

Abstract
Nanocomposite membranes based on thermosensitive, poly(N-isopropylacrylamide)-based nanogels and magnetite nanoparticles have been designed to achieve “on-demand” drug delivery upon the application of an oscillating magnetic field. On-off release of sodium fluorescein over multiple magnetic cycles has been successfully demonstrated using prototype membrane-based devices. The total drug dose delivered was directly proportional to the duration of the “on” pulse. The membranes were non-cytotoxic, biocompatible, and retained their switchable flux properties after 45 days of subcutaneous implantation.
Delivery devices that allow remote, repeatable, and reliable switching of drug flux could have a marked impact on the treatment of a variety of medical conditions. An ideal device for on-demand drug delivery should safely contain a large quantity of drug, release little or no drug in the “off” state, be repeatedly switchable to the “on” state without mechanically disrupting the device, and be triggered non-invasively to release a consistent dosage demanded by a patient (e.g. local pain relief) or prescribed by a doctor (e.g. localized chemotherapy).
Despite the clear clinical need, few such drug delivery devices have been developed and none are available for clinical use. Existing technologies are particularly limited by their inability to be effectively triggered in vivo in the absence of a local implanted heat source, their lack of reproducible release over multiple thermal cycles, their slow response times to stimuli, and/or their inability to dynamically adjust drug dosing according to patient needs. Currently, no existing device overcomes all of these limitations. For example, radio frequency-activated microchips containing drug-filled reservoirs can achieve rapid on-demand drug delivery1, 2 but deliver only fixed doses of drug and require implanted electronics. Near-IR responsive nanoparticles consisting of mixtures of PNIPAM and gold-gold sulfide nanoshells can release proteins on demand but deliver inconsistent doses upon multiple triggering cycles3. Ferrofluid-loaded polymer sheets4, liposomes5, microspheres6, 7, microcapsules8, 9, and nanospheres10-12 can be activated remotely by magnetic induction but typically achieve either single burst release events or inconsistent dosing over multiple thermal cycles due to the use of mechanical disruption of the drug-polymer matrix as the flux triggering mechanism. Hence, alternative technologies are needed.
Hydrogels13-18, gel-based microparticles19 or nanoparticles20-23, and surface-grafted polymers24-36 based on thermosensitive poly(N-isopropylacrylamide) (PNIPAM) have been frequently used in triggerable devices. With heating, PNIPAM undergoes a reversible discontinuous phase transition in water, switching from hydrophilic to hydrophobic37. In a PNIPAM-based hydrogel, this phase transition induces a deswelling response which typically reduces drug flux from the hydrogel. Alternately, when PNIPAM is used to fill the pores of a membrane, the pores are opened upon heating as the entrapped polymer shrinks, increasing drug flux through the membrane28, 38. Such membranes have been designed by grafting poly(N-isopropylacrylamide) to existing membrane networks5 or by entrapping PNIPAM microgels within a membrane matrix39. However, existing PNIPAM-based devices would be permanently “on” at physiological temperature (37°C) since their transition temperatures are ∼32°C. Existing technologies would also require use of an implanted heating system for effective in vivo activation.
Here, we developed a composite membrane based on multiple engineered smart nanoparticles which enabled rapid, repeatable, and tunable drug delivery upon the application of an external oscillating magnetic field. The membrane consisted of ethylcellulose (the membrane support), superparamagnetic magnetite nanoparticles (the triggering entity), and thermosensitive poly(N-isopropylacrylamide) (PNIPAM)-based nanogels37 (the switching entity). Membranes were prepared by co-evaporation so that the nanogel and magnetite nanoparticles were entrapped in ethylcellulose to form a presumably disordered network. Surface-etching x-ray photoelectron spectroscopy (XPS) showed that the membranes had a relatively uniform composition within the bulk but relatively less iron (ferrofluid) near the membrane surface (Figure 1a). The membrane nanogel composition determined by XPS (23% by dry weight) correlated well with the nanogel concentration in the pre-membrane suspension (25% by dry weight).
Figure 1.
