

Abstract
The hydroboration of allene 7 with (dIpc)2BH at 0 °C provides the kinetic allylborane 12Z with ≥20 : 1 selectivity. However, when the hydroboration is performed at 85 °C, the kinetically formed allylborane isomerizes to give the thermodynamic allylborane 12E with ≥12 : 1 selectivity. Subsequent treatment of 12Z or 12E with aldehydes at -78 °C, followed by oxidative workup, provides the 2-methyl-1,2-diols 8 and 9 in good yield and with 80-92% e.e.
2-Methyl-1,2-syn- and 1,2-anti-diols are common structural motifs in natural products.1 Stereocontrolled syntheses of these units mainly rely on the Sharpless asymmetric dihydroxylation reaction.2 However, when the substrate contains multiple olefins, a highly regioselective dihydroxylation can be challenging owing to the influence of the substitution pattern, steric and electronic effects of the individual double bonds.3 Consequently, a tactic frequently used in the synthesis of molecules which contain 1,2-diol units and potentially conflicting olefin functionalities is to install the diol before introduction of the olefin.4-6 Alternatively, diastereoselective 1,2-addition of a methyl group to α-hydroxy-α′,β′-unsaturated ketones or addition of a vinyl metal species to α-alkoxyl methyl ketones also provide access to these 1,2-diols subunits.7,8 In connection with an ongoing problem in natural product synthesis, we have developed and report herein a direct, one step, highly diastereo- and enantioselective synthesis of 2-methyl-1,2-syn- and 2-methyl-1,2-anti-3-butenediols via allene hydroboration-aldehyde allylboration reaction sequences.
In 1995, Brown reported the diastereo- and enantioselective synthesis of anti-1,2-diols 4 by a sequence involving the hydroboration of allenylboronate 1 with diisopinocampheylborane [(Ipc)2BH] (Figure 1).9 It is believed that hydroboration of allenylboronate 1 with (dIpc)2BH initially forms γ–boryl-(Z)-allylborane 2-Z as the kinetic product, which isomerizes rapidly through reversible 1,3-borotropic shifts to give γ–boryl-(E)-allylborane 2-E.10-14 We subsequently adopted this procedure for the synthesis of 1,5-anti- and 1,5-syn diols 5 and 6 by using the intermediate β-alkoxyallylboronate 3 in a second allylboration reaction.15 The stereochemical course of the second allylboration event depends on the structure of the boronate unit.15 The double allylboration methodology has been applied in several synthetic studies targeting natural products.16-20
Figure 1.

Hydroboration-Allylboration of Allenylboronate 1
By analogy to the results in Figure 1, we anticipated that the hydroboration of 1-methyl-allenylboronate 721 with diisopinocampheylborane [(dIpc)2BH] followed by (single) aldehyde allylboration and oxidative workup would provide a flexible, general synthesis of 1,2-anti diols 9, bearing a quaternary center. In initial experiments, treatment of allenylboronate 7 with (dIpc)2BH in toluene at 0 °C for 2 h followed by addition of hydrocinnamaldehyde at -78 °C (4 h) and then standard oxidative workup provided, surprisingly, the syn-1,2-diol 8a in 72% yield and 92% ee (Scheme 1). The absolute stereochemistry of the secondary hydroxyl group of 8a was assigned by using the modified Mosher ester analysis.22 The syn stereochemistry of 8a (and subsequently also of 8e) was assigned by 1H nOe studies of the corresponding acetonide derivatives (see Supporting Information). The conditions developed for the synthesis of 8a were then applied to a variety of aldehydes. 1,2-syn-Diols 8a-g were obtained in 56-82% yield with >20:1 diastereoselectivity and 85-92% ee (Scheme 1).
Scheme 1.

Synthesis of syn-1,2-Diols 8 via Kinetic Hydroboration of 7a
(a) Reactions were performed by treating 7 with (dIpc)2BH (1.0 equiv) in toluene at 0 °C followed by the addition of RCHO (1 equiv) at -78 °C. The mixture was then allowed to stir at -78 °C for 4 h. The reactions were subjected to standard oxidative workup (NaOH, H2O2) at 0 °C before product isolation. (b) Determined by Mosher ester analysis, unless noted otherwise. (c) See SI for enantiomeric purity determination.
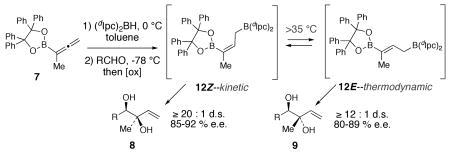
Assuming that the allylboration reaction proceeds by way of the usual chair-like transition state,23,24 the results in Scheme 1 indicate that the intermediate produced in the hydroboration of 7 is the γ–boryl-(Z)-allylic borane 12Z (Scheme 2). In contrast to the elusive intermediate 2-Z in the hydroboration of allenylboronate 1, the kinetic hydroboration product 12Z does not isomerize to the thermodynamically more stable 12E at 0 °C.
Scheme 2.

