

Abstract
Bipolar disorder (BPD) and major depressive disorder (MDD) are common, chronic, and recurrent mood disorders that affect the lives of millions of individuals worldwide. Growing evidence suggests that glutamatergic system dysfunction is directly involved in mood disorders. This article describes the role of the “tripartite glutamatergic synapse”, comprising presynaptic and postsynaptic neurons and glial cells, in the pathophysiology and therapeutics of mood disorders. Glutamatergic neurons and glia directly control synaptic and extrasynaptic glutamate levels/release through integrative effects that target glutamate excitatory amino-acid transporters, postsynaptic density proteins, ionotropic receptors (alpha-amino-3-hydroxy-5-methylisoxazole (AMPA), N-methyl-D-aspartate (NMDA), and kainate (KA)), and metabotropic receptors (mGluRs). This article also explores the glutamatergic modulators riluzole and ketamine, which are considered valuable proof of concept agents for developing the next generation of antidepressants and mood stabilizers. In therapeutically relevant paradigms, ketamine preferentially targets postsynaptic AMPA/NMDA receptors, and riluzole preferentially targets presynaptic voltage-operated channels and glia.
I. Introduction
Major depressive disorder (MDD) and Bipolar disorder (BPD) are serious, heterogeneous, and disabling psychiatric illnesses that affect millions of individuals worldwide, and have a significant negative impact on public health and productivity (Baune and others 2007; Kessler and others 2006). Indeed, mood disorders account for more bed days than any impairment except cardiovascular disease (U. S. Department of Health and Human Services 2000); the World Health Organization’s (WHO) Global Burden of Disease project estimates that by 2020, MDD will become the second leading cause of disability worldwide (Murray and Lopez 1996). Available treatments for mood disorders—in particular antidepressants and mood stabilizers—have helped millions worldwide, but a large percentage of patients taking these medications still fail to achieve response and remission. Despite adequate dosage, duration, and compliance, residual symptoms between episodes, recurrences, and functional impairment are common (Fagiolini and others 2005). For instance, the largest open-label study (STAR*D) evaluating traditional treatments in MDD found that less than one-third of patients achieved remission after up to four months of antidepressant use (Thase and others 2005). Similarly, individuals with BPD experienced symptoms almost half of the time, with a predominance of subsyndromal depressive episodes (Judd and others 2002).
One issue of particular importance is the fact that monoaminergic antidepressants take weeks to achieve their full effect. This leaves patients receiving these medications particularly vulnerable to impaired global functioning and at high risk of self-harm. High rates of mortality and morbidity are present during this latency period and, notably, worse short-term outcome predicts a poorer long-term course of the illness (Machado-Vieira and others 2008). Compounding this problem is the fact that many patients experience a lengthy trial-and-error period when attempting to find the most optimal treatment. Another key problem is our limited comprehension of the pathophysiological mechanisms involved in the therapeutic effects of antidepressants and mood stabilizers, especially the difficulty in attributing therapeutic relevance to any observed biochemical finding. The standard antidepressants are thought to exert therapeutic effects through their ability to increase monoamines in the synapses; however, given that the clinical efficacy of these traditional agents is typically observed after several weeks of treatment, it has been suggested that monoamines only represent primary effectors modulating key downstream signaling pathways responsible for their therapeutic efficacy (Manji and Lenox 2000). Therefore, new therapeutic agents that target different systems and pathways, and that work faster and more effectively, are urgently needed in the treatment of mood disorders.
Glutamate is the most abundant excitatory neurotransmitter in the brain, and acts pre- and postsynaptically by activating diverse receptors characterized by their structural properties. Glutamate ionotropic and metabotropic receptors regulate neurotransmission across excitatory synapses, and modulate several physiological brain functions such as synaptic plasticity, learning, and memory (Collingridge and Bliss 1995). Notably, diverse clinical studies have supported a key role for the glutamatergic system in the pathophysiology of mood disorders, and this system is believed to be a key target in mood regulation (reviewed in (Maeng and Zarate 2007; Zarate and others 2002). For instance, stress and neuroplasticity, which are critically regulated by the glutamatergic system, have also been shown to play an important role in MDD (Pittenger and Duman 2008). Also, emerging data indicate that glutamate is critically involved in both acute and long-term processes involved in the mode of action of mood stabilizers and antidepressants. Several glutamatergic compounds or glutamate-modulating agents have been tested in “proof of concept” studies in patients with severe mood disorders (Sanacora and others 2008; Zarate and others 2002).
This article describes and reinforces the role of the “tripartite glutamatergic synapse” in the pathophysiology and therapeutics of mood disorders (see below; Figure 1 also provides additional information). The term “tripartite” encompasses presynaptic neurons, postsynaptic neurons, and glia and includes several targets involved in the regulation of synaptic and extrasynaptic glutamate levels, such as excitatory amino acid transporters (EAATs), postsynaptic density proteins, (alpha-amino-3-hydroxy-5-methylisoxazole methylisoxazole-4-propionic acid) AMPA receptors, N-methyl-D-aspartate (NMDA) receptors, and kainate (KA) receptors. In particular, this article emphasizes the potential role of riluzole and ketamine as proof of concept agents for the development of new improved treatments with faster and sustained effects, and preferentially targeting postsynaptic AMPA/NMDA receptors (ketamine), and presynaptic voltage-operated channels and glia (riluzole) in therapeutically relevant paradigms.
Figure 1. Pathophysiological basis and potential therapeutic targets for mood disorders involving glutamatergic neurotransmission.
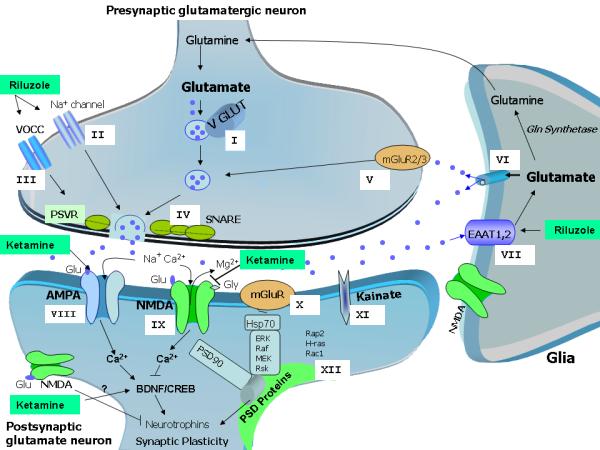
Ketamine preferentially targets postsynaptic AMPA/NMDA receptors, while riluzole’s antidepressant effects occur through direct regulation, mostly at presynaptic voltage-operated channels and glia. All potential targets for new, improved glutamatergic agents are described below.
Presynaptic targets:
Before release, Glu is packaged into vesicles by VGLUTs, which control glutamate concentration produced in the synaptic vesicles (Target I). Activation of voltage-gated sodium channels (Target II) and VOCCs (Target III) depolarizes the plasma membrane, allowing for the influx of sodium (through depolarization of axon terminals) and calcium (through interaction with the SNARE proteins) (Target IV), leading to the fusion of synaptic vesicles and the consequent release of Glu into the synaptic cleft. The type II mGluRs (mGlu2/3) are also present presynaptically (Target V), and may directly limit the synaptic release of Glu. These mGluRs also mediate feedback inhibition of Glu release, thus decreasing the activity of Glu synaptic terminals when the activation of presynaptic receptors reaches a certain level.
Glia:
Glu can also be directly released from glial cells, regulating synaptic activity pre-and post-synaptically (Target VI). After its release, large amounts of Glu are rapidly distributed across the synaptic cleft, where it can bind to glutamate receptors in the postsynaptic regions. The remaining unbound Glu is rapidly removed from the synaptic cleft to the presynaptic neuron mostly by glia and EAATs. The glial transporters EAAT1 (also known in rodents as GLAST) and EAAT2 (or GLT-1) are essential for blocking pathological increases in Glu levels (Target VII). Glial Glu transporter expression is upregulated by neuronal activity. Notably, the expression of Glu transporters by glial cells and their anatomical arrangement within the synaptic cleft can be dynamically and reversibly modified, and can directly regulate their ability to scavenge Glu.
Postsynaptic neuron:
AMPARs (GluR1-4) mediate fast Glu neurotransmission and play a major role in learning and memory through critical regulation of calcium metabolism, plasticity, and oxidative stress. When activated, these receptors open the transmembrane pore, thus allowing the influx of sodium and the consequent depolarization of the neuronal membrane (Target VIII).
The NMDAR channel includes the subunits NR1, NR2 (NR2A—NR2D), and NR3 (NR3A and NR3B). Glu’s binding sites have been described mostly in the NR2 subunit, whereas the NR1 subunit is the site for its co-agonist, glycine. NR2A and NR2B subunit receptors are both highly expressed in brain areas implicated in mood regulation, but NMDA receptors containing NR2A mediate faster neurotransmission than NR2B receptors (Target IX).
The mGluRs include eight receptor subtypes (mGluR1 to mGluR8) classified in three groups (I-III) based on their sequence homology and effectors. The mGluRs in group I, including mGluR1 and mGluR5, stimulate the breakdown of phosphoinositide phospholipids in the cell plasma membrane. mGluRs in Groups II (including mGLuR2/3) and III (including mGluRs 4, 6, 7, and 8) limit the generation of cAMP by activating inhibitory G-proteins.While Group I receptors are coupled to the phospholipase C signal transduction pathway, Group II and III mGluRs are both coupled in an inhibitory manner to the adenylyl cyclase pathway, which is involved in regulating the release of Glu or other neurotransmitters such as GABA (Target X).
KARs are involved in excitatory neurotransmission by activating postsynaptic receptors, and in inhibitory neurotransmission by modulating GABA release. There are five types of KAR subunits: GluR5 (GRIK1), GluR6 (GRIK2), GluR7 (GRIK3), KA1 (GRIK4), and KA2 (GRIK5). GluR5-7 can form heteromers; KA1 and KA2 can only form functional receptors by binding with one of the GluR5-7 subunits. KARs have a more limited distribution in the brain than AMPARs and NMDARs, and their function is not well defined; they are believed to affect synaptic signaling and plasticity less than AMPARs (Target XI).
Cytoplasmic PSD-enriched intracellular molecules (e.g. PSD93) interact with Glu receptors at the synaptic membrane to modulate receptor activity and signal transduction. PSD95 and similar scaffolding molecules link the NMDARs with intracellular enzymes that regulate signaling, and also provide a physical connection between diverse neurotransmitter systems to synchronize information from different effectors (Target XII).
II. The Tripartite Glutamatergic Synapse in Mood Disorders: Characteristics, Pathophysiology, and Potential Therapeutic Targets
Physiological activity in the glial-neuronal glutamate-glutamine cycle involves the inactivation of glutamate once its specific function as a neurotransmitter has been concluded, thus protecting surrounding neurons and glial cells from a potentially toxic insult due to overexposure to high glutamate levels. These critical effects of glutamate include diverse receptors, second-messengers, and other effectors, and changes to this circuitry have been implicated in mood disorders.