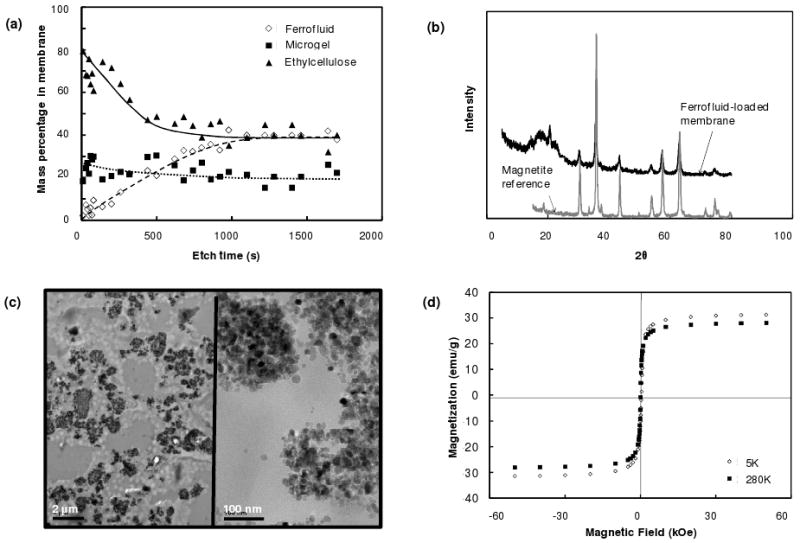
Physicochemical membrane characterization: (a) Mass percentages of ferrofluid, nanogel, and ethyl cellulose in membrane as a function of etch time, by XPS; (b) XRD spectrum of ferrofluid-loaded membranes in comparison to a magnetite-only control; (c) Transmission electron micrographs of ferrofluid distribution and size within the composite membrane; (2 μm size bar, left panel; 100nm size bar, right panel); (d) Magnetization curves for composite membranes measured at 5K and 280K.
X-ray diffraction analysis suggested that ferrofluid particles had a magnetite crystal structure and an average crystallite size of ∼12 nm (Figure 1b). Transmission electron microscopy of a membrane section (Figure 1c) confirmed the average ferrofluid particle size of 10-25 nm and suggested that local ferrofluid clusters sized between 0.1 - 3 μm were distributed throughout the membrane. The magnetic material within the membranes had a magnetic saturation value of 96.5 emu/g(Fe3O4) at 280 K (Figure 1d), similar to values previously reported for bulk magnetite (93-96 emu/g)40. Furthermore, the measured coercive field of 346 ± 4 Oe at 5 K is consistent with that of previously reported ferrofluid particles of similar size41. These results suggested that the ferrofluid particles consisted of a single magnetic domain (i.e. all iron was in magnetite form) and had the superparamagnetic properties and average particle size required for effective magnetic induction heating in an oscillating magnetic field42, 43. It should be emphasized that inductive heating of single-domain magnetic nanoparticles of ∼10-20 nm such as the magnetite particles used in this work depends strongly on the primary particle size but only weakly on particle aggregation43.
To facilitate effective in vivo triggering, the nanogels were engineered to remain swollen (i.e. in the “off” state) at physiological temperature by copolymerizing N-isopropylacrylamide (NIPAM) with N-isopropylmethacrylamide (NIPMAM) and acrylamide (AAm). The methyl group of NIPMAM sterically inhibits the phase transition44 while AAm is more hydrophilic than NIPAM45, both shifting the phase transition to higher temperatures. The ratio between the monomers was chosen to maximize the size change from the swollen to the collapsed state, in order to optimize membrane pore opening when triggered.
The ability of the membrane constituents and the composite membrane to trigger at physiologically relevant temperatures was evaluated using both thermal and magnetic stimuli. Nanogels in free suspension in PBS underwent a ∼400 nm change in diameter upon heating from physiological temperature to 50°C (Figure 2a), with >90% of the total deswelling transition completed at 43°C. Thermal triggering of the nanogel-containing membrane was tested by placing it between two chambers of a glass flow cell submerged in a water bath and evaluating the flux of sodium fluorescein across the membrane (i.e. between the chambers) as a function of time and temperature. A ∼20-fold higher flux of sodium fluorescein occurred at temperatures exceeding the volume phase transition temperature (∼40°C) of the nanogels (Figure 2a). The fluorescein flux could be switched on and off over multiple thermal cycles with high reproducibility, suggesting that the nanogel phase transition inside the membrane pores was fully reversible.