Kinetic Hydroboration of 7 and Thermodynamically Controlled Allylborane Isomerization
We were intrigued by the possibility that the diastereomeric anti-diols 9 could also be accessed if 12Z could be induced to isomerize to the γ–boryl-(E)-allylborane 12E. Indeed, when the hydroboration of allenylboronate 7 was performed at 35 °C for 16 h followed by treatment of the allylborane product mixture with hydrocinnamaldehyde gave a 3:1 mixture of diols 9a and 8a, favoring the anti-diol 9a derived from isomerization of 12Z to the thermodynamically favored allylborane 12E. Hydroboration of 7 at 65 °C for 5 h led to a 5:1 mixture of 9a and 8a. Prolonged heating of the hydroboration reaction at 65 °C (16 h), however, only led to decomposition. When the hydroboration of 7 was performed at 85 °C in toluene for 1.5 h, followed by addition of hydrocinnamaldehyde at −78 °C, anti-diol 9a was obtained with 17 : 1 d.r. (9a:8a) in 76% yield and 89% ee (Scheme 3). The stereochemistry of anti-diol 9a (and subsequently also of 9e) was assigned by 1H nOe studies of the corresponding acetonide derivatives (see SI). The hydroboration-isomerization-allylboration sequence was then applied to a variety of aldehydes (Scheme 3). In all cases, 1,2-anti-diols 9a-g were obtained in good yield with ≥12:1 diastereoselectivity and 80-89% e.e.
Scheme 3.

Synthesis of anti-1,2-Diols 9 via Hydroboration of 7 and Thermodynamically Controlled Allylborane Isomerizationa
(a) Reactions were performed by treating 7 with (dIpc)2BH (1.0 equiv) in toluene at 85-95 °C for 1.5 h followed by the addition of RCHO (1 equiv) at -78 °C. The mixture was then allowed to stir at -78 °C for 4 h. The reactions were subjected to standard oxidative workup (NaOH, H2O2) at 0 °C before product isolation. (b) Determined by Mosher ester analysis, unless indicated otherwise. (c) See SI for % e.e. determination for 9g.
Finally, double asymmetric allylboration reactions of 12Z and 12E with chiral aldehyde 14 are summarized in Scheme 4. Kinetic controlled hydroboration of allenylboronate 7 with either (dIpc)2BH or (lIpc)2BH and treatment with aldehyde 14 provided syn-diols 8h or 8i with excellent diastereoselectivity (>15:1) in 72% and 65% yield, respectively (entries 1 and 2). Alternatively, when the hydroboration of 7 was performed at 85-95 °C for 1.5 h with (dIpc)2BH [to give the thermodynamic allylborane 12E] followed by addition of 14 at -78 °C, anti-diol 9h was obtained with excellent diastereoselectivity (>15:1) (entry 3). Similarly, a 5:1 mixture of anti-diols 9i and 9h was obtained in 52% yield from 12E generated by the hydroboration of 7 with (lIpc)2BH (entry 4). The latter reaction is stereochemically mismatched.25
Scheme 4.