A. Direct regulation of glutamate release/levels at the presynaptic neuron (Targets I-V)
In human studies of individuals with mood disorders, altered glutamate levels have been observed in plasma (Altamura and others 1993; Mauri and others 1998), serum (Mitani and others 2006), and cerebrospinal fluid (CSF) (Frye and others 2007; Levine and others 2000). Imaging studies have also shown elevated glutamate levels in the occipital cortex (OCC) and reduced levels in the anterior cingulate cortex (ACC) (Hasler and others 2007; Sanacora and others 2004). MRS studies in BPD have also shown increased glutamatergic and related metabolites (Glx) in diverse cortical areas not directly related to mood state (Yildiz-Yesiloglu and Ankerst 2006). Similarly, postmortem studies have described increased levels of glutamate in the prefrontal cortex of individuals with MDD (Hashimoto and others 2007; Scarr and others 2003).
Data regarding therapeutic agents that affect glutamate levels are sparse. Riluzole is the best-studied of these compounds (see Section IV), although other riluzole-like agents have also been tested, such as agents capable of attenuating voltage-activated channel activity. Preliminary data suggest that agents that decrease exaggerated action potential, with a consequent increase in presynaptic glutamate release, may be promising novel therapeutics for mood disorders. Notably, a recent trial showed that N-acetyl cysteine (NAC) was effective as add-on therapy in the treatment of bipolar depression (Berk and others 2008). The authors postulated that NAC increases glial cystine uptake (and its levels) mediated by a cystine-glutamate antiporter, which induces glutamate release into the extracellular space. The subsequent stimulation of mGluR2 receptors blocks glutamate release, thus inducing potential neuroprotective effects (Krystal 2008). Another recent study of 35 subjects showed that cytidine as add-on therapy to valproate was associated with faster improvement in bipolar depression than placebo, probably mediated by decreased levels of cerebral glutamate and/or glutamine. The results were quantified using proton magnetic resonance spectroscopy (MRS) before and after two, four, and 12 weeks of oral cytidine administration (Yoon and others 2009).
B. EAATs and Glial Regulation (Targets VI and VII)
B1. Pathophysiology
The role of glial cells in recycling glutamate through the glutamate/glutamine cycle (Lebon and others 2002) is especially relevant to the pathophysiological processes underlying mood disorders (Kugaya and Sanacora 2005). Indeed, impaired glial cell activity may lead to increased glutamatergic activation and neural toxicity, especially at extrasynaptic sites (Soriano and Hardingham 2007). Glial cells related to glutamate homeostasis are particularly important in mood disorders because a decrease in glial cell number and volume has consistently been described in post-mortem studies of individuals with MDD and BPD (reviewed in (Rajkowska and Miguel-Hidalgo 2007)); at the same time, chronic stress decreases glial proliferation, which has been associated with depressive-like behaviors in rodents (Banasr and Duman 2008; Banasr and others 2007). Interestingly, increased levels of glutamate carboxypeptidase II (GCP II), which hydrolyzes N-acetylaspartylglutamate (NAAG) to NAA and glutamate in glial cells, were found in the hippocampus of individuals with BPD (Guilarte and others 2008). Also, elevated CSF glutamine levels that correlated positively with CSF magnesium levels have been found in individuals with mood disorders, suggesting dysregulation of the glutamate/glutamine cycle in glial cells associated with NMDA receptor (NMDAR) activity (Levine and others 2000).
With regards to glutamate transporters, decreased expression of EAAT1, EAAT2, and glutamine synthetase has been described in the frontal areas of subjects with MDD (Choudary and others 2005). Similarly, decreased levels of EAAT3, EAAT4, and mRNA expression were found in the striatum of subjects with mood disorders (McCullumsmith and Meador-Woodruff 2002). Altered hippocampal glutamate expression might also be affected by levels of neutral amino acid transporter 1 (ASCT-1), which is involved in neuronal glutamate efflux and reduced in the hippocampal neurons of individuals with BPD (Weis and others 2007). It is interesting to note that chronic, in vivo blockade of glutamate uptake by a glial/neuronal transporter antagonist in the amygdala resulted in reduced social exploratory behavior and disrupted circadian activity patterns, correlating with symptoms of mood disorders (Lee and others 2007).
Finally, studies have found mitochondrial activity dysfunction in individuals with mood disorders, suggesting energy deprivation. Experimental inhibition of glial reuptake of glutamate is known to rapidly decrease glutamate uptake and hence cause an accumulation of extrasynaptic glutamate and excitotoxicity (Jabaudon and others 1999), similar to the findings obtained in individuals with mood disorders.
B2. Therapeutics
The hypothesis that increased levels of extracellular glutamate could be related to depressive-like behaviors, and that increased glutamate uptake could have protective, anti-stress, or antidepressant-like effects suggests that agents affecting glially-regulated glutamate levels, particularly EAATs, may be promising therapeutic agents in the treatment of mood disorders. Recent studies have shown that some available pharmaceutical agents can increase EAAT activity (Mineur and others 2007; Rothstein and others 2005). For instance, beta-lactam antibiotics, used to successfully treat neurodegenerative diseases, selectively activate transcription of the gene encoding the EAAT2 glutamate transporter (Miller and Cleveland 2005; Rothstein and others 2005). In mood disorders, the antibiotic ceftriaxone, which also increases glutamate transport (Rothstein and others 2005), appears to acutely enhance expression of the gene encoding for EAAT2 and, preclinically, has significant antidepressant-like effects in the forced swim, tail suspension, and novelty suppressed feeding paradigms (Mineur and others 2007). Chronic treatment with the mood stabilizer lithium also upregulates synaptosomal uptake of glutamate in vitro and protects against glutamate-induced excitotoxicity, in part attributed to the inhibition of NMDA-receptor-mediated calcium influx (Hashimoto and others 2002). Similarly, chronic use of the mood stabilizer valproate dose-dependently increased hippocampal glutamate uptake capacity by increasing EAAT1 and EAAT2 levels in the hippocampus, as measured by uptake of [3H] glutamate. Overall, the clinical use of glial glutamate transporter enhancers may represent a valuable therapeutic tool in the treatment of mood disorders.
C. AMPA Receptor (AMPAR) Regulation (Target VIII)
C1. Pathophysiology
Decreased GluR2 and GluR3 receptor levels have been reported in the prefrontal cortex (PFC) of subjects with MDD (Hashimoto and others 2007; Scarr and others 2003). Also, decreased GluR2 levels were noted in the dorsolateral prefrontal cortex (DLPFC) of individuals with BPD (Beneyto and Meador-Woodruff 2006). Another post-mortem study found that increased AMPA binding was associated with decreased GluR1 subunit expression in the striatum of individuals with BPD (Meador-Woodruff and others 2001); no differences were found in the hippocampus or the PFC.
C2. Therapeutics
AMPARs appear to respond to chronic treatment with standard antidepressants (Paul and Skolnick 2003), which enhance AMPAR surface levels (Du and others 2004; Du and others 2007). Specifically, imipramine enhances AMPAR activity by increasing GluR1/2 surface expression and phosphorylation (Du and others 2004). Similarly, interactions of GluR1 and GluR2/3 with proteins implicated in AMPAR trafficking and with scaffolding proteins are involved in the increased membrane expression of hippocampal AMPARs after antidepressant treatment (Martinez-Turrillas and others 2007). With regards to BPD, the mood stabilizer lamotrigine significantly enhances AMPAR activity by upregulating the surface expression and phosphorylation of GluR1 and GluR2 in a time- and dose-dependent manner in cultured hippocampal neurons (Bhagwagar and Goodwin 2005; Du and others 2007). Lithium and valproate similarly downregulate GluR1 synaptic expression in hippocampus after chronic treatment both in vitro and in vivo with therapeutically relevant concentrations (Du and others 2004).
AMPAR potentiators are a new class of compounds currently being tested in mood disorders; these modulate AMPARs indirectly by decreasing the receptor desensitization rate and/or deactivation in the presence of an agonist (e.g.,AMPA and glutamate (Black 2005; Bleakman and Lodge 1998). AMPA potentiators are small benzamide compounds that produce positive allosteric effects in AMPARs. These compounds include benzoyliperidines (e.g., CX-516), benzothiazides (e.g., cyclothiazide), pyrroliddones (piracetam, aniracetam), and birylpropylsulfonamides (e.g., LY392098). Diverse AMPA potentiators have been found to produce antidepressant-like effects in different paradigms (reviewed in (Black 2005; Miu and others 2001)). In one study, the AMPA potentiator Ampalex induced a faster antidepressant response (improvement in the first week) than fluoxetine (Knapp and others 2002). Also, arylpropylsulfoname (LY392098) augmented the efficacy of antidepressants such as imipramine in a mouse model of depression, suggesting it may be useful as possible adjunctive therapy in mood disorders (Li and others 2003).
Interestingly, AMPAR potentiators can also be used to modulate and improve glutamatergic neurotransmission and cognitive function (reviewed in (Lynch 2004; O’Neill and others 2004)). Furthermore, because AMPAR potentiators are associated with antidepressant-like properties, it is possible that AMPAR antagonists could have antimanic effects. The first group of selective AMPAR antagonists to be characterized were quinoxalinedone derivatives such as 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide, which act via the AMPAR recognition site. AMPA antagonists such GYKI 52466 blocked AMPARs on the receptor-channel complex via an allosteric site (Donevan and Rogawski 1993). Also, the GYKI 52466 analog talampanel (GYKI 53773; LY 300164) had neuroprotective effects in diverse paradigms (reviewed in (Howes and Bell 2007)); talampanel has an important anticonvulsant profile and was well tolerated in other clinical trials (reviewed by (Rogawski 2006)). The competitive AMPAR antagonist NS1209 was tested in phase I/II clinical trials and was well-tolerated in addition to its CNS bioavailability; it is currently being tested for the treatment of refractory status epilepticus (reviewed in (Rogawski 2006)).
Finally, AMPAR phosphorylation and trafficking (including receptor insertion, internalization, and delivery to synaptic sites), which are believed to be involved in the antidepressant effects of AMPAR potentiators, standard antidepressants, and mood stabilizers, play a critical role in regulating activity-dependent regulation of synaptic strength, as well as various forms of neural and behavioral plasticity (Sanacora and others 2008). Interestingly, AMPAR stimulation seems to regulate the antidepressant-like effects of both ketamine (described in Section IV) and the group II mGluR antagonist MGS0039 (Karasawa and others 2005), suggesting that increased AMPA transmission may represent a shared mechanism for antidepressant action in other classes of glutamatergic modulators.
D. NMDAR Regulation (Target IX)
D1. Pathophysiology
Altered NMDAR activity and glutamatergic synaptic transmission have been described in BPD, suggesting that NMDARs may be potential therapeutic targets (Zarate and others 2003) (for a detailed review of the characterization and functional effects of NMDARs, see (Cull-Candy and others 2001)). Reduced NMDAR binding and NR1 subunit expression have also been found in the temporal cortex and two frontal brain regions of individuals with MDD (Choudary and others 2005; Nudmamud-Thanoi and Reynolds 2004). Also, reduced hippocampal NR1/2 and NMDAR expression have been described (Beneyto and others 2007; Law and Deakin 2001; McCullumsmith and others 2007; Toro and Deakin 2005). Evidence is mounting that upregulation and/or overstimulation of the NMDAR NR2A receptor subtype plays a pivotal role in the etiology of MDD (Boyce-Rustay and Holmes 2006; Sanacora and others 2008); for instance, it was recently reported that NR2A knockout mice exhibit a highly robust anxiolytic- and antidepressant-like phenotype (Boyce-Rustay and Holmes 2006).