Figure 2.
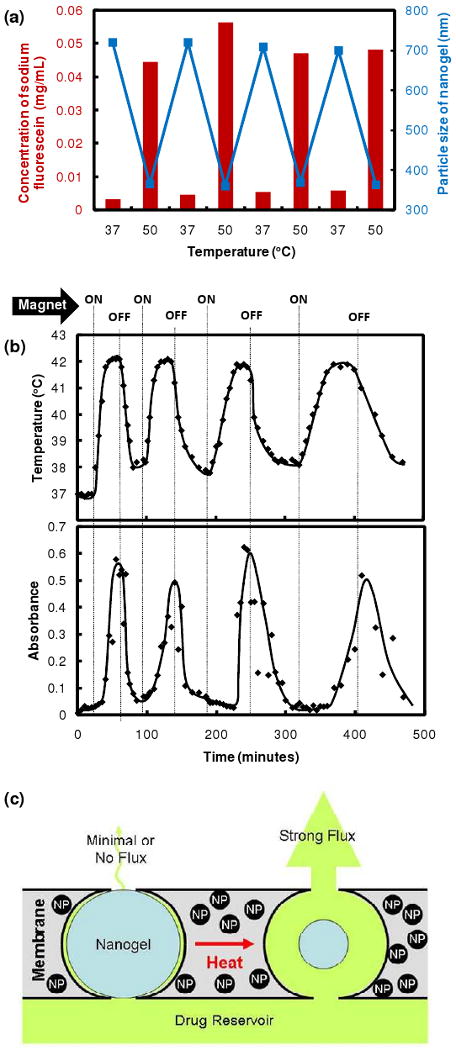
Stimulus-responsive membrane triggering in vitro: (a) Temperature-triggering: comparison of nanogel particle size in suspension (blue data, right y-axis) and differential flux of sodium fluorescein through the nanogel-loaded membranes (red data, left y-axis) as a function of temperature; (b) Magnetic triggering: temperature profile in the sample chamber and differential flux of sodium fluorescein out of membrane-capped devices as a function of time over four successive on/off cycles of the external magnetic field; (c) schema of the proposed mechanism of membrane function
No drug flux was observed through membranes containing magnetite nanoparticles but no nanogel at any temperature in the range 37°C to 50°C. Furthermore, membranes fabricated with PNIPAM-only nanogels with a transition temperature of ∼32°C 46 (i.e. the nanogel was fully collapsed at both 37°C and 50°C) showed no significant differential flux when cycling between 37°C and 50°C. FT-IR analysis also confirmed that the discontinuous increase in membrane permeability coincided with a change in the hydrogen bonding within the membrane, consistent with the occurrence of a nanogel volume phase transition. Together, these observations suggest that the observed differential drug flux is associated with the phase transition properties of the nanogel and not mechanical disruption of the membrane or temperature-enhanced free diffusion of the drug.
Magnetic triggering was evaluated in small-scale devices made by gluing two 1 cm diameter membrane disks to the ends of a 1 cm length of silicone tubing filled with a sodium fluorescein solution (Supplementary Figure S11). The devices were mounted singly inside a semi-adiabatic flow cell in a solenoid coil, with constant water flow through the flow cell to permit continuous sampling of fluorescein release. Figure 2b shows the magnetic triggering of the composite membrane. The magnetic nanoparticles embedded in the membrane heated inductively when subjected to an external oscillating magnetic field, heating previously attributed to power absorption and subsequent magnetic relaxation of single-domain nanoparticles47. At the applied magnetic frequency and field amplitude, the water inside the semi-adiabatic flow cell heated from 37°C to ∼42°C over the course of ∼10 minutes, at which point the temperature reached steady state. Heat generated by magnetite induction heating was transferred to the adjacent thermosensitive nanogels, causing the nanogels to shrink and permit drug diffusion out of the device. When the magnetic field was turned off, the device cooled, causing the nanogels to re-swell and refill the membrane pores. As a result, the drug flux returned back to a near-zero value (Figure 2c). As in the thermally-activated experiments, a 10-to-20-fold differential flux was observed between the “off” and “on” states. Furthermore, multiple on-off cycles could be performed without significantly changing the permeability of the membrane in the “off” state. This reproducibility suggests that magnetically-triggered physical distortion of the device42 plays no significant role in accelerating drug release from the membrane-based devices.