Double Asymmetric Allylboration Reactions with Aldehyde 14
The data presented herein indicate that the hydroboration of 7 with (Ipc)2BH proceeds under kinetic control at 0 °C and provides 12Z with excellent selectivity. Evidently, the normally facile 1,3-isomerization that has been documented for other allylboranes10-14 is slow in the case of 12Z owing to steric hindrance in the transition state leading to the 1,1-diboryl species 13. However, isomerization is readily achieved at higher temperatures, and a ≥12:1 mixture of 12E and 12Z is obtained at 85 °C. Thus, synthetically useful selectivity for synthesis of either the 1,2-syn or 1,2-anti diol diastereomers 8 and 9 can be achieved by appropriate control of the hydroboration conditions. Applications of this method in the synthesis of natural products will be reported in due course.
Supplementary Material
Acknowledgments
Financial support provided by the National Institutes of Health (GM038436 and GM026782) is gratefully acknowledged.
Footnotes
Supporting Information Available: Experimental procedures and spectroscopic data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Djerassi C, Halpern O. Tetrahedron. 1958;3:255. [Google Scholar]; (b) Maezawa I, Kinumaki A, Suzuki M. J Antibiot. 1974;27:84. doi: 10.7164/antibiotics.27.84. [DOI] [PubMed] [Google Scholar]; (c) Stampwala SS, Bunge RH, Hurley TR, Willmer NE, Brankiewicz AJ, Steinman CE, Smitka TA, French JC. J Antibiot. 1983;36:1601. doi: 10.7164/antibiotics.36.1601. [DOI] [PubMed] [Google Scholar]; (d) Dorta E, Diaz-Marrero AR, Cueto M, D'Croz L, Mate JL, Darias J. Tetrahedron Lett. 2004;45:7065. doi: 10.1021/ol049287l. [DOI] [PubMed] [Google Scholar]
- 2.(a) Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem Rev. 1994;94:2483. [Google Scholar]; (b) Noe MC, Letavic MA, Snow SL. Org React. 2005;66:109. [Google Scholar]
- 3.(a) Xu D, Crispino GA, Sharpless KB. J Am Chem Soc. 1992;114:7570. [Google Scholar]; (b) Français A, Bedel O, Haudrechy A. Tetrahedron. 2008;64:2495. [Google Scholar]
- 4.Miyashita K, Ikejiri M, Kawasaki H, Maemura S, Imanishi T. J Am Chem Soc. 2003;125:8238. doi: 10.1021/ja030133v. [DOI] [PubMed] [Google Scholar]
- 5.Julian LD, Newcom JS, Roush WR. J Am Chem Soc. 2005;127:6186. doi: 10.1021/ja050729d. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi Y, Yamaguchi H, Toyoshima M, Okado K, Toyo T, Shoji M. Org Lett. 2008;10:1405. doi: 10.1021/ol800195g. [DOI] [PubMed] [Google Scholar]
- 7.(a) Reetz MT. Angew Chem Int Ed Engl. 1984;23:556. [Google Scholar]; (b) Mengel A, Reiser O. Chem Rev. 1999;99:1191. doi: 10.1021/cr980379w. [DOI] [PubMed] [Google Scholar]
- 8.For representative recent examples, see: Boger DL, Ichikawa S, Zhong W. J Am Chem Soc. 2001;123:4161. doi: 10.1021/ja010195q.Chavez DE, Jacobsen EN. Angew Chem Int Ed. 2001;40:3667. doi: 10.1002/1521-3773(20011001)40:19<3667::aid-anie3667>3.0.co;2-6.Wang YG, Kobayashi Y. Org Lett. 2002;4:4615. doi: 10.1021/ol020193q.Trost BM, Frederiksen MU, Papillon JPN, Harrington PE, Shin S, Shireman BT. J Am Chem Soc. 2005;127:3666. doi: 10.1021/ja042435i.
- 9.Brown HC, Narla G. J Org Chem. 1995;60:4686. [Google Scholar]
- 10.Kramer GW, Brown HC. J Organomet Chem. 1977;132:9. [Google Scholar]
- 11.Hoffmann RW, Zeiβ HJ. J Org Chem. 1981;46:1309. [Google Scholar]
- 12.Narla G, Brown HC. Tetrahedron Lett. 1997;38:219. [Google Scholar]
- 13.Wang KK, Gu YG, Liu C. J Am Chem Soc. 1990;112:4424. [Google Scholar]
- 14.Gu YG, Wang KK. Tetrahedron Lett. 1991;32:3029. [Google Scholar]
- 15.Flamme EM, Roush WR. J Am Chem Soc. 2002;124:13644. doi: 10.1021/ja028055j. [DOI] [PubMed] [Google Scholar]
- 16.Flamme EM, Roush WR. Org Lett. 2005;7:1411. doi: 10.1021/ol050250q. [DOI] [PubMed] [Google Scholar]
- 17.Owen RM, Roush WR. Org Lett. 2005;7:3941. doi: 10.1021/ol0514303. [DOI] [PubMed] [Google Scholar]
- 18.Hicks JD, Flamme EM, Roush WR. Org Lett. 2005;7:5509. doi: 10.1021/ol052322j. [DOI] [PubMed] [Google Scholar]
- 19.Lira R, Roush WR. Org Lett. 2007;9:533. doi: 10.1021/ol0629869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hicks JD, Roush WR. Org Lett. 2008;10:681. doi: 10.1021/ol703042q. [DOI] [PubMed] [Google Scholar]
- 21.See the supporting information for the synthesis of 7.
- 22.Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092. [Google Scholar]
- 23.Roush WR. In: Comprehensive Organic Synthesis. Trost BM, editor. Vol. 2. Pergamon Press; Oxford: 1991. p. 1. [Google Scholar]
- 24.Chemler SR, Roush WR. In: Modern Carbonyl Chemistry. Otera J, editor. Wiley-VCH; Weinheim: 2000. p. 403. [Google Scholar]
- 25.Masamune S, Choy W, Petersen JS, Sita LR. Angew Chem Int Ed Engl. 1985;24:1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.