Similar decreases in NR1 and NR2A transcript expression, with no changes in hippocampal NR2B expression, have been found in individuals with BPD (McCullumsmith and others 2007). Furthermore, the density of gamma-aminobutyric acid (GABA) interneurons that express the NR2A subunit appeared to be decreased in the ACC of subjects with BPD (Woo and others 2004). With regards to genetics, polymorphisms in GRIN1, GRIN2A, and GRIN2B conferred susceptibility to BPD (Itokawa and others 2003; Martucci and others 2006; Mundo and others 2003). Specifically, two polymorphisms of the GRIN1 gene coding for the NR1 subunit have been associated with BPD through a linkage disequilibrium study (Mundo and others 2003). Two polymorphisms in the GRIN2B gene coding for NR2B were associated with BPD, particularly if psychotic features were present, with no concomitant modification in NR2B mRNA expression (Martucci and others 2006).
D2. Therapeutics
Recent evidence suggests that NMDA antagonists produce rapid antidepressant effects in diverse paradigms, further fueling interest in this model (reviewed in (Zarate and others 2003)). NMDARs are the most widespread ionotropic receptors and are considered the primary target for glutamatergic agents in mood disorders. The original observations of their efficacy date back to the 1960s, with reports of the mood-elevating effects of D-cycloserine (an NMDAR partial agonist).
Dizocilpine (MK-801), a channel blocker, and CGP 37849, an NMDAR antagonist, have antidepressant-like effects alone or in combination with standard antidepressants (Meloni and others 1993; Padovan and Guimaraes 2004; Skolnick and others 1992). A preliminary study evaluating the antidepressant efficacy of the allosteric NR2B subunit-selective antagonist CP-101,606 found significant antidepressant effects in treatment-resistant MDD, although there were prevalent psychomimetic effects during the study (Preskorn and others 2008). Besides producing potentially rapid antidepressant effects, it is important to note that downregulation in NMDAR function has also been described as an important downstream target for monoaminergic antidepressants. The period of latency necessary to achieve antidepressant action with these standard agents may encompass the time necessary to exert direct regulatory effects at the NMDAR-channel complex.
E. mGluR Regulation (Target X)
Although data linking mGluR dysfunction to mood disorders are sparse, at least one microarray study found abnormal mGluR3 expression in suicidal subjects with BPD; a subsequent study failed to replicate this finding (Devon and others 2001; Marti and others 2002). Also, olfactory bulbectomy, a well-known animal model of depression, has been shown to decrease the expression of diverse hippocampal mGluR receptors, including mGluR2/3 and mGluR7 (Wieronska and others 2008).
Studies have also suggested that mGluR1, mGluR2/3, and mGluR1/3 agonists have anxiolytic, antidepressant, and neuroprotective properties in rats (Cosford and others 2003; Maiese and others 2000; Palucha and others 2004; Schoepp and others 2003). Group I mGluR5 antagonists also have antidepressant-like effects in the modified forced swim and tail suspension tests in rodents (Li and others 2006; Wieronska and others 2002), as does the mGluR1 antagonist [3-ethyl-2-methyl-quinolin-6-yl-(4-methyoxy-cyclohexyl)-methanone methanesulfonate] (Belozertseva and others 2007). In animal models of BPD, the mGluR5-positive allosteric modulator 3-cyano-N-(1,3-diphenyl-1Hpyrazol-5-yl)benzamide reversed amphetamine-induced locomotor activity and deficits in prepulse inhibition in rats. Because both models are thought to be sensitive to antipsychotic treatment, this agent might be potentially useful in the treatment of mania (Kinney and others 2005). However, the mode of antidepressant-like activity for mGluR1 or mGluR5 antagonists is uncertain; it also remains unclear whether mGluR5 antagonists are safe for clinical use. For instance, the non-benzodiazepine anxiolytic fenobam, a potent and selective mGluR5 antagonist, is associated with significant psychostimulant effects (Porter and others 2005).
Group II mGluRs agonists (eg, LY341495) dose-dependently decrease immobility time in animal models of depression (Chaki and others 2004). Moreover, the group II mGluR antagonist MGS-0039 appears to be effective in the learned helplessness model of depression (Yoshimizu and others 2006), as well as enhanced hippocampal proliferation in mice (Yoshimizu and Chaki 2004). Interestingly, the activation of AMPARs was found to mediate, at least in part, the antidepressant-like activity of group II mGluR antagonists. For instance, the AMPA antagonist 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide blocked the antidepressant-like activity of MGS-0039 in the tail suspension test (Karasawa and others 2005).
The selective group III mGluR agonist ([1S,3R,4S]-1-aminocyclopentane-1,3,4-tricarboxylic-acid) and the mGluR8 agonist ([RS]-4-phosphonophenylglycine) both induced significant antidepressant-like effects in the forced swim test in rats (Gasparini and others 1999). Palucha and colleagues (2004) similarly reported the antidepressant-like effects of group III mGluR agonists in the behavioral despair test (Palucha and others 2004). In addition, mGluR7 knockout induced antidepressant-like effects in the forced swim and tail suspension tests in mice (Cryan and others 2003).
Taken together, the results suggest that many of these compounds may be promising for the treatment of mood disorders. So far, however, there are no published clinical studies in mood disorders demonstrating their efficacy.
F. Regulation of KA receptors (KARs) (Target XI)
A recent study with GluR6—a KAR subunit—knockout mice found that this receptor had a potential functional role in modulating the behavioral symptoms of mania (Shaltiel and others 2008); specifically, GluR6 knockout mice displayed increased risk-taking, aggressive behavior, motor activity in response to amphetamine, as well as less despair; these manifestations were decreased after chronic lithium treatment (Shaltiel and others 2008). A recent, large, family-based association study evaluating the KAR GluR7 gene (GRIK3) described significant linkage disequilibrium in MDD but not BPD (Schiffer and Heinemann 2007). Interestingly, increased GRIK3 DNA-copy number was described in individuals with BPD (Wilson and others 2006). Finally, a common variant in the 3′UTR GRIK4 gene was shown to protect against BPD (Pickard and others 2006).
To date, no KA modulator has been tested in the treatment of mood disorders. Notably, however, one recent study found that individuals with MDD who had a GRIK4 gene polymorphism (rs1954787) were more likely to respond to treatment with the antidepressant citalopram than those who did not have this allele (Paddock and others 2007).
G. Postsynaptic Density (PSD) Proteins (Target XII)
Decreased expression of NMDARs has been associated with diverse PSD signaling proteins, including NF-L, PSD95, and SAP102. Decreased transcripts of these associated intracellular proteins were found in BPD subjects (Clinton and Meador-Woodruff 2004). In individuals with MDD, a similar decrease was also observed in SAP102 levels (which primarily interact with the NR2B subunit) (Clinton and Meador-Woodruff 2004), and decreased SAP102 expression was observed in the striatum (Kristiansen and Meador-Woodruff 2005). Recently, Toro and Deakin (2005) found reduced PSD95 levels in the dentate molecular layer in individuals with BPD (Toro and Deakin 2005). Taken together, these findings suggest an important role for PSD regulation by glutamatergic turnover in the pathophysiology of mood disorders.
To date, no specific treatment selectively targeting PSD proteins has been developed.
IV. Glutamatergic Modulators and Mood Disorders: Riluzole and Ketamine as the Prototypes for the Development of the Next Generation of Antidepressants and Mood Stabilizers
The in-depth study of particular agents can lead to novel insights into the pathophysiology of mood disorders, as well as the development of biomarkers and improved therapeutics. Here we review riluzole and ketamine, with a particular emphasis on how increased understanding of the manner in which these agents exert their antidepressant effects can have profound implications for the development of novel therapeutics.
A. Riluzole
Riluzole (2-amino-6-trifluoromethoxy benzothiazole) is a blood-brain-penetrant glutamatergic modulator with neuroprotective and anticonvulsant properties approved by the US Food and Drug Administration (FDA) for the treatment of amyotrophic lateral sclerosis (ALS). Riluzole inhibits glutamate release and increases AMPA trafficking by enhancing membrane insertion of GluR1 and GluR2. It also increases glutamate reuptake and activates neurotrophic factor synthesis (Frizzo and others 2004; Mizuta and others 2001). Riluzole exerts its anti-glutamatergic effects by inhibiting voltage-dependent sodium channels in neurons and by reducing glutamate.
Enhanced glutamate uptake in glial cells is thought to underlie riluzole’s cellular mechanism for antidepressant action (Frizzo and others 2004; Fumagalli and others 2008). It is also possible that riluzole can prevent the overstimulation of extrasynaptic glutamate receptors via EAATs, which may prevent activation of cellular excitotoxicity (Hardingham 2006). Increased 13C-glucose metabolism was observed after chronic (21 days) treatment with riluzole, as assessed by 13C labeling from [1-13C]glucose infused intravenously in prefrontal cortex and hippocampus; this suggests enhanced glutamatergic metabolism rather than decreased glutamate release in these areas (Chowdhury and others 2008).
Notably, long-term administration of riluzole in animal models was recently found to induce antidepressant-like behaviors in the forced swim test (Banasr and others 2008). In mice, pretreatment with riluzole significantly decreased amphetamine-induced hyperlocomotion, suggesting antimanic properties. It has similarly been shown to protect glial cells against glutamate excitotoxicity (Dagci and others 2007).
Diverse clinical studies have been conducted with riluzole in mood disorders. In an open label trial of 19 patients with treatment-resistant MDD who received riluzole monotherapy (100-200 mg/day), riluzole had significant antidepressant effects—as measured by Montgomery-Asberg Depression Rating Scale (MADRS) scores on weeks three through six—with few side effects (Zarate and others 2004). Thirteen patients (68%) completed the trial, and all patients experienced a significant (33%) improvement in MADRS scores at week six. Notably, a similar anti-anxiety effect was observed, with a 29% decrease in total Hamilton Anxiety Rating Scale (HAM-A) scores.
Another eight-week, open-label trial investigated the effects of riluzole (100-200 mg/day) as an add-on to lithium in 14 patients with bipolar depression (Zarate and others 2005); of the 57% of patients who completed this trial, significant improvement in MADRS scores was observed by week five, with a 60% overall reduction in MADRS scores across the eight weeks of treatment. Riluzole (50mg/twice daily) has also been used as adjunctive therapy in 13 individuals with MDD with significant residual symptoms (Sanacora and others 2007), 10 of whom completed at least six weeks of treatment. A significant decrease (36%) in Hamilton Depression Rating Scale (HAM-D) scores was observed among those who completed six weeks of treatment. Similarly, two open-label trials of riluzole in generalized anxiety disorder (GAD) (Mathew and others 2005) and obsessive-compulsive disorder (OCD) (Coric and others 2005) found that riluzole had significant therapeutic effects. Taken together, these results are particularly promising when considering that these studies included patients who had failed to respond to multiple previous attempts at pharmacotherapy, yet showed significant early response to riluzole. Double-blind, placebo-controlled trials are necessary to confirm these data.