The membrane-based devices also permitted precise control of the amount of drug released as a function of the duration of the magnetic pulse. Table 1 shows the dose of fluorescein delivered for each of the four magnetically-activated cycles shown in Figure 2, calculated by integrating the area under the absorbance vs. time curve for each cycle. The mass of compound released over each triggering cycle varied directly with the duration of the magnetic pulse (R2 = 0.995), with the rate of drug release varying by less than 10% in each cycle. Thus, drug release could be controlled by modulating both the frequency and duration of magnetic pulse.
Table 1.
Total mass of sodium fluorescein release and rate of drug release during each magnetic cycle shown in Figure 2b
| Cycle | Duration of “on” cycle (minutes) |
Total mass released (mg) |
Rate of drug release (mg/min) |
|---|---|---|---|
| 1 | 35 | 0.43 | 0.012 |
| 2 | 40 | 0.47 | 0.012 |
| 3 | 57 | 0.69 | 0.012 |
| 4 | 75 | 0.83 | 0.011 |
The devices turned “on” with only a 1-2 minute time lag after the solution temperature reached 40°C and turn “off” with a ∼5-10 minute lag from the cooling temperature profile (Figure 2b). This response rate was much more rapid than that seen with bulk, interpenetrating hydrogel networks, which can exhibit swelling kinetics on the order of hours48.
To confirm that the membranes are sufficiently mechanically strong to remain coherent in vivo and safely contain a depot concentration of drug, the mechanical stability of the membrane was assayed using tensile and compression testing. Wetted membranes can tolerate applied tensile stresses of 11±1 MPa and tensile strains of 12.6±2.7% prior to fracture and can be compressed by >90% without fracturing. These mechanical properties are comparable to those of a human tendon49, suggesting that the membranes would be mechanically stable against fracture in the in vivo environment. This is particularly likely given that the device would typically be embedded in connective tissue which experiences low shear.
To evaluate potential applicability of the membranes for in vivo drug delivery, we first evaluated the cytotoxicity of the membranes to a broad range of cell types (differentiated myoblasts, fibroblasts, macrophages, fibroblasts, macrophages, and mesothelial cells). Figure 3a shows the viability of cells in media exposed to a composite membrane or its components (an ethylcellulose film, a 5 mg/mL copolymer nanogel suspension, and a nanogel-loaded membrane), expressed as a ratio to cell survival in media alone as measured by the MTT assay. No significant decrease in cell viability was observed in any cell line upon exposure to the composite membrane or its individual components.
Figure 3.
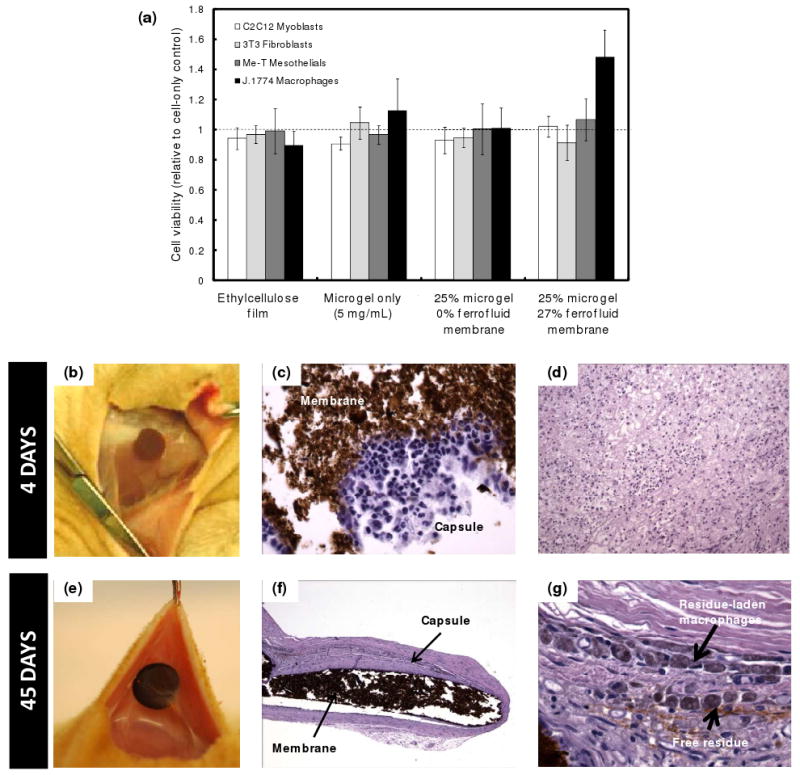
