | R3 = Et | 76% | >10:1 | |
| 3c | R3 = OMe | 65% | 5:1 | |
| 4 | R3 = SEt | 88% | >10:1 | |
| 5c | 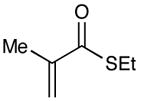 |
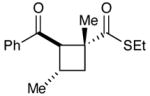 |
57% | 5:1 |
| 6 | 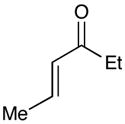 |
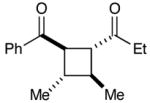 |
9% | -- |
| 7c,d | 35% | >10:1 | ||
Unless otherwise noted, reactions conducted using 1 equiv of 1, 2.5 equiv of Michael acceptor, and 5 mol% Ru(bipy)3Cl2 in the presence of LiBF4 and i-Pr2NEt in MeCN. Irradiation time using a 23 W compact fluorescent bulb at 30 cm was 4 h.
Isolated yields and diastereomer ratios are the averaged results of two reproducible experiments.
6 equiv of Michael acceptor and 12 h irradiation time.
The homodimerization product of 1 was isolated in 65% yield.
An important feature of this process is that the reaction is initiated by photoexcitation of the ruthenium catalyst and does not access electronically excited states of the enone. Thus, this method avoids some of the synthetic limitations of cycloadditions conducted under standard UV photolysis conditions. A solution of enones 1 and 2 irradiated in a Rayonet reactor (300 nm) fails to produce any observable cycloaddition products; the only new products observed in the reaction mixture arise from E/Z isomerization of enone 1 (eq 2). On the other hand, in the presence of the Ru(bipy)32+ photocatalyst, this reaction proceeds upon irradiation with any visible light source. In order to demonstrate this principle, we conducted a gram-scale cycloaddition experiment on the roof of our laboratory building (eq 3). The [2+2] cycloaddition between 1 and 2 was equally efficient in ambient sunlight as it was in our laboratory experiments and produced a single diastereomer of the crossed cycloadduct 3 in 84% yield.
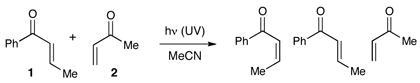 |
(2) |
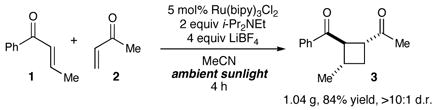 |
(3) |
In summary, we have developed an efficient method for crossed [2+2] cycloadditions of acyclic enones promoted by visible light. The excellent chemo- and stereoselectivity observed in this reaction represents a considerable advance in the construction of strained four-membered rings and should have a significant impact on the approach towards synthesis of cyclobutane-containing structures. Furthermore, the success of this method provides corroborative evidence for our mechanistic hypothesis and establishes a solid framework for the further development of our research program in visible light photocatalysis.
Supplementary Material
Acknowledgments
We thank the Beckman Foundation and the Research Corporation for financial support. The NMR facilities at UW-Madison are funded by the NSF (CHE-9208463, CHE-9629688) and NIH (RR08389-01).
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds (PDF format) are provided. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Ciamician G, Silber P. Chem Ber. 1908;41:1928–1935. [Google Scholar]
- (2).For recent reviews of [2+2] enone photocycloadditions, see: Baldwin SW. In: Organic Photochemistry. Padwa A, editor. Vol. 5. Marcel Dekker; New York: 1981. pp. 123–225.Crimmins MT. Chem Rev. 1988;88:1453–1473.Schuster DI, Lem G, Kaprinidis NA. Chem Rev. 1993;93:3–22.Bach T. Synthesis. 1998:683–703.Iriondo-Alberdi J, Greaney MF. Eur J Org Chem. 2007:4801–4815.Hoffmann N. Chem Rev. 2008;108:1052–1103. doi: 10.1021/cr0680336.
- (3).Morrison H, Rodriguez O. J Photochem. 1974;3:471–474. [Google Scholar]
- (4).For reviews of transition metal-mediated photocatalysis in organic synthesis, see: Salomon RG. Tetrahedron. 1983;39:485–575.Hennig H. Coord Chem Rev. 1999;182:101–123.
- (5).Ischay MA, Anzovino ME, Du J, Yoon TP. J Am Chem Soc. 2008;130:12886–12887. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]
- 6.(a) Baik TG, Luis AL, Wang LC, Krische MJ. J Am Chem Soc. 2001;123:6716–6717. doi: 10.1021/ja010800p. [DOI] [PubMed] [Google Scholar]; (b) Wang LC, Jang HY, Roh Y, Lynch V, Schultz AJ, Wang X, Krische MJ. J Am Chem Soc. 2002;124:9448–9453. doi: 10.1021/ja020223k. [DOI] [PubMed] [Google Scholar]; (c) Roh Y, Jang HY, Lynch V, Bauld NL, Krische MJ. Org Lett. 2002;4:611–613. doi: 10.1021/ol0172065. [DOI] [PubMed] [Google Scholar]; (d) Yang J, Cauble DF, Berro AJ, Bauld NL, Krische MJ. J Org Chem. 2004;69:7979–7984. doi: 10.1021/jo048499t. [DOI] [PubMed] [Google Scholar]; (e) Yang J, Felton GAN, Bauld NL, Krische MJ. J Am Chem Soc. 2004;126:1634–1635. doi: 10.1021/ja030543j. [DOI] [PubMed] [Google Scholar]
- (7).Although LiBF4 and iPr2NEt are not consumed in the reaction, highly diastereoselective cycloaddition is only observed when these additives are present in excess. The reason for this dependence is not clear at this time and is the subject of ongoing investigation in our lab.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.