B. Ketamine
Ketamine (dl2-(o-chlorophenyl)-2-(methylamino) cyclohexanone hydrochloride) is a non-competitive NMDA antagonist (Harrison and Simmonds 1985) and a derivative of phencyclidine (PCP). First isolated in 1961 as CI581, ketamine acts by antagonizing NMDARs, thus preventing excess calcium influx and cellular damage. Ketamine has a high affinity for the NMDAR, with slow open-channel blocking/unblocking kinetics, and a specific type of channel closure (called “trapping block”). Simultaneously, it induces a substantial presynaptic release of glutamate by increasing the firing rate of glutamatergic neurons after disinhibiting GABAergic inputs (Moghaddam and others 1997). Some of these properties are believed to be involved in ketamine’s antidepressant effects.
One initial clinical study described improved depressive symptoms within 72 hours after ketamine infusion in seven subjects with treatment-resistant MDD (Berman and others 2000). Another randomized, double-blind, placebo-controlled, crossover study described a rapid (within the first two hours after infusion), robust, and relatively sustained antidepressant effect (lasting one to two weeks) after a single infusion of a subanesthetic dose of ketamine (0.5 mg/kg for 40 minutes) in patients with treatment-resistant MDD (Zarate and others 2006a). More than 70% of patients met criteria for response (50% improvement) at 24 hours after infusion and 35% maintained a sustained response after one week. Patients were rated 60 minutes before infusion and at 40, 80, 110, and 230 minutes, as well as one, two, three, and seven days after the single intravenous dose. Significant improvement in the 21-item HAM-D with ketamine over placebo was observed from 110 minutes through seven days. Notably, the response rates with ketamine after 24 hours (71%) were similar to those described after six to eight weeks of treatment with traditional monoaminergic-based antidepressants (65%) (Entsuah and others 2001; Thase and others 2005). It is important to mention that the antidepressant efficacy of ketamine has been noted much beyond its short half-life (Zarate and others 2006a). Two patients maintained their response for at least two weeks in the ketamine group. In contrast, no patient had a sustained response one day following infusion in the placebo group. Mild perceptual disturbances were observed in most patients only in the first hour after infusion; no serious adverse events occurred.
Further reports of ketamine’s antidepressant effects concur with the studies described above. Ketamine infusion had similar therapeutic effects in depressed patients during pre- and post-operative states, and in patients with MDD and concurrent pain syndrome (Goforth and Holsinger 2007; Kudoh and others 2002). Ketamine was also effective when administered concurrently with ECT, and case reports of its rapid antidepressant efficacy have been described in treatment-resistant MDD comorbid with alcohol dependence and pain syndrome (Liebrenz and others 2007; Ostroff and others 2005). In addition, many animal models have noted ketamine’s significant antidepressant and anxiolytic effects (Aguado and others 1994; Garcia and others 2008; Maeng and others 2008; Mickley and others 1998; Silvestre and others 1997; Zarate and others 2002).
Despite ketamine’s safety profile and lack of physical dependence (Britt and McCance-Katz 2005; Green and others 1998), one issue of particular import is its sedative and psychotomimetic side effects, which will probably continue to limit its clinical use in larger samples. Relatedly, an increased propensity to psychotomimetic effects and tolerance to ketamine’s antidepressant effects might occur after repeated doses. For instance, a recent case report noted that a second infusion of ketamine showed limited effect one month after an initial infusion (Liebrenz and others 2007). Similarly, repeated exposure to ketamine increases the risk of severe psychosis, dissociative episodes, and severe emotional distress or euphoria in patients and healthy subjects (Carpenter 1999; Perry and others 2007).
Despite ketamine’s sedative and psychotomimetic side effects, it does induce a consistently reproducible antidepressant effect within a short period of time. Thus, its true worth may be as a research tool. For instance, in contrast to ketamine, memantine is a more selective, non-competitive NMDA antagonist with both anticonvulsant and neuroprotective properties. Studies have found that memantine has no antidepressant response in MDD (Zarate and others 2006b). Thus, the data suggest that high affinity NMDA antagonism is key to producing rapid improvement of depressive symptoms. Currently, our laboratory is using ketamine to develop biomarkers of response in order to develop the next generation of antidepressants. Such work is important because identifying the biological correlates associated with ketamine’s rapid antidepressant effects may help identify valuable, specific biomarkers and novel targets for the development of new compounds that can similarly produce rapid antidepressant effects, but without ketamine’s side effects.
B1. Family history of alcoholism
In the past few years there has been increased interest in the joint study of the pathophysiology of MDD and risk of alcoholism, as the glutamate system appears to play a major role in both conditions. One recent study found that alcohol-dependent individuals had marked reductions to the subjective intoxicating effects of ketamine compared to healthy controls (Krystal and others 2003). The same group subsequently found that healthy individuals with a positive family history of alcohol dependence had fewer perceptual alterations and lower dysphoric mood after receiving ketamine than those without a family history (Petrakis and others 2004). Another recent study assessed antidepressant response in patients with treatment-resistant MDD and found that subjects with MDD and a family history of alcohol dependence showed significantly greater improvement after ketamine infusion than those who had no family history of alcohol dependence (Phelps and others 2008).
The precise reasons underlying the better response of patients with treatment-resistant MDD with a positive family history of alcohol dependence is essentially unknown. However, emerging data suggest that genetically determined alterations in NMDAR subunits may be associated with alcohol dependence. For instance, a recent study found evidence of an association between NR2A and alcohol dependence (Schumann and others 2008), and alcohol has been shown to regulate NR2A expression in brain regions implicated in addiction-related neurobiological processes, including the amygdala and hippocampus (Boyce-Rustay and Holmes 2006). Thus, it is possible that genetic variations in NMDA subunits, particularly NR2A, increase vulnerability to alcohol dependence by altering the sensitivity of the NMDA complex. Because ketamine acts as a partial NR2A antagonist (Petrenko and others 2004), it is possible that this difference in NR2A sensitivity may impart a greater response to ketamine’s antidepressant effects.
B2. ACC activity
In a study using standard antidepressants, Mayberg and colleagues (1997) were the first to show that higher rostral ACC (BA 24a/b) metabolism differentiated treatment responders from non-responders at six weeks (Mayberg and others 1997). A recent functional MRI study found that faster improvement of depressive symptoms with fluoxetine was predicted by more positive functional activation of the ACC during the face emotional task (Chen and others 2007). Similarly, patients with better antidepressant response to nortriptyline also had hyperactivity (higher theta activity) in the rostral ACC (Brodmann’s area 24/32), thus supporting the role of ACC activation as a predictor of antidepressant response (Pizzagalli and others 2001). Interestingly, the antidepressant effects of ECT also appear to be correlated with increased metabolism in the left subgenual ACC (McCormick and others 2007). Likewise, high pretreatment ACC activity predicted better response to repetitive transcranial magnetic stimulation (rTMS) (Langguth and others 2007). A consistent association between increased subgenual cingulate (Cg25) activity and acute sadness has also been noted using positron emission tomography (PET); in this study, application of chronic deep brain stimulation (DBS) close to the subgenual cingulate region (Brodmann’s area 25) reduced the elevated activity in this area, and this reduction was associated with a significant and sustained remission of depressive symptoms in four out of six patients (Mayberg and others 2005). Other antidepressants, as well as sleep deprivation, have also been shown to produce similar effects (Saxena and others 2003; Wu and others 1999). Taken together, these findings support the role of ACC activation as a predictor of improved antidepressant response (Pizzagalli and others 2001), but its specific role in the fast-acting agents needs to be further evaluated.
One recent study tested the hypothesis that magnetoencephalography (MEG) recordingscould provide a neurophysiologic biomarker associated with ketamine’s rapid antidepressant effects. Increased pretreatment rostral ACC activity in MDD patients was indeed positively correlated with rapid antidepressant response to ketamine infusion (Salvadore and others 2008). The ACC activity in response to rapid exposure to fearful faces was measured in eleven drug-free MDD subjects matched with healthy controls. In this same study, amygdala response to fearful faces was negatively correlated with antidepressant response to ketamine 230 minutes after infusion (Salvadore and others 2008). Interestingly, Deakin and colleagues (2008) also observed a direct regulation of orbitofrontal and subgenual ACC blood oxygenation level dependent (BOLD) activity by ketamine in healthy individuals. In addition, they observed three neuroanatomical systems potentially related to ketamine’s side effects, especially psychosis: (1) deactivations in the ventral anterior limbic cortex; (2) activation in the mid-posterior cingulate; and (3) activations in the temporal lobe hippocampus/parahippocampal cortex and superior, middle, and inferior temporal cortices (Deakin and others 2008).
B3. Brain-derived neurotrophic factor (BDNF)
BDNF is the most studied neurotrophin in the pathophysiology of MDD. One disease model suggested that the pathophysiology of mood disorders was associated with decreased neurotrophin levels during mood episodes. Because of this association, one recent study investigated whether changes in BDNF levels were associated with the initial antidepressant effects of ketamine, but found that these antidepressant effects were not mediated by BDNF. BDNF levels showed no changes from baseline after ketamine infusion and up to 230 minutes post-infusion (a time point when antidepressant effects and response were manifest). The study did, however, confirm ketamine’s efficacy as a fast-acting antidepressant (Machado-Vieira and others in press).
Intriguingly, both ketamine and standard monoaminergic antidepressants seem to act directly at the NMDARs and AMPARs (Du and others 2004; Du and others 2007; Maeng and others 2008); however, it appears that ketamine’s glutamatergic targets may rapidly activate early neuroplastic changes and synaptic potentiation through AMPARs without being related to BDNF effects. In contrast, traditional antidepressants seem to activate long neurotrophic signaling cascades, which may be the reason for their delayed therapeutic effects.
B4. AMPA relative to NMDA throughput
Preclinical data suggest a relevant interaction between AMPARs and NMDARs in ketamine’s rapid antidepressant efficacy (Maeng and Zarate 2007; Maeng and others 2008). For instance, ketamine decreased immobility time in the forced swim test, and this antidepressant effect was abolished when the AMPA antagonist NBQX was given prior to its infusion. Conversely, AMPA inhibition with NBQX did not regulate imipramine’s antidepressant-like effects (Maeng and others 2008), thus reinforcing ketamine’s selectivity for this effect, although it may also extend to other glutamatergic modulators.
Recent studies from our laboratory found that Ro 25-6981, a selective NR2B subunit antagonist, had antidepressant-like properties in rodents. As with ketamine, these effects appear to be largely mediated through AMPARs (Maeng and others 2008). This issue is particularly interesting when considering that synaptic potentiation is thought to be involved in ketamine’s acute antidepressant effects (Salvadore and others 2008). As noted above, ketamine produces rapid AMPA-mediated synaptic potentiation, whereas traditional antidepressants do so in a delayed manner through a cascade of intracellular signaling changes (Sanacora and others 2008), which might explain their differential time of onset for antidepressant effects. Sleep deprivation, which also induces rapid antidepressant effects, has similarly has been shown to increase AMPA-mediated synaptic plasticity (Faraguna and others 2008). Indeed, ketamine seems to increase synaptic efficacy in the amygdala-accumbens pathway (Kessal and others 2005). Notably, synaptic potentiation is known to involve AMPA trafficking, which enhances AMPA throughput. Therefore, it is possible that enhanced glutamatergic throughput of AMPARs relative to NMDARs after ketamine treatment may result in increased synaptic potentiation and activation of early neuroplastic genes (possibly not related to increased BDNF levels). The consequent increase in glutamate release then preferentially favors AMPARs over NMDARs because the latter are blocked by ketamine; thus, the net effect of ketamine’s antidepressant effect on a cellular level is increased glutamatergic throughput.