Biological testing of membranes: (a) Cell viability (relative to a cell-only control well) for differentiated myoblasts, fibroblasts, mesothelial cells, and macrophages in the presence of membrane components and membranes; (b-g) Tissue response to implanted nanogel-loaded membrane (25% nanogel, 27% ferrofluid) after 4 and 45 days of implantation: (b) top view, 4 days post-implantation; (c) histological section of membrane-tissue interface, 400× magnification; (d) histological section of capsule inflammatory response, 100× magnification; (e) top view, 45 days post-implantation; (f) histological section of membrane-tissue interface, 40× magnification; (g) histological section of capsule inflammatory response, 400× magnification.
Biocompatibility of 1 cm diameter composite membrane disks was tested by subcutaneously implanting 1 cm diameter membrane disks in Sprague-Dawley rats. Rats were sacrificed at predetermined intervals, at which point the membrane and surrounding tissues were removed and analyzed by histology. Representative tissue responses at 4 and 45 days post-implantation are shown in Figures 3b-g. After 4 days, the membrane was not significantly walled off from the surrounding tissues, with only minimal tissue adhering to the membrane (Figure 3b). The gross appearance of the implant at 4 days was bland with only mild erythema consistent with the recent implantation. Microscopically, there was acute and early chronic inflammation around the implant, as would be expected at this time point (Figures 3c and 3d). At 45 days, there was a thin translucent tissue capsule around the implant (easily separable from the membrane by gentle dissection) with no evidence of tissue damage (Figures 3e and 3f). The implants were grossly intact at both time points. The sections showed a mature fibrous capsule with macrophages and occasional foreign body giant cells at the material-tissue interface (Figure 3g). Occasional macrophages containing implant material were present in the tissue capsule, along with some residual free membrane material (Figure 3g). There was no apparent amplification of an inflammatory response and no evidence of ongoing acute inflammation.
To assess whether membranes retained their inducible drug-releasing properties in vivo, a membrane was excised after 45 days of subcutaneous implantation, the thin tissue capsule was removed, and the thermally-triggered fluorescein flux was measured using a glass flow cell apparatus. The flux response of the excised membrane was compared to that of a fresh, non-implanted membrane with the same composition, as shown in Figure 4. No significant difference was observed in the flux differential between the “on” and “off” states or the absolute magnitude of fluorescein flux across the membrane before or after implantation. This result suggests that protein adsorption or biofilm formation in vivo does not significantly impact the functionality of the membrane. Based on the residual bulk magnetic properties of the explanted membrane, the proven record of ferrofluid biocompatibility in vivo50, the macroscopic nature of the device, and the direct correlations we have observed between thermal and magnetic triggering, it is reasonable to expect that magnetic triggering would yield similar results.
Figure 4.

Comparison of thermally-triggered membrane flux of a freshly-prepared membrane to a membrane explanted from a Sprague-Dawley rat after 45 days of implantation.