IV. Summary and Conclusions
Recent advances in our understanding of glutamatergic dysfunction in the pathophysiology of severe mood disorders—and in particular the role played by the tripartite glutamatergic synapse (presynaptic neuron, postsynaptic neuron, and glia)—have provided a better understanding of the importance of this complex and critical cross-talk mediated by glutamate in the physiological and pathological conditions associated with mood disorders. These insights have fueled the development of new and potentially more effective compounds as therapeutics for severe mood disorders.
Several glutamatergic compounds have been tested in “proof-of-concept” studies in severe mood disorders. As described above, ketamine and riluzole merit continued study as putative archetypes for improved therapeutics. They offer a unique opportunity to better delineate the exact cellular mechanisms involved in therapeutic response. Finally, the promising field of biomarkers of response may ultimately expand our understanding of what bio-signatures are necessary to determine the clinical relevance of specific regulatory effects induced by other glutamatergic modulators targeting specific receptors/subunits in the development of the next generation of improved therapeutics for severe mood disorders. Pharmacological treatments that could exert faster and sustained therapeutic effect in mood disorders could significantly impact care for our patients as well as public health worldwide.
Acknowledgements
Funding for this work was supported by the Intramural Research Program of the National Institute of Mental Health (NIMH) and a NARSAD Award (CAZ). Ioline Henter provided outstanding editorial assistance. A patent application for the use of ketamine in depression has been submitted listing Dr. Zarate among the inventors; he has assigned his rights on the patent to the US government.
Abbreviations
- AMPA
(-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid)
- cAMP
(cyclic adenosine monophosphate)
- EAAT
(excitatory amino-acid transporters)
- GABA
(gamma-aminobutyric acid)
- Glu
(Glutamate)
- Gly
(Glycine)
- KA
(kainate)
- NMDA
(N-methyl-D-aspartate)
- PSD
(postsynaptic density)
- PSVR
(presynaptic voltage-operated release)
- (mGluRs)
metabotropic Glu receptors
- SNARE
(soluble N-ethylmaleimide-sensitive factor attachment receptor)
- VGLUTs
(vesicular Glu transporters)
REFERENCES
- Aguado L, San Antonio A, Perez L, del Valle R, Gomez J. Effects of the NMDA receptor antagonist ketamine on flavor memory: conditioned aversion, latent inhibition, and habituation of neophobia. Behav Neural Biol. 1994;61:271–81. doi: 10.1016/s0163-1047(05)80010-x. [DOI] [PubMed] [Google Scholar]
- Altamura CA, Mauri MC, Ferrara A, Moro AR, D’Andrea G, Zamberlan F. Plasma and platelet excitatory amino acids in psychiatric disorders. Am J Psychiatry. 1993;150:1731–3. doi: 10.1176/ajp.150.11.1731. [DOI] [PubMed] [Google Scholar]
- Banasr M, Chowdhury GM, Terwilliger R, Newton SS, Duman RS, Behar KL, Sanacora G. Glial pathology in an animal model of depression: reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol Psychiatry. 2008 doi: 10.1038/mp.2008.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banasr M, Duman RS. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol Psychiatry. 2008;64:863–70. doi: 10.1016/j.biopsych.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banasr M, Valentine GW, Li XY, Gourley SL, Taylor JR, Duman RS. Chronic unpredictable stress decreases cell proliferation in the cerebral cortex of the adult rat. Biol Psychiatry. 2007;62:496–504. doi: 10.1016/j.biopsych.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Baune BT, Adrian I, Jacobi F. Medical disorders affect health outcome and general functioning depending on comorbid major depression in the general population. J Psychosom Res. 2007;62:109–18. doi: 10.1016/j.jpsychores.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Belozertseva IV, Kos T, Popik P, Danysz W, Bespalov AY. Antidepressant-like effects of mGluR1 and mGluR5 antagonists in the rat forced swim and the mouse tail suspension tests. Eur Neuropsychopharmacol. 2007;17:172–9. doi: 10.1016/j.euroneuro.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Beneyto M, Kristiansen LV, Oni-Orisan A, McCullumsmith RE, Meador-Woodruff JH. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology. 2007;32:1888–902. doi: 10.1038/sj.npp.1301312. [DOI] [PubMed] [Google Scholar]
- Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of AMPA receptor trafficking and signaling molecule transcripts in the prefrontal cortex in schizophrenia. Synapse. 2006;60:585–98. doi: 10.1002/syn.20329. [DOI] [PubMed] [Google Scholar]
- Berk M, Copolov DL, Dean O, Lu K, Jeavons S, Schapkaitz I, Anderson-Hunt M, Bush AI. N-acetyl cysteine for depressive symptoms in bipolar disorder--a double-blind randomized placebo-controlled trial. Biol Psychiatry. 2008;64:468–75. doi: 10.1016/j.biopsych.2008.04.022. [DOI] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–4. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Bhagwagar Z, Goodwin GM. Lamotrigine in the treatment of bipolar disorder. Expert Opin Pharmacother. 2005;6:1401–8. doi: 10.1517/14656566.6.8.1401. [DOI] [PubMed] [Google Scholar]
- Black MD. Therapeutic potential of positive AMPA modulators and their relationship to AMPA receptor subunits. A review of preclinical data. Psychopharmacology (Berl) 2005;179:154–63. doi: 10.1007/s00213-004-2065-6. [DOI] [PubMed] [Google Scholar]
- Bleakman D, Lodge D. Neuropharmacology of AMPA and kainate receptors. Neuropharmacology. 1998;37:1187–204. doi: 10.1016/s0028-3908(98)00139-7. [DOI] [PubMed] [Google Scholar]
- Boyce-Rustay JM, Holmes A. Genetic inactivation of the NMDA receptor NR2A subunit has anxiolytic- and antidepressant-like effects in mice. Neuropsychopharmacology. 2006;31:2405–14. doi: 10.1038/sj.npp.1301039. [DOI] [PubMed] [Google Scholar]
- Britt GC, McCance-Katz EF. A brief overview of the clinical pharmacology of “club drugs”. Subst Use Misuse. 2005;40:1189–201. doi: 10.1081/JA-200066730. [DOI] [PubMed] [Google Scholar]
- Carpenter WT., Jr. The schizophrenia ketamine challenge study debate. Biol Psychiatry. 1999;46:1081–91. doi: 10.1016/s0006-3223(99)00194-8. [DOI] [PubMed] [Google Scholar]
- Chaki S, Yoshikawa R, Hirota S, Shimazaki T, Maeda M, Kawashima N, Yoshimizu T, Yasuhara A, Sakagami K, Okuyama S. MGS0039: a potent and selective group II metabotropic glutamate receptor antagonist with antidepressant-like activity. Neuropharmacology. 2004;46:457–67. doi: 10.1016/j.neuropharm.2003.10.009. others. [DOI] [PubMed] [Google Scholar]
- Chen CH, Ridler K, Suckling J, Williams S, Fu CH, Merlo-Pich E, Bullmore E. Brain imaging correlates of depressive symptom severity and predictors of symptom improvement after antidepressant treatment. Biol Psychiatry. 2007;62:407–14. doi: 10.1016/j.biopsych.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Choudary PV, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP, Myers RM, Bunney WE, Jr., Akil H, Watson SJ. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc Natl Acad Sci U S A. 2005;102:15653–8. doi: 10.1073/pnas.0507901102. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury GM, Banasr M, de Graaf RA, Rothman DL, Behar KL, Sanacora G. Chronic riluzole treatment increases glucose metabolism in rat prefrontal cortex and hippocamus. J Cereb Blood Flow Metab. 2008;28:1892–1897. doi: 10.1038/jcbfm.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinton SM, Meador-Woodruff JH. Abnormalities of the NMDA Receptor and Associated Intracellular Molecules in the Thalamus in Schizophrenia and Bipolar Disorder. Neuropsychopharmacology. 2004;29:1353–62. doi: 10.1038/sj.npp.1300451. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Bliss TV. Memories of NMDA receptors and LTP. Trends Neurosci. 1995;18:54–6. [PubMed] [Google Scholar]
- Coric V, Taskiran S, Pittenger C, Wasylink S, Mathalon DH, Valentine G, Saksa J, Wu YT, Gueorguieva R, Sanacora G. Riluzole augmentation in treatment-resistant obsessive-compulsive disorder: an open-label trial. Biol Psychiatry. 2005;58:424–8. doi: 10.1016/j.biopsych.2005.04.043. others. [DOI] [PubMed] [Google Scholar]
- Cosford ND, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson J, Bristow L, Brodkin J, Jiang X, McDonald I. 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine: a potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J Med Chem. 2003;46:204–6. doi: 10.1021/jm025570j. others. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Kelly PH, Neijt HC, Sansig G, Flor PJ, van Der Putten H. Antidepressant and anxiolytic-like effects in mice lacking the group III metabotropic glutamate receptor mGluR7. Eur J Neurosci. 2003;17:2409–17. doi: 10.1046/j.1460-9568.2003.02667.x. [DOI] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327–35. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- Dagci T, Yilmaz O, Taskiran D, Peker G. Neuroprotective agents: is effective on toxicity in glial cells? Cell Mol Neurobiol. 2007;27:171–7. doi: 10.1007/s10571-006-9082-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin JF, Lees J, McKie S, Hallak JE, Williams SR, Dursun SM. Glutamate and the neural basis of the subjective effects of ketamine: a pharmaco-magnetic resonance imaging study. Arch Gen Psychiatry. 2008;65:154–64. doi: 10.1001/archgenpsychiatry.2007.37. [DOI] [PubMed] [Google Scholar]
- Devon RS, Anderson S, Teague PW, Burgess P, Kipari TM, Semple CA, Millar JK, Muir WJ, Murray V, Pelosi AJ. Identification of polymorphisms within Disrupted in Schizophrenia 1 and Disrupted in Schizophrenia 2, and an investigation of their association with schizophrenia and bipolar affective disorder. Psychiatr Genet. 2001;11:71–8. doi: 10.1097/00041444-200106000-00003. others. [DOI] [PubMed] [Google Scholar]
- Donevan SD, Rogawski MA. GYKI 52466, a 2,3-benzodiazepine, is a highly selective, noncompetitive antagonist of AMPA/kainate receptor responses. Neuron. 1993;10:51–9. doi: 10.1016/0896-6273(93)90241-i. [DOI] [PubMed] [Google Scholar]
- Du J, Gray NA, Falke CA, Chen W, Yuan P, Szabo ST, Einat H, Manji HK. Modulation of synaptic plasticity by antimanic agents: the role of AMPA glutamate receptor subunit 1 synaptic expression. J Neurosci. 2004;24:6578–89. doi: 10.1523/JNEUROSCI.1258-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Suzuki K, Wei Y, Wang Y, Blumenthal R, Chen Z, Falke C, Zarate CA, Jr., Manji HK. The anticonvulsants lamotrigine, riluzole, and valproate differentially regulate AMPA receptor membrane localization: relationship to clinical effects in mood disorders. Neuropsychopharmacology. 2007;32:793–802. doi: 10.1038/sj.npp.1301178. [DOI] [PubMed] [Google Scholar]
- Entsuah AR, Huang H, Thase ME. Response and remission rates in different subpopulations with major depressive disorder administered venlafaxine, selective serotonin reuptake inhibitors, or placebo. J Clin Psychiatry. 2001;62:869–77. doi: 10.4088/jcp.v62n1106. [DOI] [PubMed] [Google Scholar]
- Fagiolini A, Kupfer DJ, Masalehdan A, Scott JA, Houck PR, Frank E. Functional impairment in the remission phase of bipolar disorder. Bipolar Disord. 2005;7:281–5. doi: 10.1111/j.1399-5618.2005.00207.x. [DOI] [PubMed] [Google Scholar]
- Faraguna U, Vyazovskiy VV, Nelson AB, Tononi G, Cirelli C. A causal role for brain-derived neurotrophic factor in the homeostatic regulation of sleep. J Neurosci. 2008;28:4088–95. doi: 10.1523/JNEUROSCI.5510-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frizzo ME, Dall’Onder LP, Dalcin KB, Souza DO. Riluzole enhances glutamate uptake in rat astrocyte cultures. Cell Mol Neurobiol. 2004;24:123–8. doi: 10.1023/B:CEMN.0000012717.37839.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye MA, Tsai GE, Huggins T, Coyle JT, Post RM. Low cerebrospinal fluid glutamate and glycine in refractory affective disorder. Biol Psychiatry. 2007;61:162–6. doi: 10.1016/j.biopsych.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Fumagalli E, Funicello M, Rauen T, Gobbi M, Mennini T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol. 2008;578:171–6. doi: 10.1016/j.ejphar.2007.10.023. [DOI] [PubMed] [Google Scholar]
- Garcia LS, Comim CM, Valvassori SS, Reus GZ, Barbosa LM, Andreazza AC, Stertz L, Fries GR, Gavioli EC, Kapczinski F. Acute administration of ketamine induces antidepressant-like effects in the forced swimming test and increases BDNF levels in the rat hippocampus. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:140–4. doi: 10.1016/j.pnpbp.2007.07.027. others. [DOI] [PubMed] [Google Scholar]
- Gasparini F, Bruno V, Battaglia G, Lukic S, Leonhardt T, Inderbitzin W, Laurie D, Sommer B, Varney MA, Hess SD. (R,S)-4-phosphonophenylglycine, a potent and selective group III metabotropic glutamate receptor agonist, is anticonvulsive and neuroprotective in vivo. J Pharmacol Exp Ther. 1999;289:1678–87. others. [PubMed] [Google Scholar]
- Goforth HW, Holsinger T. Rapid relief of severe major depressive disorder by use of preoperative ketamine and electroconvulsive therapy. J Ect. 2007;23:23–5. doi: 10.1097/01.yct.0000263257.44539.23. [DOI] [PubMed] [Google Scholar]
- Green SM, Rothrock SG, Lynch EL, Ho M, Harris T, Hestdalen R, Hopkins GA, Garrett W, Westcott K. Intramuscular ketamine for pediatric sedation in the emergency department: safety profile in 1,022 cases. Ann Emerg Med. 1998;31:688–97. doi: 10.1016/s0196-0644(98)70226-4. [DOI] [PubMed] [Google Scholar]
- Guilarte TR, Hammoud DA, McGlothan JL, Caffo BS, Foss CA, Kozikowski AP, Pomper MG. Dysregulation of glutamate carboxypeptidase II in psychiatric disease. Schizophr Res. 2008;99:324–32. doi: 10.1016/j.schres.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE. Pro-survival signalling from the NMDA receptor. Biochem Soc Trans. 2006;34:936–8. doi: 10.1042/BST0340936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison NL, Simmonds MA. Quantitative studies on some antagonists of N-methyl D-aspartate in slices of rat cerebral cortex. Br J Pharmacol. 1985;84:381–91. doi: 10.1111/j.1476-5381.1985.tb12922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Sawa A, Iyo M. Increased levels of glutamate in brains from patients with mood disorders. Biol Psychiatry. 2007;62:1310–6. doi: 10.1016/j.biopsych.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Hashimoto R, Hough C, Nakazawa T, Yamamoto T, Chuang DM. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J Neurochem. 2002;80:589–97. doi: 10.1046/j.0022-3042.2001.00728.x. [DOI] [PubMed] [Google Scholar]
- Hasler G, van der Veen JW, Tumonis T, Meyers N, Shen J, Drevets WC. Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 2007;64:193–200. doi: 10.1001/archpsyc.64.2.193. [DOI] [PubMed] [Google Scholar]
- Howes JF, Bell C. Talampanel. Neurotherapeutics. 2007;4:126–9. doi: 10.1016/j.nurt.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itokawa M, Yamada K, Iwayama-Shigeno Y, Ishitsuka Y, Detera-Wadleigh S, Yoshikawa T. Genetic analysis of a functional GRIN2A promoter (GT)n repeat in bipolar disorder pedigrees in humans. Neurosci Lett. 2003;345:53–6. doi: 10.1016/s0304-3940(03)00501-9. [DOI] [PubMed] [Google Scholar]
- Jabaudon D, Shimamoto K, Yasuda-Kamatani Y, Scanziani M, Gahwiler BH, Gerber U. Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc Natl Acad Sci U S A. 1999;96:8733–8. doi: 10.1073/pnas.96.15.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judd LL, Akiskal HS, Schettler PJ, Endicott J, Maser J, Solomon DA, Leon AC, Rice JA, Keller MB. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry. 2002;59:530–7. doi: 10.1001/archpsyc.59.6.530. [DOI] [PubMed] [Google Scholar]
- Karasawa J, Shimazaki T, Kawashima N, Chaki S. AMPA receptor stimulation mediates the antidepressant-like effect of a group II metabotropic glutamate receptor antagonist. Brain Res. 2005;1042:92–8. doi: 10.1016/j.brainres.2005.02.032. [DOI] [PubMed] [Google Scholar]
- Kessal K, Chessel A, Spennato G, Garcia R. Ketamine and amphetamine both enhance synaptic transmission in the amygdala-nucleus accumbens pathway but with different time-courses. Synapse. 2005;57:61–5. doi: 10.1002/syn.20154. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Akiskal HS, Ames M, Birnbaum H, Greenberg P, Hirschfeld RM, Jin R, Merikangas KR, Simon GE, Wang PS. Prevalence and effects of mood disorders on work performance in a nationally representative sample of U.S. workers. Am J Psychiatry. 2006;163:1561–8. doi: 10.1176/appi.ajp.163.9.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney GG, O’Brien JA, Lemaire W, Burno M, Bickel DJ, Clements MK, Chen TB, Wisnoski DD, Lindsley CW, Tiller PR. A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J Pharmacol Exp Ther. 2005;313:199–206. doi: 10.1124/jpet.104.079244. others. [DOI] [PubMed] [Google Scholar]
- Knapp RJ, Goldenberg R, Shuck C, Cecil A, Watkins J, Miller C, Crites G, Malatynska E. Antidepressant activity of memory-enhancing drugs in the reduction of submissive behavior model. Eur J Pharmacol. 2002;440:27–35. doi: 10.1016/s0014-2999(02)01338-9. [DOI] [PubMed] [Google Scholar]
- Kristiansen LV, Meador-Woodruff JH. Abnormal striatal expression of transcripts encoding NMDA interacting PSD proteins in schizophrenia, bipolar disorder and major depression. Schizophr Res. 2005;78:87–93. doi: 10.1016/j.schres.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Krystal JH. Capitalizing on extrasynaptic glutamate neurotransmission to treat antipsychotic-resistant symptoms in schizophrenia. Biol Psychiatry. 2008;64:358–60. doi: 10.1016/j.biopsych.2008.06.011. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Petrakis IL, Krupitsky E, Schutz C, Trevisan L, D’Souza DC. NMDA receptor antagonism and the ethanol intoxication signal: from alcoholism risk to pharmacotherapy. Ann N Y Acad Sci. 2003;1003:176–84. doi: 10.1196/annals.1300.010. [DOI] [PubMed] [Google Scholar]
- Kudoh A, Takahira Y, Katagai H, Takazawa T. Small-dose ketamine improves the postoperative state of depressed patients. Anesth Analg. 2002;95:114–8. doi: 10.1097/00000539-200207000-00020. table of contents. [DOI] [PubMed] [Google Scholar]
- Kugaya A, Sanacora G. Beyond monoamines: glutamatergic function in mood disorders. CNS Spectr. 2005;10:808–19. doi: 10.1017/s1092852900010403. [DOI] [PubMed] [Google Scholar]
- Langguth B, Wiegand R, Kharraz A, Landgrebe M, Marienhagen J, Frick U, Hajak G, Eichhammer P. Pre-treatment anterior cingulate activity as a predictor of antidepressant response to repetitive transcranial magnetic stimulation (rTMS) Neuro Endocrinol Lett. 2007;28:633–638. [PubMed] [Google Scholar]
- Law AJ, Deakin JF. Asymmetrical reductions of hippocampal NMDAR1 glutamate receptor mRNA in the psychoses. Neuroreport. 2001;12:2971–4. doi: 10.1097/00001756-200109170-00043. [DOI] [PubMed] [Google Scholar]
- Lebon V, Petersen KF, Cline GW, Shen J, Mason GF, Dufour S, Behar KL, Shulman GI, Rothman DL. Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J Neurosci. 2002;22:1523–31. doi: 10.1523/JNEUROSCI.22-05-01523.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Gaskins D, Anand A, Shekhar A. Glia mechanisms in mood regulation: a novel model of mood disorders. Psychopharmacology (Berl) 2007;191:55–65. doi: 10.1007/s00213-006-0652-4. [DOI] [PubMed] [Google Scholar]
- Levine J, Panchalingam K, Rapoport A, Gershon S, McClure RJ, Pettegrew JW. Increased cerebrospinal fluid glutamine levels in depressed patients. Biol Psychiatry. 2000;47:586–93. doi: 10.1016/s0006-3223(99)00284-x. [DOI] [PubMed] [Google Scholar]
- Li X, Need AB, Baez M, Witkin JM. Metabotropic glutamate 5 receptor antagonism is associated with antidepressant-like effects in mice. J Pharmacol Exp Ther. 2006;319:254–9. doi: 10.1124/jpet.106.103143. [DOI] [PubMed] [Google Scholar]
- Li X, Witkin JM, Need AB, Skolnick P. Enhancement of antidepressant potency by a potentiator of AMPA receptors. Cell Mol Neurobiol. 2003;23:419–30. doi: 10.1023/A:1023648923447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebrenz M, Stohler R, Borgeat A. Repeated intravenous ketamine therapy in a patient with treatment-resistant major depression. World J Biol Psychiatry. 2007:1–4. doi: 10.1080/15622970701420481. [DOI] [PubMed] [Google Scholar]