The composite nanogel-ferrofluid membrane described here meets the important criteria for “on-demand” drug delivery devices. It can undergo a sharp, discontinuous volume phase transition at ≥40°C and so can be switched from the “off” state at normal physiological temperature to the “on” state at a temperature where it would not typically be triggered by fever, local inflammation, etc. The membrane could be switched on and off rapidly by the application and removal of an external oscillating magnetic field. Thus, on-demand drug delivery could be triggered non-invasively without implanted electronics. Furthermore, the membrane remained stable during multiple magnetic triggering cycles and over extended in vivo implantation, making reproducible, multi-cycle drug delivery possible. In each case, the functionality of the membrane was directly attributable to the nanoparticle properties; specifically, the rapid swelling kinetics and engineered phase transition behavior of the nanogel and the surface chemistry and optimized size of the magnetite nanoparticles for magnetic induction heating enabled the rapid, repeatable, and tunable drug release properties observed under physiological conditions. Membrane-based devices could also contain very large quantities of drug limited only by the reasonable dimensions of an implanted device, offering the potential for long-term delivery applications and repeated triggering cycles.
Reproducibility will clearly be a key consideration in devices of this type, especially with drugs with narrow therapeutic indices. We have shown excellent reproducibility over four magnetically-induced cycles. The maximum number of cycles over which that reproducibility can be maintained remains to be determined, as does the number of cycles over which it needs to be maintained. The latter will depend to a large extent on the specific clinical indication and the expected duration of therapy. Some devices might only need to be triggered a few times, while others (e.g. for a chronic condition requiring treatment several times a day) might require reproducible triggering over thousands of cycles. This issue will be of great importance in the downstream development of the device. Indeed, the ultimate design of a clinical drug delivery device based on this membrane technology, including the specific materials of which it will be composed, is yet to be determined.
Composite membrane-based drug delivery devices have the potential to greatly increase the flexibility of pharmacotherapy and improve the quality of patients' lives by providing repeated, long-term, on-demand drug delivery for a variety of medical applications, including the treatment of pain (local or systemic anesthetic delivery), local chemotherapy, and insulin delivery. Clinical implementation of magnetic membranes is possible using existing AC magnetic power supplies currently used in a variety of industries and coil geometries optimized for the specific area of the body to be treated. Indeed, prototype devices with even higher output powers than required for triggering membrane-based drug delivery devices have been developed for use in thermoablation oncology trials51. Modulation of the magnetic field could allow for fine-tuning of the rate of drug release, in addition to the frequency and duration of treatments. Additionally, the ability of the membranes to remotely and reversibly control chemical permeation may be applied in the design of triggered bioseparation modules, selective chemosensors, or externally activated microreactors.
Supplementary Material
Acknowledgments
This research was funded by NIH grant GM073626 to DSK. TH acknowledges post-doctoral funding from the Natural Sciences and Engineering Research Council of Canada. SI and GFG acknowledge support from the Spanish MEC through the Ramon y Cajal program. We would like to thank Dr. Mikhail Shapiro for helpful discussions and insights relating to this research.
Footnotes
Supporting Information Available: Descriptions of the materials and methods used, extensive physicochemical characterization of the membranes, thermal phase transition profiles of the nanogels, and drug release kinetics from the composite membranes are all reported in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Santini JT, Cima MJ, Langer R. Nature. 1999;397(6717):335–338. doi: 10.1038/16898. [DOI] [PubMed] [Google Scholar]
- 2.Grayson ACR, Choi IS, Tyler BM, Wang PP, Brem H, Cima MJ, Langer R. Nature Materials. 2003;2(11):767–772. doi: 10.1038/nmat998. [DOI] [PubMed] [Google Scholar]
- 3.Sershen SR, Westcott SL, Halas NJ, West JL. Journal of Biomedical Materials Research. 2000;51(3):293–298. doi: 10.1002/1097-4636(20000905)51:3<293::aid-jbm1>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 4.Edelman ER, Kost J, Bobeck H, Langer R. Journal of Biomedical Materials Research. 1985;19(1):67–83. doi: 10.1002/jbm.820190107. [DOI] [PubMed] [Google Scholar]
- 5.Muller-Schulte D. Thermosensitive, biocompatible polymer carriers with changeable physical structure for therapy, diagnostics, and analytics. 2007 [Google Scholar]
- 6.Muller-Schulte D, Schmitz-Rode T. Journal of Magnetism and Magnetic Materials. 2006;302(1):267–271. [Google Scholar]
- 7.Zhang J, Misra RDK. Acta Biomaterialia. 2007;3(6):838–850. doi: 10.1016/j.actbio.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 8.Hu SH, Tsai CH, Liao CF, Liu DM, Chen SY. Langmuir. 2008;24(20):11811–11818. doi: 10.1021/la801138e. [DOI] [PubMed] [Google Scholar]
- 9.Sukhorukov GB, Rogach AL, Garstka M, Springer S, Parak WJ, Munoz-Javier A, Kreft O, Skirtach AG, Susha AS, Ramaye Y, Palankar R, Winterhalter M. Small. 2007;3(6):944–955. doi: 10.1002/smll.200600622. [DOI] [PubMed] [Google Scholar]
- 10.Hu SH, Chen SY, Liu DM, Hsiao CS. Advanced Materials. 2008;20(14):2690–2695. doi: 10.1002/adma.200800193. [DOI] [PubMed] [Google Scholar]
- 11.Hu SH, Liu TY, Huang HY, Liu DM, Chen SY. Langmuir. 2008;24(1):239–244. doi: 10.1021/la701570z. [DOI] [PubMed] [Google Scholar]
- 12.Liu TY, Hu SH, Liu DM, Chen SY, Chen IW. Nano Today. 2009;4(1):52–65. [Google Scholar]
- 13.Alexander C. Nature Materials. 2008;7(10):767–768. doi: 10.1038/nmat2281. [DOI] [PubMed] [Google Scholar]
- 14.Ehrick JD, Deo SK, Browning TW, Bachas LG, Madou MJ, Daunert S. Nature Materials. 2005;4(4):298–302. doi: 10.1038/nmat1352. [DOI] [PubMed] [Google Scholar]
- 15.Ehrbar M, Schoenmakers R, Christen EH, Fussenegger M, Weber W. Nature Materials. 2008;7(10):800–804. doi: 10.1038/nmat2250. [DOI] [PubMed] [Google Scholar]
- 16.Kikuchi A, Okano T. Advanced Drug Delivery Reviews. 2002;54(1):53–77. doi: 10.1016/s0169-409x(01)00243-5. [DOI] [PubMed] [Google Scholar]
- 17.Okuyama Y, Yoshida R, Sakai K, Okano T, Sakurai Y. Journal of Biomaterials Science-Polymer Edition. 1993;4(5):545–556. doi: 10.1163/156856293x00195. [DOI] [PubMed] [Google Scholar]
- 18.Qiu Y, Park K. Advanced Drug Delivery Reviews. 2001;53(3):321–339. doi: 10.1016/s0169-409x(01)00203-4. [DOI] [PubMed] [Google Scholar]
- 19.Ichikawa H, Fukumori Y. Journal of Controlled Release. 2000;63(12):107–119. doi: 10.1016/s0168-3659(99)00181-9. [DOI] [PubMed] [Google Scholar]
- 20.Sahiner N, Alb AM, Graves R, Mandal T, McPherson GL, Reed WF, John VT. Polymer. 2007;48(3):704–711. [Google Scholar]
- 21.Eichenbaum GM, Kiser PF, Dobrynin AV, Simon SA, Needham D. Macromolecules. 1999;32(15):4867–4878. doi: 10.1021/ma970897t. [DOI] [PubMed] [Google Scholar]