- Lynch G. AMPA receptor modulators as cognitive enhancers. Curr Opin Pharmacol. 2004;4:4–11. doi: 10.1016/j.coph.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Machado-Vieira R, Salvadore G, Luckenbaugh DA, Manji HK, Zarate CA., Jr. Rapid onset of antidepressant action: a new paradigm in the research and treatment of major depressive disorder. J Clin Psychiatry. 2008;69:946–58. doi: 10.4088/jcp.v69n0610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado-Vieira R, Yuan P, Brutsche N, DiazGranados N, Luckenbaugh D, Manji HK, Zarate CA. Brain derived neurotrophic factor and initial antidepressant response to an NMDA antagonist. J Clin Psychiatry. doi: 10.4088/JCP.08m04659. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeng S, Zarate CA., Jr. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9:467–74. doi: 10.1007/s11920-007-0063-1. [DOI] [PubMed] [Google Scholar]
- Maeng S, Zarate CA, Jr., Du J, Schloesser RJ, McCammon J, Chen G, Manji HK. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63:349–52. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Maiese K, Vincent A, Lin SH, Shaw T. Group I and group III metabotropic glutamate receptor subtypes provide enhanced neuroprotection. J Neurosci Res. 2000;62:257–72. doi: 10.1002/1097-4547(20001015)62:2<257::AID-JNR10>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Manji HK, Lenox RH. The nature of bipolar disorder. J Clin Psychiatry. 2000;61(Supp 13):42–57. [PubMed] [Google Scholar]
- Marti SB, Cichon S, Propping P, Nothen M. Metabotropic glutamate receptor 3 (GRM3) gene variation is not associated with schizophrenia or bipolar affective disorder in the German population. Am J Med Genet. 2002;114:46–50. doi: 10.1002/ajmg.1624. [DOI] [PubMed] [Google Scholar]
- Martinez-Turrillas R, Del Rio J, Frechilla D. Neuronal proteins involved in synaptic targeting of AMPA receptors in rat hippocampus by antidepressant drugs. Biochem Biophys Res Commun. 2007;353:750–5. doi: 10.1016/j.bbrc.2006.12.078. [DOI] [PubMed] [Google Scholar]
- Martucci L, Wong AH, De Luca V, Likhodi O, Wong GW, King N, Kennedy JL. N-methyl-D-aspartate receptor NR2B subunit gene GRIN2B in schizophrenia and bipolar disorder: Polymorphisms and mRNA levels. Schizophr Res. 2006;84:214–21. doi: 10.1016/j.schres.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Mathew SJ, Amiel JM, Coplan JD, Fitterling HA, Sackeim HA, Gorman JM. Open-label trial of riluzole in generalized anxiety disorder. Am J Psychiatry. 2005;162:2379–81. doi: 10.1176/appi.ajp.162.12.2379. [DOI] [PubMed] [Google Scholar]
- Mauri MC, Ferrara A, Boscati L, Bravin S, Zamberlan F, Alecci M, Invernizzi G. Plasma and platelet amino acid concentrations in patients affected by major depression and under fluvoxamine treatment. Neuropsychobiology. 1998;37:124–9. doi: 10.1159/000026491. [DOI] [PubMed] [Google Scholar]
- Mayberg HS, Brannan SK, Mahurin RK, Jerabek PA, Brickman JS, Tekell JL, Silva JA, McGinnis S, Glass TG, Martin CC. Cingulate function in depression: a potential predictor of treatment response. Neuroreport. 1997;8:1057–61. doi: 10.1097/00001756-199703030-00048. others. [DOI] [PubMed] [Google Scholar]
- Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C, Schwalb JM, Kennedy SH. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45:651–60. doi: 10.1016/j.neuron.2005.02.014. [DOI] [PubMed] [Google Scholar]
- McCormick LM, Ponto LL Boles, Pierson RK, Johnson HJ, Magnotta V, Brumm MC. Metabolic correlates of antidepressant and antipsychotic response in patients with psychotic depression undergoing electroconvulsive therapy. J ECT. 2007;23:265–73. doi: 10.1097/yct.0b013e318150d56d. [DOI] [PubMed] [Google Scholar]
- McCullumsmith RE, Kristiansen LV, Beneyto M, Scarr E, Dean B, Meador-Woodruff JH. Decreased NR1, NR2A, and SAP102 transcript expression in the hippocampus in bipolar disorder. Brain Res. 2007;1127:108–18. doi: 10.1016/j.brainres.2006.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullumsmith RE, Meador-Woodruff JH. Striatal excitatory amino acid transporter transcript expression in schizophrenia, bipolar disorder, and major depressive disorder. Neuropsychopharmacology. 2002;26:368–75. doi: 10.1016/S0893-133X(01)00370-0. [DOI] [PubMed] [Google Scholar]
- Meador-Woodruff JH, Hogg AJ, Jr., Smith RE. Striatal ionotropic glutamate receptor expression in schizophrenia, bipolar disorder, and major depressive disorder. Brain Res Bull. 2001;55:631–40. doi: 10.1016/s0361-9230(01)00523-8. [DOI] [PubMed] [Google Scholar]
- Meloni D, Gambarana C, De Montis MG, Dal Pra P, Taddei I, Tagliamonte A. Dizocilpine antagonizes the effect of chronic imipramine on learned helplessness in rats. Pharmacol Biochem Behav. 1993;46:423–6. doi: 10.1016/0091-3057(93)90374-3. [DOI] [PubMed] [Google Scholar]
- Mickley GA, Schaldach MA, Snyder KJ, Balogh SA, Len T, Neimanis K, Goulis P, Hug J, Sauchak K, Remmers-Roeber DR. Ketamine blocks a conditioned taste aversion (CTA) in neonatal rats. Physiol Behav. 1998;64:381–90. doi: 10.1016/s0031-9384(98)00097-3. others. [DOI] [PubMed] [Google Scholar]
- Miller TM, Cleveland DW. Medicine. Treating neurodegenerative diseases with antibiotics. Science. 2005;307:361–2. doi: 10.1126/science.1109027. [DOI] [PubMed] [Google Scholar]
- Mineur YS, Picciotto MR, Sanacora G. Antidepressant-like effects of ceftriaxone in male C57BL/6J mice. Biol Psychiatry. 2007;61:250–2. doi: 10.1016/j.biopsych.2006.04.037. [DOI] [PubMed] [Google Scholar]
- Mitani H, Shirayama Y, Yamada T, Maeda K, Ashby CR, Jr., Kawahara R. Correlation between plasma levels of glutamate, alanine and serine with severity of depression. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:1155–8. doi: 10.1016/j.pnpbp.2006.03.036. [DOI] [PubMed] [Google Scholar]
- Miu P, Jarvie KR, Radhakrishnan V, Gates MR, Ogden A, Ornstein PL, Zarrinmayeh H, Ho K, Peters D, Grabell J. Novel AMPA receptor potentiators LY392098 and LY404187: effects on recombinant human AMPA receptors in vitro. Neuropharmacology. 2001;40:976–83. doi: 10.1016/s0028-3908(01)00027-2. others. [DOI] [PubMed] [Google Scholar]
- Mizuta I, Ohta M, Ohta K, Nishimura M, Mizuta E, Kuno S. Riluzole stimulates nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor synthesis in cultured mouse astrocytes. Neurosci Lett. 2001;310:117–20. doi: 10.1016/s0304-3940(01)02098-5. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–7. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundo E, Tharmalingham S, Neves-Pereira M, Dalton EJ, Macciardi F, Parikh SV, Bolonna A, Kerwin RW, Arranz MJ, Makoff AJ. Evidence that the N-methyl-D-aspartate subunit 1 receptor gene (GRIN1) confers susceptibility to bipolar disorder. Mol Psychiatry. 2003;8:241–5. doi: 10.1038/sj.mp.4001218. others. [DOI] [PubMed] [Google Scholar]
- Murray CJ, Lopez AD. Evidence-based health policy--lessons from the Global Burden of Disease Study. Science. 1996;274:740–3. doi: 10.1126/science.274.5288.740. [DOI] [PubMed] [Google Scholar]
- Nudmamud-Thanoi S, Reynolds GP. The NR1 subunit of the glutamate/NMDA receptor in the superior temporal cortex in schizophrenia and affective disorders. Neurosci Lett. 2004;372:173–7. doi: 10.1016/j.neulet.2004.09.035. [DOI] [PubMed] [Google Scholar]
- O’Neill MJ, Bleakman D, Zimmerman DM, Nisenbaum ES. AMPA receptor potentiators for the treatment of CNS disorders. Curr Drug Targets CNS Neurol Disord. 2004;3:181–94. doi: 10.2174/1568007043337508. [DOI] [PubMed] [Google Scholar]
- Ostroff R, Gonzales M, Sanacora G. Antidepressant effect of ketamine during ECT. Am J Psychiatry. 2005;162:1385–6. doi: 10.1176/appi.ajp.162.7.1385. [DOI] [PubMed] [Google Scholar]
- Paddock S, Laje G, Charney D, Rush AJ, Wilson AF, Sorant AJ, Lipsky R, Wisniewski SR, Manji H, McMahon FJ. Association of GRIK4 with outcome of antidepressant treatment in the STAR*D cohort. Am J Psychiatry. 2007;164:1181–8. doi: 10.1176/appi.ajp.2007.06111790. [DOI] [PubMed] [Google Scholar]
- Padovan CM, Guimaraes FS. Antidepressant-like effects of NMDA-receptor antagonist injected into the dorsal hippocampus of rats. Pharmacol Biochem Behav. 2004;77:15–9. doi: 10.1016/j.pbb.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Palucha A, Tatarczynska E, Branski P, Szewczyk B, Wieronska JM, Klak K, Chojnacka-Wojcik E, Nowak G, Pilc A. Group III mGlu receptor agonists produce anxiolytic- and antidepressant-like effects after central administration in rats. Neuropharmacology. 2004;46:151–9. doi: 10.1016/j.neuropharm.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Paul IA, Skolnick P. Glutamate and depression: clinical and preclinical studies. Ann N Y Acad Sci. 2003;1003:250–72. doi: 10.1196/annals.1300.016. [DOI] [PubMed] [Google Scholar]
- Perry EB, Jr., Cramer JA, Cho HS, Petrakis IL, Karper LP, Genovese A, O’Donnell E, Krystal JH, D’Souza DC. Psychiatric safety of ketamine in psychopharmacology research. Psychopharmacology (Berl) 2007;192:253–60. doi: 10.1007/s00213-007-0706-2. [DOI] [PubMed] [Google Scholar]
- Petrakis IL, Limoncelli D, Gueorguieva R, Jatlow P, Boutros NN, Trevisan L, Gelernter J, Krystal JH. Altered NMDA glutamate receptor antagonist response in individuals with a family vulnerability to alcoholism. Am J Psychiatry. 2004;161:1776–82. doi: 10.1176/ajp.161.10.1776. [DOI] [PubMed] [Google Scholar]
- Petrenko AB, Yamakura T, Fujiwara N, Askalany AR, Baba H, Sakimura K. Reduced sensitivity to ketamine and pentobarbital in mice lacking the N-methyl-D-aspartate receptor GluRepsilon1 subunit. Anesth Analg. 2004;99:1136–40. doi: 10.1213/01.ANE.0000131729.54986.30. table of contents. [DOI] [PubMed] [Google Scholar]
- Phelps LE, Brutsche N, Moral JR, Luckenbaugh DA, Manji HK, Zarate CA., Jr. Family History of Alcohol Dependence and Initial Antidepressant Response to an N-methyl-D-aspartate Antagonist. Biol Psychiatry. 2008 doi: 10.1016/j.biopsych.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard BS, Malloy MP, Christoforou A, Thomson PA, Evans KL, Morris SW, Hampson M, Porteous DJ, Blackwood DH, Muir WJ. Cytogenetic and genetic evidence supports a role for the kainate-type glutamate receptor gene, GRIK4, in schizophrenia and bipolar disorder. Mol Psychiatry. 2006;11:847–57. doi: 10.1038/sj.mp.4001867. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Duman RS. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology. 2008;33:88–109. doi: 10.1038/sj.npp.1301574. [DOI] [PubMed] [Google Scholar]
- Pizzagalli D, Pascual-Marqui RD, Nitschke JB, Oakes TR, Larson CL, Abercrombie HC, Schaefer SM, Koger JV, Benca RM, Davidson RJ. Anterior cingulate activity as a predictor of degree of treatment response in major depression: evidence from brain electrical tomography analysis. Am J Psychiatry. 2001;158:405–15. doi: 10.1176/appi.ajp.158.3.405. [DOI] [PubMed] [Google Scholar]
- Porter RH, Jaeschke G, Spooren W, Ballard TM, Buttelmann B, Kolczewski S, Peters JU, Prinssen E, Wichmann J, Vieira E. Fenobam: a clinically validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J Pharmacol Exp Ther. 2005;315:711–21. doi: 10.1124/jpet.105.089839. others. [DOI] [PubMed] [Google Scholar]
- Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol. 2008;28:631–7. doi: 10.1097/JCP.0b013e31818a6cea. [DOI] [PubMed] [Google Scholar]
- Rajkowska G, Miguel-Hidalgo JJ. Gliogenesis and glial pathology in depression. CNS Neurol Disord Drug Targets. 2007;6:219–33. doi: 10.2174/187152707780619326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA. Diverse mechanisms of antiepileptic drugs in the development pipeline. Epilepsy Res. 2006;69:273–94. doi: 10.1016/j.eplepsyres.2006.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Hoberg M Dykes, Vidensky S, Chung DS. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–7. doi: 10.1038/nature03180. others. [DOI] [PubMed] [Google Scholar]
- Salvadore G, Cornwell BR, Colon-Rosario V, Coppola R, Grillon C, Zarate CA, Jr., Manji HK. Increased Anterior Cingulate Cortical Activity in Response to Fearful Faces: A Neurophysiological Biomarker that Predicts Rapid Antidepressant Response to Ketamine. Biol Psychiatry. 2008 doi: 10.1016/j.biopsych.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL, Krystal JH, Mason GF. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry. 2004;61:705–13. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Kendell SF, Levin Y, Simen AA, Fenton LR, Coric V, Krystal JH. Preliminary evidence of riluzole efficacy in antidepressant-treated patients with residual depressive symptoms. Biol Psychiatry. 2007;61:822–5. doi: 10.1016/j.biopsych.2006.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;7:426–37. doi: 10.1038/nrd2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Brody AL, Ho ML, Zohrabi N, Maidment KM, Baxter LR., Jr. Differential brain metabolic predictors of response to paroxetine in obsessive-compulsive disorder versus major depression. Am J Psychiatry. 2003;160:522–32. doi: 10.1176/appi.ajp.160.3.522. [DOI] [PubMed] [Google Scholar]
- Scarr E, Pavey G, Sundram S, MacKinnon A, Dean B. Decreased hippocampal NMDA, but not kainate or AMPA receptors in bipolar disorder. Bipolar Disord. 2003;5:257–64. doi: 10.1034/j.1399-5618.2003.00024.x. [DOI] [PubMed] [Google Scholar]
- Schiffer HH, Heinemann SF. Association of the human kainate receptor GluR7 gene (GRIK3) with recurrent major depressive disorder. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:20–6. doi: 10.1002/ajmg.b.30374. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Wright RA, Levine LR, Gaydos B, Potter WZ. LY354740, an mGlu2/3 receptor agonist as a novel approach to treat anxiety/stress. Stress. 2003;6:189–97. doi: 10.1080/1025389031000146773. [DOI] [PubMed] [Google Scholar]
- Schumann G, Johann M, Frank J, Preuss U, Dahmen N, Laucht M, Rietschel M, Rujescu D, Lourdusamy A, Clark TK. Systematic analysis of glutamatergic neurotransmission genes in alcohol dependence and adolescent risky drinking behavior. Arch Gen Psychiatry. 2008;65:826–838. doi: 10.1001/archpsyc.65.7.826. others. [DOI] [PubMed] [Google Scholar]
- Shaltiel G, Maeng S, Malkesman O, Pearson B, Schloesser RJ, Tragon T, Rogawski M, Gasior M, Luckenbaugh D, Chen G. Evidence for the involvement of the kainate receptor subunit GluR6 (GRIK2) in mediating behavioral displays related to behavioral symptoms of mania. Mol Psychiatry. 2008;13:858–72. doi: 10.1038/mp.2008.20. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestre JS, Nadal R, Pallares M, Ferre N. Acute effects of ketamine in the holeboard, the elevated-plus maze, and the social interaction test in Wistar rats. Depress Anxiety. 1997;5:29–33. [PubMed] [Google Scholar]
- Skolnick P, Miller R, Young A, Boje K, Trullas R. Chronic treatment with 1-aminocyclopropanecarboxylic acid desensitizes behavioral responses to compounds acting at the N-methyl-D-aspartate receptor complex. Psychopharmacology (Berl) 1992;107:489–96. doi: 10.1007/BF02245261. [DOI] [PubMed] [Google Scholar]
- Soriano FX, Hardingham GE. Compartmentalized NMDA receptor signalling to survival and death. J Physiol. 2007;584:381–7. doi: 10.1113/jphysiol.2007.138875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thase ME, Haight BR, Richard N, Rockett CB, Mitton M, Modell JG, VanMeter S, Harriett AE, Wang Y. Remission rates following antidepressant therapy with bupropion or selective serotonin reuptake inhibitors: a meta-analysis of original data from 7 randomized controlled trials. J Clin Psychiatry. 2005;66:974–81. doi: 10.4088/jcp.v66n0803. [DOI] [PubMed] [Google Scholar]
- Toro C, Deakin JF. NMDA receptor subunit NRI and postsynaptic protein PSD-95 in hippocampus and orbitofrontal cortex in schizophrenia and mood disorder. Schizophr Res. 2005;80:323–30. doi: 10.1016/j.schres.2005.07.003. [DOI] [PubMed] [Google Scholar]
- U. S. Department of Health and Human Services . Mental health: A report of the Surgeon General. Department of Health and Human Services, Center for Mental Health Services, National Institutes of Health, National Institute of Mental Health (Department of Health and Human Services; Rockville, Maryland: 2000. [Google Scholar]
- Weis S, Llenos IC, Dulay JR, Verma N, Sabunciyan S, Yolken RH. Changes in region- and cell type-specific expression patterns of neutral amino acid transporter 1 (ASCT-1) in the anterior cingulate cortex and hippocampus in schizophrenia, bipolar disorder and major depression. J Neural Transm. 2007;114:261–71. doi: 10.1007/s00702-006-0544-0. [DOI] [PubMed] [Google Scholar]
- Wieronska JM, Legutko B, Dudys D, Pilc A. Olfactory bulbectomy and amitriptyline treatment influences mGlu receptors expression in the mouse brain hippocampus. Pharmacol Rep. 2008;60:844–55. [PubMed] [Google Scholar]
- Wieronska JM, Szewczyk B, Branski P, Palucha A, Pilc A. Antidepressant-like effect of MPEP, a potent, selective and systemically active mGlu5 receptor antagonist in the olfactory bulbectomized rats. Amino Acids. 2002;23:213–6. doi: 10.1007/s00726-001-0131-5. [DOI] [PubMed] [Google Scholar]
- Wilson GM, Flibotte S, Chopra V, Melnyk BL, Honer WG, Holt RA. DNA copy-number analysis in bipolar disorder and schizophrenia reveals aberrations in genes involved in glutamate signaling. Hum Mol Genet. 2006;15:743–9. doi: 10.1093/hmg/ddi489. [DOI] [PubMed] [Google Scholar]
- Woo TU, Walsh JP, Benes FM. Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-D-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch Gen Psychiatry. 2004;61:649–57. doi: 10.1001/archpsyc.61.7.649. [DOI] [PubMed] [Google Scholar]
- Wu J, Buchsbaum MS, Gillin JC, Tang C, Cadwell S, Wiegand M, Najafi A, Klein E, Hazen K, Bunney WE., Jr. Prediction of antidepressant effects of sleep deprivation by metabolic rates in the ventral anterior cingulate and medial prefrontal cortex. Am J Psychiatry. 1999;156:1149–58. doi: 10.1176/ajp.156.8.1149. others. [DOI] [PubMed] [Google Scholar]
- Yildiz-Yesiloglu A, Ankerst DP. Neurochemical alterations of the brain in bipolar disorder and their implications for pathophysiology: a systematic review of the in vivo proton magnetic resonance spectroscopy findings. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:969–95. doi: 10.1016/j.pnpbp.2006.03.012. [DOI] [PubMed] [Google Scholar]
- Yoon SJ, Lyoo IK, Haws C, Kim TS, Cohen BM, Renshaw PF. Decreased Glutamate/Glutamine Levels May Mediate Cytidine’s Efficacy in Treating Bipolar Depression: A Longitudinal Proton Magnetic Resonance Spectroscopy Study. Neuropsychopharmacology. 2009 doi: 10.1038/npp.2009.2. [DOI] [PubMed] [Google Scholar]
- Yoshimizu T, Chaki S. Increased cell proliferation in the adult mouse hippocampus following chronic administration of group II metabotropic glutamate receptor antagonist, MGS0039. Biochem Biophys Res Commun. 2004;315:493–6. doi: 10.1016/j.bbrc.2004.01.073. [DOI] [PubMed] [Google Scholar]
- Yoshimizu T, Shimazaki T, Ito A, Chaki S. An mGluR2/3 antagonist, MGS0039, exerts antidepressant and anxiolytic effects in behavioral models in rats. Psychopharmacology (Berl) 2006;186:587–93. doi: 10.1007/s00213-006-0390-7. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Jr., Du J, Quiroz J, Gray NA, Denicoff KD, Singh J, Charney DS, Manji HK. Regulation of cellular plasticity cascades in the pathophysiology and treatment of mood disorders: role of the glutamatergic system. Ann N Y Acad Sci. 2003;1003:273–91. doi: 10.1196/annals.1300.017. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Jr., Payne JL, Quiroz J, Sporn J, Denicoff KK, Luckenbaugh D, Charney DS, Manji HK. An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry. 2004;161:171–4. doi: 10.1176/appi.ajp.161.1.171. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Jr., Quiroz JA, Singh JB, Denicoff KD, De Jesus G, Luckenbaugh DA, Charney DS, Manji HK. An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry. 2005;57:430–2. doi: 10.1016/j.biopsych.2004.11.023. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Jr., Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006a;63:856–64. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Jr., Singh JB, Quiroz JA, De Jesus G, Denicoff KK, Luckenbaugh DA, Manji HK, Charney DS. A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J Psychiatry. 2006b;163:153–5. doi: 10.1176/appi.ajp.163.1.153. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Quiroz J, Payne J, Manji HK. Modulators of the glutamatergic system: implications for the development of improved therapeutics in mood disorders. Psychopharmacol Bull. 2002;36:35–83. [PubMed] [Google Scholar]