- 22.Hoare T, Pelton R. Langmuir. 2008;24(3):1005–1012. doi: 10.1021/la7024507. [DOI] [PubMed] [Google Scholar]
- 23.Snowden MJ. Journal of the Chemical Society-Chemical Communications. 1992;(11):803–804. [Google Scholar]
- 24.Alem H, Duwez AS, Lussis P, Lipnik P, Jonas AM, Demoustier-Champagne S. Journal of Membrane Science. 2008;308(12):75–86. [Google Scholar]
- 25.Fu Q, Rao GVR, Ward TL, Lu YF, Lopez GP. Langmuir. 2007;23(1):170–174. doi: 10.1021/la062770f. [DOI] [PubMed] [Google Scholar]
- 26.Okahata Y, Noguchi H, Seki T. Macromolecules. 1986;19(2):493–494. [Google Scholar]
- 27.Yoshida M, Asano M, Safranj A, Omichi H, Spohr R, Vetter J, Katakai R. Macromolecules. 1996;29(27):8987–8989. [Google Scholar]
- 28.Yoshida R, Kaneko Y, Sakai K, Okano T, Sakurai Y, Bae YH, Kim SW. Journal of Controlled Release. 1994;32(1):97–102. [Google Scholar]
- 29.Choi YJ, Yamaguchi T, Nakao S. Industrial & Engineering Chemistry Research. 2000;39(7):2491–2495. [Google Scholar]
- 30.Hesampour M, Huuhilo T, Makinen K, Manttari M, Nystrom M. Journal of Membrane Science. 2008;310(12):85–92. [Google Scholar]
- 31.Lee YM, Shim JK. Polymer. 1997;38(5):1227–1232. [Google Scholar]
- 32.Liang L, Feng XD, Peurrung L, Viswanathan V. Journal of Membrane Science. 1999;162(12):235–246. [Google Scholar]
- 33.Akerman S, Viinikka P, Svarfvar B, Putkonen K, Jarvinen K, Kontturi K, Nasman J, Urtti A, Paronen P. International Journal of Pharmaceutics. 1998;164(12):29–36. [Google Scholar]
- 34.Iwata H, Oodate M, Uyama Y, Amemiya H, Ikada Y. Journal of Membrane Science. 1991;55(12):119–130. [Google Scholar]
- 35.Wang WY, Chen L. Journal of Applied Polymer Science. 2007;104(3):1482–1486. [Google Scholar]
- 36.Wang WY, Chen L, Yu X. Journal of Applied Polymer Science. 2006;101(2):833–837. [Google Scholar]
- 37.Schild HG. Progress in Polymer Science. 1992;17:163–249. [Google Scholar]
- 38.Dinarvand R, Demanuele A. Journal of Controlled Release. 1995;36(3):221–227. [Google Scholar]
- 39.Zhang K, Wu XY. Biomaterials. 2004;25(22):5281–5291. doi: 10.1016/j.biomaterials.2003.12.032. [DOI] [PubMed] [Google Scholar]
- 40.Moussy JB, Gota S, Bataille A, Guittet MJ, Gautier-Soyer M, Delille F, Dieny B, Ott F, Doan TD, Warin P, Bayle-Guillemaud P, Gatel C, Snoeck E. Physical Review B. 2004;70(17):174448. [Google Scholar]
- 41.Goya GF, Berquo TS, Fonseca FC, Morales MP. Journal of Applied Physics. 2003;94(5):3520–3528. [Google Scholar]
- 42.Edelman ER, Langer R. Biomaterials. 1993;14(8):621–626. doi: 10.1016/0142-9612(93)90182-2. [DOI] [PubMed] [Google Scholar]
- 43.Goya GF, Lima E, Arelaro AD, Torres T, Rechenberg HR, Rossi L, Marquina C, Ibarra MR. Ieee Transactions on Magnetics. 2008;44(11):4444–4447. [Google Scholar]
- 44.Keerl M, Smirnovas V, Winter R, Richtering W. Macromolecules. 2008;41(18):6830–6836. [Google Scholar]
- 45.Wang Q, Zhao YB, Yang YJ, Xu HB, Yang XL. Colloid and Polymer Science. 2007;285(5):515–521. [Google Scholar]
- 46.Pelton RH, Chibante P. Colloids and Surfaces. 1986;20(3):247–256. [Google Scholar]
- 47.Jackson DK, Leeb SB, Mitwalli AH, Narvaez P, Fusco D, Lupton EC. Ieee Transactions on Industrial Electronics. 1997;44(2):217–225. [Google Scholar]
- 48.Matsuo ES, Tanaka T. Journal of Chemical Physics. 1988;89(3):1695–1703. [Google Scholar]
- 49.Soden PD, Kershaw I. Medical and Biological Engineering. 1974;12:510–518. doi: 10.1007/BF02478609. [DOI] [PubMed] [Google Scholar]
- 50.Zhao H, Saatchi K, Hafeli UO. Journal of Magnetism and Magnetic Materials. 2009;321(10):1356–1363. [Google Scholar]
- 51.MagForce. A New Dimension: Effective cancer therapy with minimal side-effects. www.magforce.com.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.